Gut-Pancreas-Liver Axis as a Target for Treatment of NAFLD/NASH

 , and
, and 
Abstract
1. Introduction
2. Physiological Effects of Incretin Hormones
2.1. Incretin Secretion and Physiological Effects
2.2. Incretin Effects on Liver Enzymes, Steatosis, Inflammation and Fibrosis
2.3. Effect of GLP-1 and GLP-1RA on Hepatic Glucose and Lipid Metabolism
2.4. Impact of DPP-4 and Its Inhibition on NAFLD
2.5. GLP-1 Co-Agonists in the Treatment of Lipid Disorders and NAFLD/NASH
3. Gut Microbiota Dysbiosis and the Gut-Liver Axis in NAFLD
Alterations of Lipid and Glucose Metabolism by Gut Bacteria
4. Bile Acid Signaling in NAFLD
Targeting BA Receptors as a Therapeutic Option for NAFLD
5. Gut Permeability
5.1. Gut Permeability and Bacterial Translocations
5.2. Gut-Derived Inflammatory Mediators Triggering NAFLD/NASH
6. Targeting the Gut Permeability to Improve NAFLD/NASH
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALT | Alanine aminotransferase |
| AMPK | AMP-activated protein kinase |
| APRI | AST to platelet ratio index |
| AST | Aspartate aminotransferase |
| BA | Bile acids |
| BMI | Body mass index |
| CM | Chylomicron |
| CYP | Cytochrome P450 |
| DCA | Deoxycholic acid |
| DIO | Diet-induced obesity |
| DPP-4 | Dipeptidyl peptidase 4 |
| FXR | Farnesoid X receptor |
| FGF | Fibroblast growth factor |
| FFA | Free fatty acid |
| GCDCA | Glycolchenodeoxycholic acid |
| GIP | Glucose-dependent insulinotropic peptide |
| GLP-1 | Glucagon-like peptide-1 |
| GLP-1RA | GLP-1 receptor agonists |
| GPCR | G-protein-coupled receptors |
| HbA1c | Glycosylated hemoglobin |
| HCC | Hepatocellular carcinoma |
| HFD | High-fat diet |
| IL | Interleukin |
| LCA | Lithocholic acid |
| LPS | Lipopolysaccharide |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| OCA | Obeticholic acid |
| PPAR | Peroxisome proliferator-activated receptor |
| RLP | Remnant lipoprotein |
| SCFA | Short-chain fatty acids |
| STAT3 | Signal transducer and activator of transcription 3 |
| T2D | Type-2 diabetes |
| TCDCA | Taurochenodeoxycholic acid |
| TG | Triglycerides |
| T2D | Type-2 diabetes |
| TLR | Toll-Like Receptors |
| TNF | Tumor necrosis factor |
| VLDL | Very low-density lipoprotein particles |
| ZO-1 | Zonula occludens-1 |
References
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Tordjman, J.; Aron-Wisnewsky, J.; Poitou, C.; Oppert, J.M.; Torcivia, A.; Bouillot, J.L.; Paradis, V.; Ratziu, V.; Clement, K. Systematic review of bariatric surgery liver biopsies clarifies the natural history of liver disease in patients with severe obesity. Gut 2017, 66, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Souto, K.P.; Meinhardt, N.G.; Ramos, M.J.; Ulbrich-Kulkzynski, J.M.; Stein, A.T.; Damin, D.C. Nonalcoholic fatty liver disease in patients with different baseline glucose status undergoing bariatric surgery: Analysis of intraoperative liver biopsies and literature review. Surg. Obes. Relat. Dis. 2018, 14, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Sun, E.W.L.; Martin, A.M.; Young, R.L.; Keating, D.J. The Regulation of Peripheral Metabolism by Gut-Derived Hormones. Front. Endocrinol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Herrmann, C.; Goke, R.; Richter, G.; Fehmann, H.C.; Arnold, R.; Goke, B. Glucagon-like peptide-1 and glucose-dependent insulin-releasing polypeptide plasma levels in response to nutrients. Digestion 1995, 56, 117–126. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- McCaughan, G.W.; Gorrell, M.D.; Bishop, G.A.; Abbott, C.A.; Shackel, N.A.; McGuinness, P.H.; Levy, M.T.; Sharland, A.F.; Bowen, D.G.; Yu, D.; et al. Molecular pathogenesis of liver disease: An approach to hepatic inflammation, cirrhosis and liver transplant tolerance. Immunol. Rev. 2000, 174, 172–191. [Google Scholar] [CrossRef]
