Novel Comprehensive Bioinformatics Approaches to Determine the Molecular Genetic Susceptibility Profile of Moderate and Severe Asthma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. DEG Identification
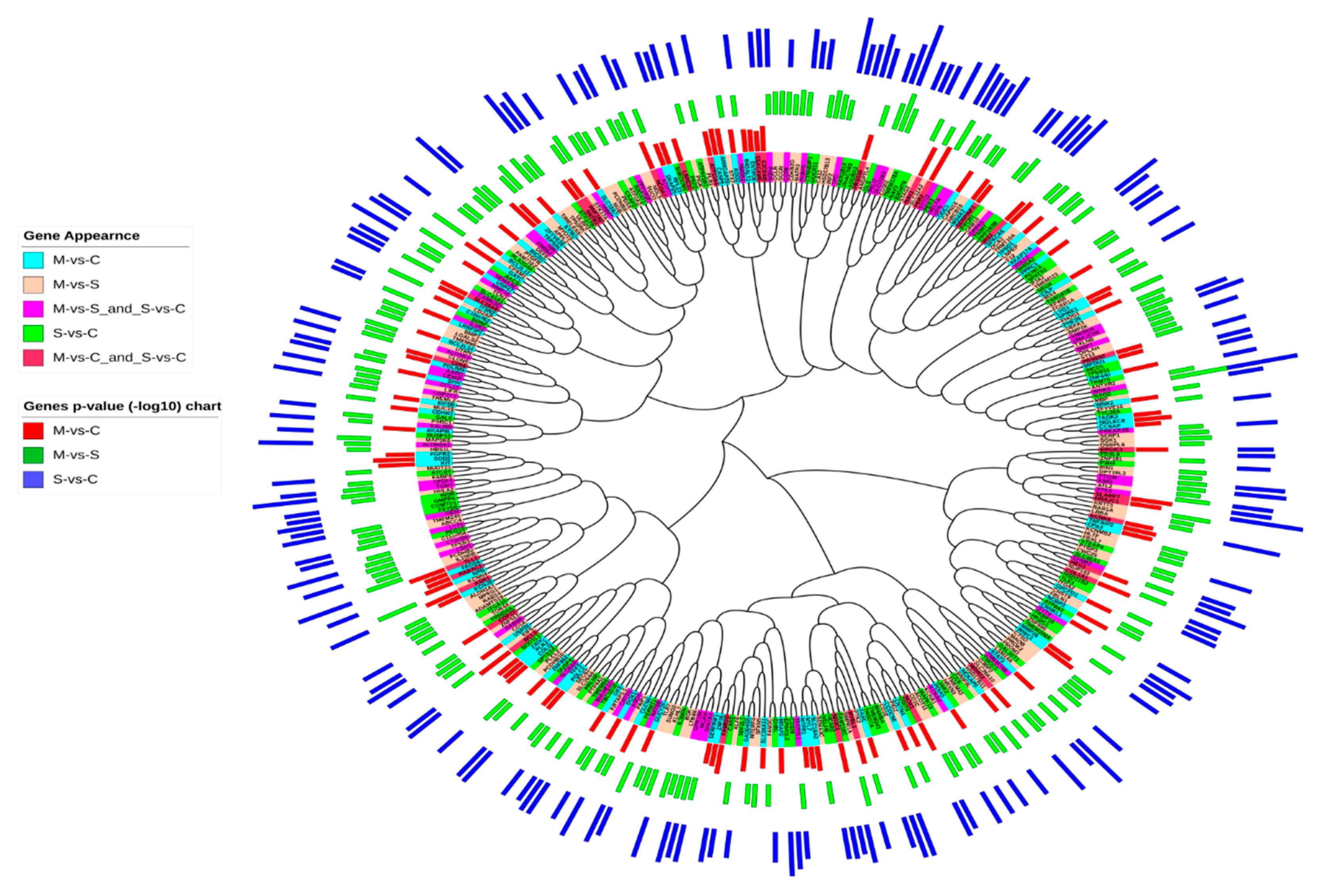
2.2. Pathogenic SNP Analysis among DEGs in Asthmatic Patients
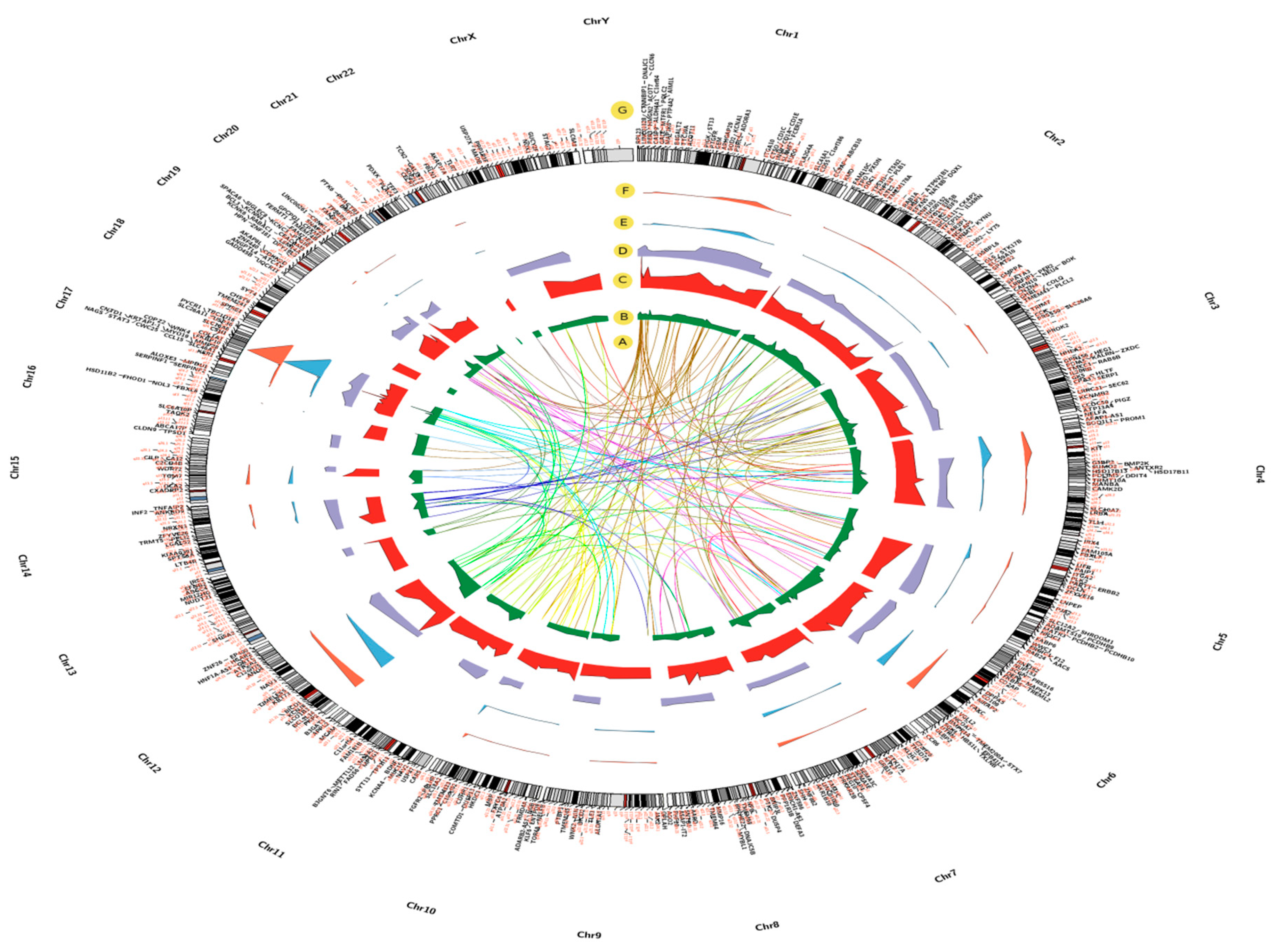
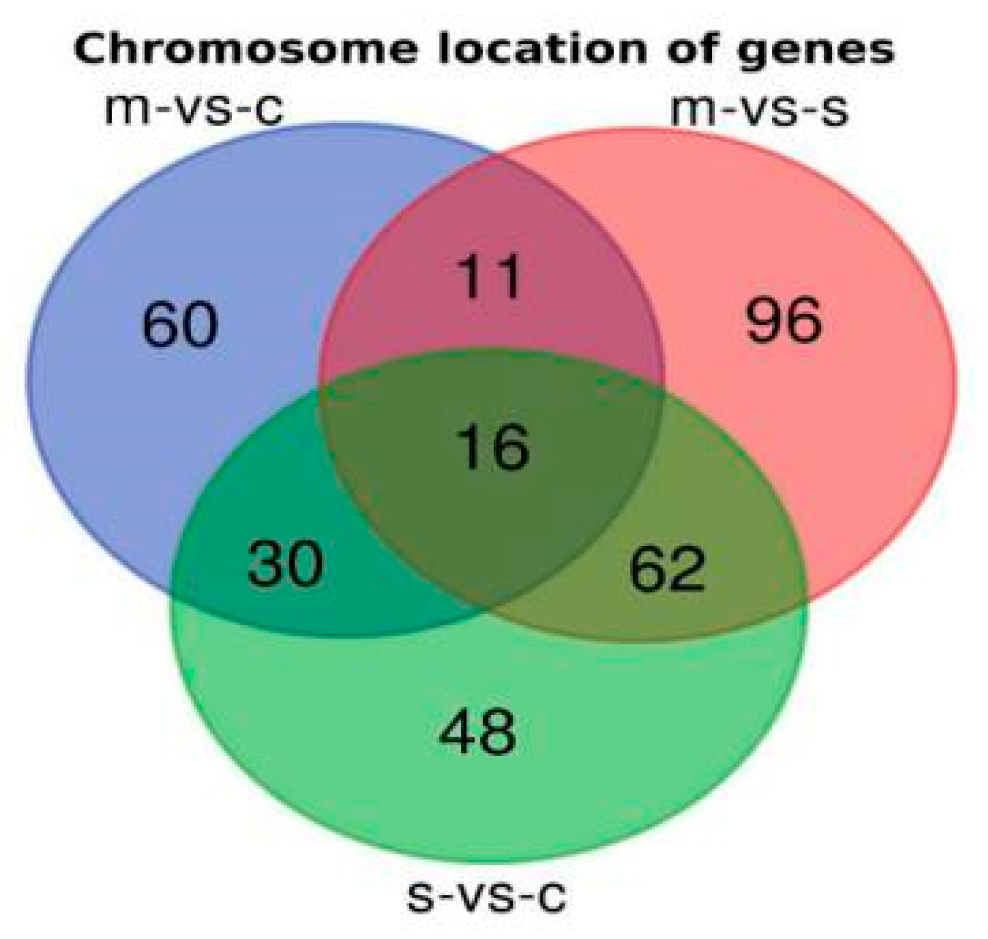
2.3. Chromosomal Locations of DEGs among Asthma Patients
2.4. Sequence Similarities between DEGs in Asthmatic Patients
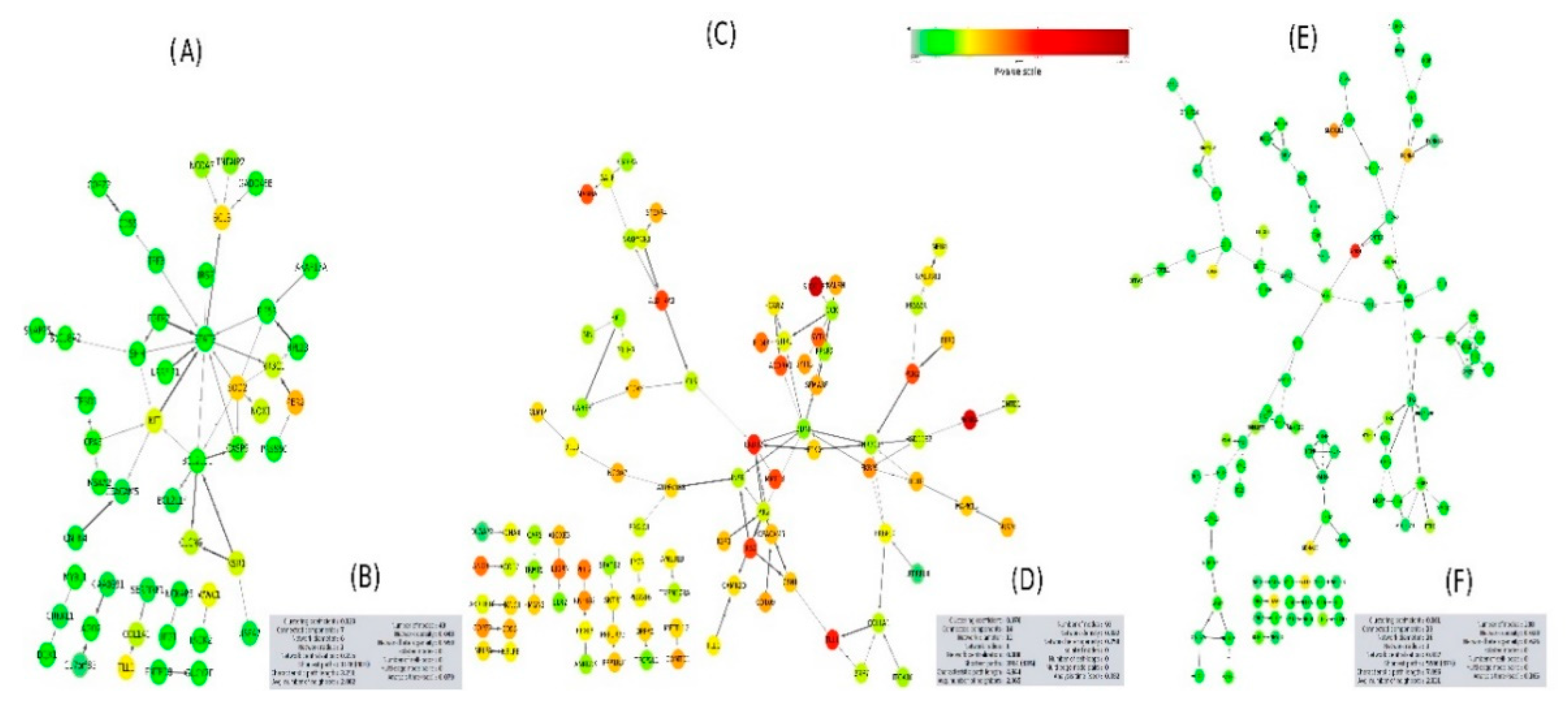
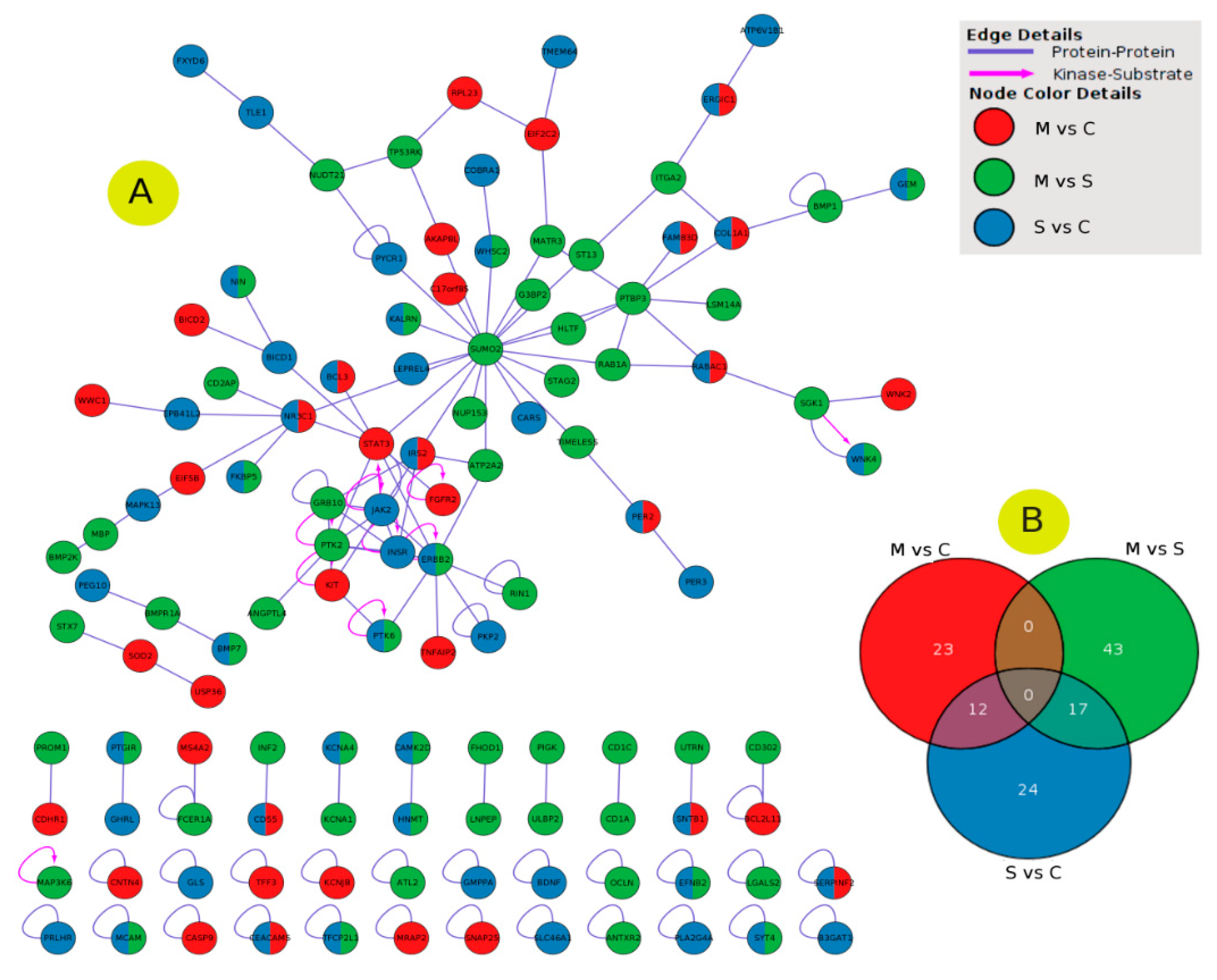
2.5. PPI Network Interaction
2.6. Enrichment Analysis
2.6.1. Comparative Pathways and Gene Ontology Process Analysis between Moderate and Severe Asthma Gene Sets
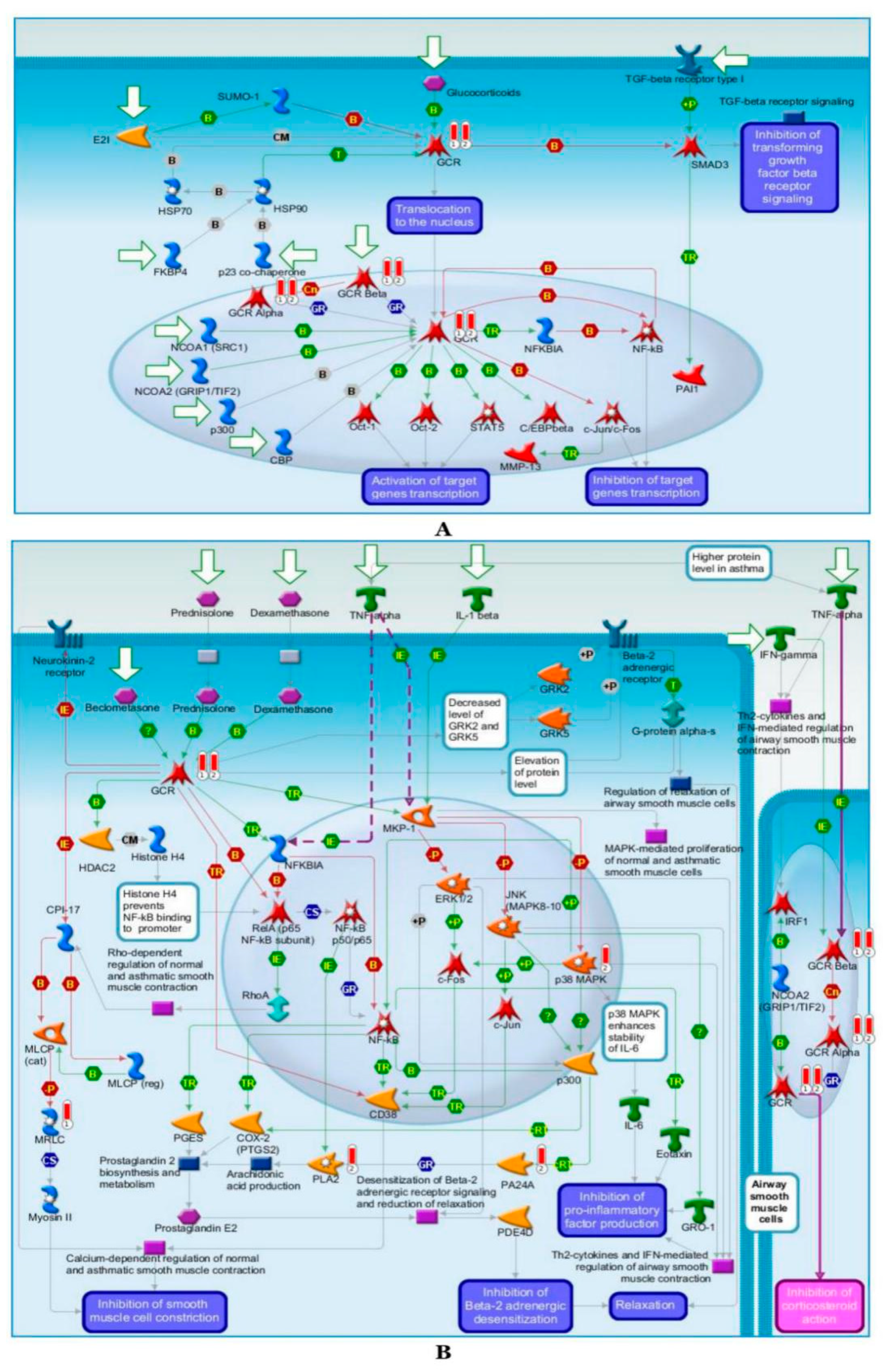
2.6.2. Pathway Map Analysis
2.6.3. Process Network Analysis
3. Discussion
4. Materials and Methods
4.1. Data Retrieval
4.2. Gene Ontology Enrichment and Protein–Protein Interaction Network Analysis
4.3. Genes and Single Nucleotide Polymorphism (SNP) Analysis
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Loftus, P.A.; Wise, S.K. Epidemiology of Asthma. Curr. Opin. Otolaryngol. Head Neck Surg. 2016, 24, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Illig, T.; Wjst, M. Genetics of asthma and related phenotypes. Paediatr. Respir. Rev. 2002, 3, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Ramratnam, S.K.; Bacharier, L.B.; Guilbert, T.W. Severe asthma in children. J. Allergy Clin. Immunol. Pract. 2017, 5, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.M.; Moore, W.C. Severe asthma phenotypes—how should they guide evaluation and treatment? J. Allergy Clin. Immunol. 2017, 5, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Raundhal, M.; Oriss, T.B.; Ray, P.; Wenzel, S.E. Current concepts of severe asthma. J. Clin. Investig. 2016, 126, 2394–2403. [Google Scholar] [CrossRef]
- Longo, G.; Strinati, R.; Poli, F.; Fumi, F. Genetic factors in nonspecific bronchial hyperreactivity. An epidemiologic study. Am. J. Dis. Child. 1987, 141, 331–334. [Google Scholar] [CrossRef]
- Sibbald, B.; E Horn, M.; Gregg, I. A family study of the genetic basis of asthma and wheezy bronchitis. Arch. Dis. Child. 1980, 55, 354–357. [Google Scholar] [CrossRef]
- Ramachandra, N.B.; Davoodi, P.; Mahesh, P.A.; Holla, A.D. Family history & the risk for adult onset asthma. Indian J. Med Res. 2015, 141, 361–363. [Google Scholar] [CrossRef]
- Murphy, T.; Wong, C.; Arseneault, L.; Burrage, J.; Macdonald, R.; Hannon, E.; Fisher, H.L.; Ambler, A.; Moffitt, T.E.; Caspi, A.; et al. Methylomic markers of persistent childhood asthma: A longitudinal study of asthma-discordant monozygotic twins. Clin. Epigenetics 2015, 7, 130. [Google Scholar] [CrossRef]
- Ullemar, V.; Magnusson, P.K.E.; Lundholm, C.; Zettergren, A.; Melén, E.; Lichtenstein, P.; Almqvist, C. Heritability and confirmation of genetic association studies for childhood asthma in twins. Allergy 2015, 71, 230–238. [Google Scholar] [CrossRef]