- Mentzel, S.; Dijkman, H.B.; Van Son, J.P.; Koene, R.A.; Assmann, K.J. Organ distribution of aminopeptidase A and dipeptidyl peptidase IV in normal mice. J. Histochem. Cytochem. 1996, 44, 445–461. [Google Scholar] [CrossRef]
- Kikuchi, M.; Fukuyama, K.; Epstein, W.L. Soluble dipeptidyl peptidase IV from terminal differentiated rat epidermal cells: Purification and its activity on synthetic and natural peptides. Arch. Biochem. Biophys. 1988, 266, 369–376. [Google Scholar] [CrossRef]
- Parker, H.E.; Reimann, F.; Gribble, F.M. Molecular mechanisms underlying nutrient-stimulated incretin secretion. Expert Rev. Mol. Med. 2010, 12, e1. [Google Scholar] [CrossRef] [PubMed]
- Fava, G.E.; Dong, E.W.; Wu, H. Intra-islet glucagon-like peptide 1. J. Diabetes Complicat. 2016, 30, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Dupre, J.; Ross, S.A.; Watson, D.; Brown, J.C. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J. Clin. Endocrinol. Metab. 1973, 37, 826–828. [Google Scholar] [CrossRef]
- Kreymann, B.; Williams, G.; Ghatei, M.A.; Bloom, S.R. Glucagon-like peptide-1 7-36: A physiological incretin in man. Lancet 1987, 2, 1300–1304. [Google Scholar] [CrossRef]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef]
- Holst, J.J. Glucagon-like peptide-1: From extract to agent. The Claude Bernard Lecture, 2005. Diabetologia 2006, 49, 253–260. [Google Scholar] [CrossRef]
- Nauck, M.A.; Heimesaat, M.M.; Behle, K.; Holst, J.J.; Nauck, M.S.; Ritzel, R.; Hüfner, M.; Schmiegel, W.H. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J. Clin. Endocrinol. Metab. 2002, 87, 1239–1246. [Google Scholar] [CrossRef]
- Meier, J.J.; Gallwitz, B.; Siepmann, N.; Holst, J.J.; Deacon, C.F.; Schmidt, W.E.; Nauck, M.A. Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 2003, 46, 798–801. [Google Scholar] [CrossRef]
- Vilsboll, T.; Krarup, T.; Madsbad, S.; Holst, J.J. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul. Pept. 2003, 114, 115–121. [Google Scholar] [CrossRef]
- Nauck, M.A.; Bartels, E.; Orskov, C.; Ebert, R.; Creutzfeldt, W. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7-36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J. Clin. Endocrinol. Metab. 1993, 76, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: Physiology, pathophysiology, and response to therapeutic interventions. Lancet. Diabetes Endocrinol. 2016, 4, 525–536. [Google Scholar] [CrossRef]
- Nauck, M.A.; Heimesaat, M.M.; Orskov, C.; Holst, J.J.; Ebert, R.; Creutzfeldt, W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Investig. 1993, 91, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Gaggini, M.; Daniele, G.; Ciociaro, D.; Cersosimo, E.; Tripathy, D.; Triplitt, C.; Fox, P.; Musi, N.; DeFronzo, R.; et al. Exenatide improves both hepatic and adipose tissue insulin resistance: A dynamic positron emission tomography study. Hepatology 2016, 64, 2028–2037. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Wollschlager, D.; Werner, J.; Holst, J.J.; Orskov, C.; Creutzfeldt, W.; Willms, B. Effects of subcutaneous glucagon-like peptide 1 (GLP-1 [7-36 amide]) in patients with NIDDM. Diabetologia 1996, 39, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Daniele, G.; Iozzo, P.; Molina-Carrion, M.; Lancaster, J.; Ciociaro, D.; Cersosimo, E.; Tripathy, D.; Triplitt, C.; Fox, P.; Musi, N.; et al. Exenatide Regulates Cerebral Glucose Metabolism in Brain Areas Associated With Glucose Homeostasis and Reward System. Diabetes 2015, 64, 3406–3412. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. The incretin system in healthy humans: The role of GIP and GLP-1. Metabolism 2019, 96, 46–55. [Google Scholar] [CrossRef]
- Muscogiuri, G.; DeFronzo, R.A.; Gastaldelli, A.; Holst, J.J. Glucagon-like Peptide-1 and the Central/Peripheral Nervous System: Crosstalk in Diabetes. Trends Endocrinol. Metab. 2017, 28, 88–103. [Google Scholar] [CrossRef]
- Flint, A.; Raben, A.; Astrup, A.; Holst, J.J. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J. Clin. Investig. 1998, 101, 515–520. [Google Scholar] [CrossRef]
- Gasbjerg, L.S.; Helsted, M.M.; Hartmann, B.; Jensen, M.H.; Gabe, M.B.N.; Sparre-Ulrich, A.H.; Veedfald, S.; Stensen, S.; Lanng, A.R.; Bergmann, N.C.; et al. Separate and Combined Glucometabolic Effects of Endogenous Glucose-Dependent Insulinotropic Polypeptide and Glucagon-like Peptide 1 in Healthy Individuals. Diabetes 2019, 68, 906–917. [Google Scholar] [CrossRef]
- Vilsboll, T.; Krarup, T.; Sonne, J.; Madsbad, S.; Volund, A.; Juul, A.G.; Holst, J.J. Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2003, 88, 2706–2713. [Google Scholar] [CrossRef] [PubMed]
- Iepsen, E.W.; Lundgren, J.; Holst, J.J.; Madsbad, S.; Torekov, S.S. Successful weight loss maintenance includes long-term increased meal responses of GLP-1 and PYY3-36. Eur. J. Endocrinol. 2016, 174, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.Y.; Yoshikawa, T.; Katsura, Y.; Usui, T.; Fujimoto, S. Comparable effects of moderate intensity exercise on changes in anorectic gut hormone levels and energy intake to high intensity exercise. J. Endocrinol. 2009, 203, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Vilsboll, T.; Christensen, M.; Junker, A.E.; Knop, F.K.; Gluud, L.L. Effects of glucagon-like peptide-1 receptor agonists on weight loss: Systematic review and meta-analyses of randomised controlled trials. BMJ 2012, 344, d7771. [Google Scholar] [CrossRef] [PubMed]
- Bunck, M.C.; Corner, A.; Eliasson, B.; Heine, R.J.; Shaginian, R.M.; Taskinen, M.R.; Smith, U.; Yki-Jarvinen, H.; Diamant, M. Effects of exenatide on measures of beta-cell function after 3 years in metformin-treated patients with type 2 diabetes. Diabetes Care 2011, 34, 2041–2047. [Google Scholar] [CrossRef]
- Nauck, M.; Frid, A.; Hermansen, K.; Thomsen, A.B.; During, M.; Shah, N.; Tankova, T.; Mitha, I.; Matthews, D.R. Long-term efficacy and safety comparison of liraglutide, glimepiride and placebo, all in combination with metformin in type 2 diabetes: 2-year results from the LEAD-2 study. Diabetesobesity Metab. 2013, 15, 204–212. [Google Scholar] [CrossRef]
- Buse, J.B.; Nauck, M.; Forst, T.; Sheu, W.H.; Shenouda, S.K.; Heilmann, C.R.; Hoogwerf, B.J.; Gao, A.; Boardman, M.K.; Fineman, M.; et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION-6): A randomised, open-label study. Lancet 2013, 381, 117–124. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Triplitt, C.; Qu, Y.; Lewis, M.S.; Maggs, D.; Glass, L.C. Effects of exenatide plus rosiglitazone on beta-cell function and insulin sensitivity in subjects with type 2 diabetes on metformin. Diabetes Care 2010, 33, 951–957. [Google Scholar] [CrossRef]
- Grimm, M.; Han, J.; Weaver, C.; Griffin, P.; Schulteis, C.T.; Dong, H.; Malloy, J. Efficacy, safety, and tolerability of exenatide once weekly in patients with type 2 diabetes mellitus: An integrated analysis of the DURATION trials. Postgrad. Med. 2013, 125, 47–57. [Google Scholar] [CrossRef]