- Moffatt, M.F.; Gut, I.G.; Demenais, F.; Strachan, D.P.; Bouzigon, E.; Heath, S.; Von Mutius, E.; Farrall, M.; Lathrop, M.; Cookson, W.O.; et al. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 2010, 363, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Alrashoudi, R.H. Gene Expression Data Analysis Identifies Multiple Deregulated Pathways in Patients with Asthma, Bioscience Reports. Biosci. Rep. 2018, 38, BSR20180548. [Google Scholar] [CrossRef] [PubMed]
- Willis-Owen, S.A.; Cookson, W.O.; Moffatt, M.F. The Genetics and Genomics of Asthma. Annu. Rev. Genom. Hum. Genet. 2018, 19, 223–246. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, S.F. Genetics of asthma: An introduction for the clinician. Eur. Clin. Respir. J. 2015, 2, 215. [Google Scholar] [CrossRef] [PubMed]
- Bunyavanich, S.; Schadt, E.E. Systems biology of asthma and allergic diseases: A multiscale approach. J. Allergy Clin. Immunol. 2014, 135, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Son, S.-W.; Kim, S.C.; Kim, Y.J.; Jeong, H.; Lee, D.-J. A protein interaction network associated with asthma. J. Theor. Boil. 2008, 252, 722–731. [Google Scholar] [CrossRef]
- Sweeney, J.; Patterson, C.C.; Menzies-Gow, A.; Niven, R.M.; Mansur, A.H.; Bucknall, C.; Chaudhuri, R.; Price, D.; Brightling, C.E.; Heaney, L.G. Comorbidity in severe asthma requiring systemic corticosteroid therapy: Cross-sectional data from the Optimum Patient Care Research Database and the British Thoracic Difficult Asthma Registry. Thorax 2016, 71, 339–346. [Google Scholar] [CrossRef]