- Choi, I.Y.; Kim, J.K.; Lee, J.S.; Park, E.; Kim, Y.H.; Jung, S.Y.; Kim, S.J. Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in a NASH and Fibrosis Animal Model. Diabetes 2018, 67, 1106-P. [Google Scholar] [CrossRef]
- Choi, J.; Kim, J.K.; Lee, S.M.; Kwon, H.; Lee, J.; Bae, S.; Kim, D.; Choi, I.Y. 1830-P: Therapeutic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in CDHFD-Induced NASH and Fibrosis Mice. Diabetes 2020, 69, 1830-P. [Google Scholar] [CrossRef]
- Choi, J.D.; Baek, S.; Kim, Y.; Eun, K.; Kwon, S.C.; Morrow, L.; Hompesch, M.; Kang, J. 982-P: A Double-Blinded, Placebo Controlled, Single Ascending Dose Study for Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics after Subcutaneous Administration of Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in Healthy Obese Subjects. Diabetes 2019, 68, 982-P. [Google Scholar] [CrossRef]
- DePaoli, A.; Bashir, M.; Phung, V.; Yan, A.; Ling, L.; Baxter, B.; Tian, H. PS-108-NGM, a novel activator of beta-Klotho/FGFR1c: A single dose significantly reduces steatosis (liver fat by MRI-PDFF), inflammation (ALT, AST) and fibrogenic activity (Pro-C3) in NAFLD subjects. J. Hepatol. 2019, 70, e68–e69. [Google Scholar] [CrossRef]
- Depaoli, A.; Phung, V.A.N.; Bashir, M.R.; Morrow, L.; Beysen, C.; Yan, A.; Ling, L.E.I.; Baxter, B.; Luskey, K.L.; Olefsky, J.M. 140-LB: NGM313, a Novel Activator of b-Klotho/FGFR1c, Improves Insulin Resistance and Reduces Hepatic Fat in Obese, Nondiabetic Subjects. Diabetes 2019, 68, 140-LB. [Google Scholar] [CrossRef]
- Heise, T.; Morrow, L.; Hompesch, M.; Haring, H.U.; Kapitza, C.; Abt, M.; Ramsauer, M.; Magnone, M.C.; Fuerst-Recktenwald, S. Safety, efficacy and weight effect of two 11beta-HSD1 inhibitors in metformin-treated patients with type 2 diabetes. Diabetes Obes. Metab. 2014, 16, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Ramsauer, M.; Jordan, P.; Nowotny, B.; Kantartzis, K.; Machann, J.; Hwang, J.H.; Nowotny, P.; Kahl, S.; Harreiter, J.; et al. Inhibition of 11beta-HSD1 with RO5093151 for non-alcoholic fatty liver disease: A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014, 2, 406–416. [Google Scholar] [CrossRef]
- Acton, J.J., 3rd; Akiyama, T.E.; Chang, C.H.; Colwell, L.; Debenham, S.; Doebber, T.; Einstein, M.; Liu, K.; McCann, M.E.; Moller, D.E.; et al. Discovery of (2R)-2-(3-{3-[(4-Methoxyphenyl)carbonyl]-2-methyl-6-(trifluoromethoxy)-1H-indol-1 -yl}phenoxy)butanoic acid (MK-0533): A novel selective peroxisome proliferator-activated receptor gamma modulator for the treatment of type 2 diabetes mellitus with a reduced potential to increase plasma and extracellular fluid volume. J. Med. Chem. 2009, 52, 3846–3854. [Google Scholar] [CrossRef]
- Ida, S.; Kaneko, R.; Murata, K. Efficacy and safety of pemafibrate administration in patients with dyslipidemia: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2019, 18, 38. [Google Scholar] [CrossRef]
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P.; Jaiswal, A. New dual peroxisome proliferator activated receptor agonist-Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: Integrated analysis of the real world evidence. Cardiovasc. Diabetol. 2019, 18, 80. [Google Scholar] [CrossRef]
- Roy, S.; Ghosh, A. Significant Reduction of Elevated Triglycerides and Liver Fibrosis in Diabetic Dyslipidemia with Saroglitazar: A Case Report. Cureus 2019, 11, e6361. [Google Scholar] [CrossRef]
- Zydus Cadila. World’s First Drug for the Treatment of Non-Cirrhotic NASH. Press Release March 5. 2020. Available online: https://zyduscadila.com/public/pdf/pressrelease/Zydus_announces_world’s_first_drug_for_the_treatment_of_Non_Cirrhotic_NASH.pdf (accessed on 4 August 2020).