- Deokar, A.A.; Tar’An, B. Genome-Wide Analysis of the Aquaporin Gene Family in Chickpea (Cicer arietinum L.). Front. Plant Sci. 2016, 7, 1802. [Google Scholar] [CrossRef]
- Cowley, M.J.; Pinese, M.; Kassahn, K.; Waddell, N.; Pearson, J.V.; Grimmond, S.; Biankin, A.V.; Hautaniemi, S.; Wu, J. PINA v2.0: Mining interactome modules. Nucleic Acids Res. 2011, 40, D862–D865. [Google Scholar] [CrossRef]
- Galeone, C.; Scelfo, C.; Bertolini, F.; Caminati, M.; Ruggiero, P.; Facciolongo, N.; Menzella, F. Precision Medicine in Targeted Therapies for Severe Asthma: Is There Any Place for “Omics” Technology? BioMed Res. Int. 2018, 2018, 1–15. [Google Scholar] [CrossRef]
- Zayed, H. The Arab genome: Health and wealth. Gene 2016, 592, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Zayed, H. The Qatar genome project: Translation of whole-genome sequencing into clinical practice. Int. J. Clin. Pr. 2016, 70, 832–834. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, H.; Zuo, L.; Tao, A. Key genes and co-expression modules involved in asthma pathogenesis. PeerJ 2020, 8, e8456. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.F.; Eckols, T.; Tuvim, M.; Dickey, B.; Tweardy, D. Selective Small Molecule STAT3 Inhibitor to Treat Eosinophilic and Neutrophilic Asthma in Mice. In A21. Advances in Asthma Pathogenes; American Thoracic Society: New York, NY, USA, 2019; p. 7298. [Google Scholar] [CrossRef]