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) alpha/gamma/delta Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef] [PubMed]
- Inventiva. Lanifibranor Meets the Primary and Key Secondary Endpoints in the Phase IIb NATIVE Cinical Trial in Non-alcoholic Steatohepatitis (NASH). Press Release June 15. 2020. Available online: https://www.globenewswire.com/news-release/2020/06/15/2048284/0/en/Inventiva-s-lanifibranor-meets-the-primary-and-key-secondary-endpoints-in-the-Phase-IIb-NATIVE-clinical-trial-in-non-alcoholic-steatohepatitis-NASH.html (accessed on 4 August 2020).
- Lucas, K.J.; Lopez, P.; Lawitz, E.; Sheikh, A.; Aizenberg, D.; Hsia, S.; Bee, G.G.B.; Vierling, J.; Frias, J.; White, J.; et al. Tropifexor, a highly potent FXR agonist, produces robust and dose-dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy: FLIGHT-FXR Part C interim results. Dig. Liver Dis. 2020, 52, e38. [Google Scholar] [CrossRef]
- Tully, D.C.; Rucker, P.V.; Chianelli, D.; Williams, J.; Vidal, A.; Alper, P.B.; Mutnick, D.; Bursulaya, B.; Schmeits, J.; Wu, X.; et al. Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). J. Med. Chem. 2017, 60, 9960–9973. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Harrison, S.A.; Elkashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Non-Cirrhotic Patients with Nonalcoholic Steatohepatitis: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef]
- DePaoli, A.M.; Zhou, M.; Kaplan, D.D.; Hunt, S.C.; Adams, T.D.; Learned, R.M.; Tian, H.; Ling, L. FGF19 Analog as a Surgical Factor Mimetic That Contributes to Metabolic Effects Beyond Glucose Homeostasis. Diabetes 2019, 68, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rossi, S.J.; Paredes, A.H.; Trotter, J.F.; Bashir, M.R.; Guy, C.D.; Banerjee, R.; Jaros, M.J.; Owers, S.; Baxter, B.A.; et al. NGM282 Improves Liver Fibrosis and Histology in 12 Weeks in Patients With Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 1198–1212. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Chazouilleres, O.; Drenth, J.P.; Thorburn, D.; Harrison, S.A.; Landis, C.S.; Mayo, M.J.; Muir, A.J.; Trotter, J.F.; Leeming, D.J.; et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial. J. Hepatol. 2019, 70, 483–493. [Google Scholar] [CrossRef]
- Rinella, M.E.; Trotter, J.F.; Abdelmalek, M.F.; Paredes, A.H.; Connelly, M.A.; Jaros, M.J.; Ling, L.; Rossi, S.J.; DePaoli, A.M.; Harrison, S.A. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. J. Hepatol. 2019, 70, 735–744. [Google Scholar] [CrossRef]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; Tian, H.; DePaoli, A.M.; Ling, L. Therapeutic FGF19 promotes HDL biogenesis and transhepatic cholesterol efflux to prevent atherosclerosis. J. Lipid Res. 2019, 60, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Ambery, P.; Parker, V.E.; Stumvoll, M.; Posch, M.G.; Heise, T.; Plum-Moerschel, L.; Tsai, L.F.; Robertson, D.; Jain, M.; Petrone, M.; et al. MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: A randomised, controlled, double-blind, ascending dose and phase 2a study. Lancet 2018, 391, 2607–2618. [Google Scholar] [CrossRef]
- Parker, V.E.R.; Robertson, D.; Wang, T.; Hornigold, D.C.; Petrone, M.; Cooper, A.T.; Posch, M.G.; Heise, T.; Plum-Moerschel, L.; Schlichthaar, H.; et al. Efficacy, Safety, and Mechanistic Insights of Cotadutide, a Dual Receptor Glucagon-Like Peptide-1 and Glucagon Agonist. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Skarbaliene, J.; Madsen, A.N.; Mouritzen, U.; Bak, H.H.; Just, R. Exploring the therapeutic potential of Glucagon/GLP-1 dual agonist ZP2929 in a mouse model of diet induced and biopsy-confirmed non-alcoholic steatohepatitis. J. Hepatol. 2017, 66, S102. [Google Scholar] [CrossRef]