- Simeone-Penney, M.C.; Severgnini, M.; Tu, P.; Homer, R.; Mariani, T.J.; Cohn, L.; Simon, A.R. Airway epithelial STAT3 is required for allergic inflammation in a murine model of asthma. J. Immunol. 2007, 178, 6191–6199. [Google Scholar] [CrossRef]
- Yang, X.; Wang, F. The effect of astragaloside IV on JAK2-STAT6 signalling pathway in mouse model of ovalbumin-induced asthma. J. Anim. Physiol. Anim. Nutr. 2019, 103, 1578–1584. [Google Scholar] [CrossRef]
- Inoue, H. Dysfunctional ErbB2, an EGF receptor family member, in asthmatic airway epithelial cells critically hinders repair after mechanical wounding. J. Allergy Clin. Immunol. 2019, 143, 2075. [Google Scholar] [CrossRef]
- Yan, Y.-R. MiR-216a inhibits proliferation and promotes apoptosis of human airway smooth muscle cells by targeting JAK2. J. Asthma 2019, 56, 938–946. [Google Scholar] [CrossRef]
- Keskin, Ö.; Farzan, N.; Birben, E.; Akel, H.; Karaaslan, C.; Ahmadizar, F.; Wechsler, M.E.; Vijverberg, S.J.; Kalayci, O. Genetic associations of the response to inhaled corticosteroids in asthma: A systematic review. Clin. Transl. Allergy 2019, 9, 2. [Google Scholar] [CrossRef]
- Pascoe, C.D.; Obeidat, M.; Arsenault, B.A.; Nie, Y.; Warner, S.; Stefanowicz, D.; Wadsworth, S.J.; A Hirota, J.; Yang, S.J.; Dorscheid, D.R.; et al. Gene expression analysis in asthma using a targeted multiplex array. BMC Pulm. Med. 2017, 17, 189. [Google Scholar] [CrossRef]
- Panek, M.; Pietras, T.; Antczak, A.; Górski, P.; Kuna, P.; Szemraj, J. The role of functional single nucleotide polymorphisms of the human glucocorticoid receptor gene NR3C1 in Polish patients with bronchial asthma. Mol. Boil. Rep. 2011, 39, 4749–4757. [Google Scholar] [CrossRef]
- Tsai, M.-J. Common MicroRNA-mediated Pathobiological Changes in Bronchial Epithelial Cells of Both Asthma and Chronic Obstructive Pulmonary Diseases. In B32. Asthma: Mechanisma of Disease I; American Thoracic Society: New York, NY, USA, 2019; p. A2920. [Google Scholar]
- Wu, T.D.; Brigham, E.P.; Keet, C.A.; Brown, T.T.; Hansel, N.N.; McCormack, M.C. Association Between Prediabetes/Diabetes and Asthma Exacerbations in a Claims-Based Obese Asthma Cohort. J. Allergy Clin. Immunol. Pract. 2019, 7, 1868–1873.e5. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J. Networks of DNA Methylation Loci in Lung Tissue and Peripheral Blood Highlight Cross-Tissue Epigenetic Signatures of COPD. In C43. Copd and Population Health; American Thoracic Society: New York, NY, USA, 2019; p. A4865. [Google Scholar]
- Feng, H. Sample Size Estimation and Type I Error Correction in Genetic Association Studies; University of Idaho: Pocatello, ID, USA, 2016. [Google Scholar]
- Mohapatra, S.S.; Lima, J.J. Materials and Methods for Diagnosis of Asthma. U.S. Patent 20130029870A1, 2010. Available online: https://patents.google.com/patent/US20130029870A1/en (accessed on 10 March 2020).