- Brandt, S.J.; Gotz, A.; Tschop, M.H.; Muller, T.D. Gut hormone polyagonists for the treatment of type 2 diabetes. Peptides 2018, 100, 190–201. [Google Scholar] [CrossRef]
- He, Y.L.; Haynes, W.; Meyers, C.D.; Amer, A.; Zhang, Y.; Mahling, P.; Mendonza, A.E.; Ma, S.; Chutkow, W.; Bachman, E. The effects of licogliflozin, a dual SGLT1/2 inhibitor, on body weight in obese patients with or without diabetes. Diabetes Obes. Metab. 2019, 21, 1311–1321. [Google Scholar] [CrossRef]
- Yokote, K.; Sano, M.; Tsumiyama, I.; Keefe, D. Dose-dependent reduction in body weight with LIK066 (licogliflozin) treatment in Japanese patients with obesity. Diabetes Obes. Metab. 2020, 22, 1102–1110. [Google Scholar] [CrossRef]
- Rosenstock, M.; Ayalon, M.; Mansbach, H.; Liu, Y.; Margalit, M. LBP29 BIO89–100, a novel PEG-FGF21 analogue, is efficacious following weekly and every 2-week subcutaneous dosing in spontaneous diabetic cynomolgus monkeys. J. Hepatol. 2019, 70, e141–e382. [Google Scholar] [CrossRef]
- Calle, R.; Bergman, A.; Somayaji, V.; Chidsey, K.; Kazierad, D. PS-110-Ketohexokinase inhibitor PF-06835919 administered for 6 weeks reduces whole liver fat as measured by magnetic resonance imaging-proton density fat fraction in subjects with non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, e69–e70. [Google Scholar] [CrossRef]
- Syed-Abdul, M.M.; Parks, E.J.; Gaballah, A.H.; Bingham, K.; Hammoud, G.M.; Kemble, G.; Buckley, D.; McCulloch, W.; Manrique-Acevedo, C. Fatty Acid Synthase Inhibitor TVB-2640 Reduces Hepatic de Novo Lipogenesis in Males With Metabolic Abnormalities. Hepatology 2019, 72, 103–118. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Coste, A.; Poordad, F.; Alkhouri, N.; Loo, N.; McColgan, B.J.; Tarrant, J.M.; Nguyen, T.; Han, L.; Chung, C.; et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2018, 16, 1983–1991.e3. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Colca, J.R.; McDonald, W.G.; Adams, W.J. MSDC-0602K, a metabolic modulator directed at the core pathology of non-alcoholic steatohepatitis. Expert Opin. Investig. Drugs 2018, 27, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Alkhouri, N.; Davison, B.A.; Sanyal, A.; Edwards, C.; Colca, J.R.; Lee, B.H.; Loomba, R.; Cusi, K.; Kolterman, O.; et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J. Hepatol. 2020, 72, 613–626. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.J.; Cho, Y.M.; Lee, B.W. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness. J. Korean Med. Sci. 2017, 32, 60–69. [Google Scholar] [CrossRef]
- DePaoli, A.M.; Higgins, L.S.; Henry, R.R.; Mantzoros, C.; Dunn, F.L.; Group, I.N.T.S. Can a selective PPARgamma modulator improve glycemic control in patients with type 2 diabetes with fewer side effects compared with pioglitazone? Diabetes Care 2014, 37, 1918–1923. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef]
- Genfit. Results from Interim Analysis of RESOLVE-IT Phase 3 Trial of Elafibranor in Adults with NASH and Fibrosis. Press Release May 11. 2020. Available online: https://ml-eu.globenewswire.com/Resource/Download/38e085e1-66f5-4251-8abe-648d0e7b9ed1 (accessed on 4 August 2020).