- Jiang, X. The Emerging Role of MicroRNAs in Asthma, Molecular and Cellular Biochemistry; Springer: Berlin/Heidelberg, Germany, 2011; Volume 353, pp. 35–40. [Google Scholar]
- McCarthy, N.; Jones, H.A.; Marks, N.A.; Shiner, R.J.; Ind, P.W.; Al-Hassi, H.O.; English, N.R.; Murray, C.; Lambert, J.R.; Knight, S.C.; et al. Inhaled allergen-driven CD1c up-regulation and enhanced antigen uptake by activated human respiratory-tract dendritic cells in atopic asthma. Clin. Exp. Allergy 2007, 37, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Møller-Larsen, S.; Nyegaard, M.; Haagerup, A.; Vestbo, J.; A Kruse, T.; Børglum, A.D. Association analysis identifies TLR7 and TLR8 as novel risk genes in asthma and related disorders. Thorax 2008, 63, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Broekema, M.; Volbeda, F.; Timens, W.; Dijkstra, A.; Lee, N.; Lee, J.; Lodewijk, M.; Postma, D.; Hylkema, M.; Hacken, N.H.T.T. Airway eosinophilia in remission and progression of asthma: Accumulation with a fast decline of FEV1. Respir. Med. 2010, 104, 1254–1262. [Google Scholar] [CrossRef]
- Fu, G.; Fu, L.; Cai, Y.; Zhao, H.; Fu, W. Association between polymorphisms of glucocorticoid receptor genes and asthma: A meta-analysis. Cell. Mol. Boil. 2018, 64, 13–23. [Google Scholar] [CrossRef]
- Schofield, J.P.R.; Bigler, J.; Boedigheimer, M.; Affleck, K.; Taylor, A.; Pavlidis, S.; Riley, J.H.; Strazzeri, F.; Roberts, G.; Brandsma, J.; et al. Topological data analysis (TDA) of U-BIOPRED paediatric peripheral blood gene expression identified asthma phenotypes characterised by alternative splicing of glucocorticoid receptor (GR) mRNA. Paediatr. Asthma Allergy 2018, 52, PA5435. [Google Scholar] [CrossRef]
- Dautel, S.E.; Kyle, J.E.; Clair, G.; Sontag, R.L.; Weitz, K.K.; Shukla, A.K.; Nguyen, S.N.; Kim, Y.-M.; Zink, E.M.; Luders, T.; et al. Lipidomics reveals dramatic lipid compositional changes in the maturing postnatal lung. Sci. Rep. 2017, 7, 40555. [Google Scholar] [CrossRef]
- Hayakawa, M.; Ishida, N.; Takeuchi, K.; Shibamoto, S.; Hori, T.; Oku, N.; Ito, F.; Tsujimoto, M. Arachidonic acid-selective cytosolic phospholipase A2 is crucial in the cytotoxic action of tumor necrosis factor. J. Boil. Chem. 1993, 268, 11290–11295. [Google Scholar]
- Vermeulen, C.J.; Xu, C.-J.; Vonk, J.M.; Hacken, N.H.T.T.; Timens, W.; Heijink, I.H.; Nawijn, M.C.; Boekhoudt, J.; Van Oosterhout, A.J.; Affleck, K.; et al. Differential DNA methylation in bronchial biopsies between persistent asthma and asthma in remission. Eur. Respir. J. 2019, 55, 1901280. [Google Scholar] [CrossRef]
- Anathy, V. Airway Epithelium-Specific Deletion of Leptin Receptor Worsens Murine Asthma. In D101. Allergy and Asthma: Novel Regulatory Pathways; American Thoracic Society: New York, NY, USA, 2016; p. 7562. [Google Scholar]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. In Frontiers of Medicine; Springer: Berlin/Heidelberg, Germany, 2013; Volume 7, pp. 207–222. [Google Scholar]
- Hisada, T.; Aoki-Saito, H.; Koga, Y. Are specialized pro-resolving mediators promising therapeutic agents for severe bronchial asthma? J. Thorac. Dis. 2017, 9, 4266. [Google Scholar] [CrossRef]
- Tliba, O.; Amrani, Y. Airway Smooth Muscle Cell as an Inflammatory Cell. Proc. Am. Thorac. Soc. 2008, 5, 106–112. [Google Scholar] [CrossRef]
- Kim, D.W.; Chang, C.; Chen, X.; Doran, A.C.; Gaudreault, F.; Wager, T.; Demarco, G.J.; Kim, J.K. Systems approach reveals photosensitivity and PER 2 level as determinants of clock-modulator efficacy. Mol. Syst. Boil. 2019, 15, e8838. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, K.; Durrington, H.J. The Role of the Body Clock in Asthma and COPD: Implication for Treatment. Pulm. Ther. 2018, 4, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Ceriotti, S. Assessment of pulmonary vascular smooth muscle remodeling in severe equine asthma. Int. J. Health Anim. Sci. Food Saf. 2017, 4. [Google Scholar] [CrossRef]
- Sethi, G.S.; Dharwal, V.; Naura, A.S. Immunological Basis of Oxidative Stress-Induced Lung Inflammation in Asthma and COPD. In Oxidative Stress in Lung Diseases; Springer Science and Business Media LLC: Berlin, Germany, 2019; pp. 195–223. [Google Scholar]
- Wang, W.; Ji, H.-L. Epithelial Sodium and Chloride Channels and Asthma. Chin. Med. J. 2015, 128, 2242–2249. [Google Scholar] [CrossRef]
- Valverde, M.A.; Cantero-Recasens, G.; Vicente, R.; Jung, C.; Carreras-Sureda, A.; Vicente, R. Ion Channels in Asthma. J. Boil. Chem. 2011, 286, 32877–32882. [Google Scholar] [CrossRef]
- Van Der Velden, V.H. Glucocorticoids: Mechanisms of action and anti-inflammatory potential in asthma. Mediat. Inflamm. 1998, 7, 229–237. [Google Scholar] [CrossRef]
- Gagliardo, R.; Vignola, A.M.; Mathieu, M. Is there a role for glucocorticoid receptor beta in asthma? Respiratory research. BioMed Cent. 2000, 2, 1. [Google Scholar]
- Tohda, Y. Role of GABA receptors in the bronchial response: Studies in sensitized guinea-pigs. Clin. Exp. Allergy J. Br. Soc. Clin. Immunol. 1998, 28, 772–777. [Google Scholar] [CrossRef]
- Shrine, N.; A Portelli, M.; John, C.; Artigas, M.S.; Bennett, N.; Hall, R.; Lewis, J.; Henry, A.P.; Billington, C.K.; Ahmad, A.; et al. Moderate-to-severe asthma in individuals of European ancestry: A genome-wide association study. Lancet Respir. Med. 2018, 7, 20–34. [Google Scholar] [CrossRef]
- Johansson, A.; Rask-Andersen, M.; Karlsson, T.; E Ek, W. Genome-wide association analysis of 350 000 Caucasians from the UK Biobank identifies novel loci for asthma, hay fever and eczema. Hum. Mol. Genet. 2019, 28, 4022–4041. [Google Scholar] [CrossRef] [PubMed]
- Pividori, M.; Schoettler, N.; Nicolae, D.L.; Ober, C.; Im, H.K. Shared and distinct genetic risk factors for childhood-onset and adult-onset asthma: Genome-wide and transcriptome-wide studies. Lancet Respir. Med. 2019, 7, 509–522. [Google Scholar] [CrossRef]
- Tantisira, K.G.; Lazarus, R.; Litonjua, A.; Klanderman, B.; Weiss, S.T. Chromosome 17: Association of a large inversion polymorphism with corticosteroid response in asthma. Pharm. Genom. 2008, 18, 733–737. [Google Scholar] [CrossRef]
- Ylä-Anttila, P.; Eskelinen, E.-L. Roles for RAB24 in autophagy and disease. Small GTPases 2017, 9, 57–65. [Google Scholar] [CrossRef]
- Voraphani, N. An airway epithelial Inos-DUOX2-thyroid peroxidase metabolome drives Th1/Th2 nitrative stress in human severe asthma. In Mucosal Immunology; Nature Publishing Group: Berlin, Germany, 2014; Volume 7, p. 1175. [Google Scholar]
- Barrett, T. NCBI GEO: Archive for Functional Genomics Data Sets—Update, Nucleic Acids Research; Oxford University Press: Oxford, UK, 2012; Volume 41, pp. D991–D995. [Google Scholar]
- Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources, Nature Protocols; Nature Publishing Group: London, UK, 2009; Volume 4, pp. 44–57. [Google Scholar]
- Szklarczyk, D.; Morris, J.H.; Cook, H.V.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2006, 23, 127–128. [Google Scholar] [CrossRef]
- Hubbard, T. The Ensembl Genome Database Project, Nucleic Acids Research; Oxford University Press: Oxford, UK, 2002; Volume 30, pp. 38–41. [Google Scholar]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J. ClinVar: Public Archive of Interpretations of Clinically Relevant Variants, Nucleic Acids Research; Oxford University Press: Oxford, UK, 2015; Volume 44, pp. D862–D868. [Google Scholar]
- Altschul, S.F. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. In Nucleic Acids Research; Oxford University Press: Oxford, UK, 1997; Volume 25, pp. 3389–3402. [Google Scholar]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.M.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]








© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayed, H. Novel Comprehensive Bioinformatics Approaches to Determine the Molecular Genetic Susceptibility Profile of Moderate and Severe Asthma. Int. J. Mol. Sci. 2020, 21, 4022. https://doi.org/10.3390/ijms21114022
Zayed H. Novel Comprehensive Bioinformatics Approaches to Determine the Molecular Genetic Susceptibility Profile of Moderate and Severe Asthma. International Journal of Molecular Sciences. 2020; 21(11):4022. https://doi.org/10.3390/ijms21114022
Chicago/Turabian StyleZayed, Hatem. 2020. "Novel Comprehensive Bioinformatics Approaches to Determine the Molecular Genetic Susceptibility Profile of Moderate and Severe Asthma" International Journal of Molecular Sciences 21, no. 11: 4022. https://doi.org/10.3390/ijms21114022
APA StyleZayed, H. (2020). Novel Comprehensive Bioinformatics Approaches to Determine the Molecular Genetic Susceptibility Profile of Moderate and Severe Asthma. International Journal of Molecular Sciences, 21(11), 4022. https://doi.org/10.3390/ijms21114022