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Frias, J.P.; Nauck, M.A.; Van, J.; Benson, C.; Bray, R.; Cui, X.; Milicevic, Z.; Urva, S.; Haupt, A.; Robins, D.A. Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: A 12-week, randomized, double-blind, placebo-controlled study to evaluate different dose-escalation regimens. Diabetes Obes. Metab. 2020, 22, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Frias, J.P.; Bastyr, E.J., 3rd; Vignati, L.; Tschop, M.H.; Schmitt, C.; Owen, K.; Christensen, R.H.; DiMarchi, R.D. The Sustained Effects of a Dual GIP/GLP-1 Receptor Agonist, NNC0090-2746, in Patients with Type 2 Diabetes. Cell Metab. 2017, 26, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Charles, E.D.; Neuschwander-Tetri, B.A.; Pablo Frias, J.; Kundu, S.; Luo, Y.; Tirucherai, G.S.; Christian, R. Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obes. Silver Spring 2019, 27, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Iruarrizaga-Lejarreta, M.; Varela-Rey, M.; Fernandez-Ramos, D.; Martinez-Arranz, I.; Delgado, T.C.; Simon, J.; Juan, V.G.; delaCruz-Villar, L.; Azkargorta, M.; Lavin, J.L.; et al. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 911–927. [Google Scholar] [CrossRef]
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R.; Group, F. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2014, 12, 2085–2091.e1. [Google Scholar] [CrossRef]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients With Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2018, 16, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Berria, R.; Cornell, J.; Cusi, K. Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, M.; Jafari, S.; Sajedi, B.; Gohari, S.; Akbarieh, S.; Heydari, A.H.; Jameshoorani, M. Comparison of fenofibrate and pioglitazone effects on patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2017, 29, 1385–1388. [Google Scholar] [CrossRef]
- Fernandez-Miranda, C.; Perez-Carreras, M.; Colina, F.; Lopez-Alonso, G.; Vargas, C.; Solis-Herruzo, J.A. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef]
- Corpechot, C.; Chazouilleres, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef]
- Karhapaa, P.; Uusitupa, M.; Voutilainen, E.; Laakso, M. Effects of bezafibrate on insulin sensitivity and glucose tolerance in subjects with combined hyperlipidemia. Clin. Pharm. 1992, 52, 620–626. [Google Scholar] [CrossRef]
- Day, A.P.; Feher, M.D.; Chopra, R.; Mayne, P.D. The effect of bezafibrate treatment on serum alkaline phosphatase isoenzyme activities. Metabolism 1993, 42, 839–842. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Team, L.T.; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Matikainen, N.; Soderlund, S.; Bjornson, E.; Pietilainen, K.; Hakkarainen, A.; Lundbom, N.; Taskinen, M.R.; Boren, J. Liraglutide treatment improves postprandial lipid metabolism and cardiometabolic risk factors in humans with adequately controlled type 2 diabetes: A single-centre randomized controlled study. Diabetesobesity Metab. 2019, 21, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.; Francque, S.; Harrison, S.; Ratziu, V.; Van Gaal, L.; Calanna, S.; Hansen, M.; Linder, M.; Sanyal, A. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment. Pharm. 2019, 50, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Sattar, N.; Garcia-Perez, L.E.; Pavo, I.; Yu, M.; Robertson, K.E.; Karanikas, C.A.; Haupt, A. Dulaglutide decreases plasma aminotransferases in people with Type 2 diabetes in a pattern consistent with liver fat reduction: A post hoc analysis of the AWARD programme. Diabet Med. 2018, 35, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Sumida, Y.; Tanaka, S.; Mori, K.; Taketani, H.; Ishiba, H.; Hara, T.; Okajima, A.; Umemura, A.; Nishikawa, T.; et al. Effect of 12-week dulaglutide therapy in Japanese patients with biopsy-proven non-alcoholic fatty liver disease and type 2 diabetes mellitus. Hepatol. Res. 2017, 47, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Zhang, B.; Xu, W.; Yang, H.; Feng, W.; Li, C.; Tong, G.; Li, M.; Wang, X.; Shen, S.; et al. Effects of exenatide, insulin, and pioglitazone on liver fat content and body fat distributions in drug-naive subjects with type 2 diabetes. Acta Diabetol. 2014, 51, 865–873. [Google Scholar] [CrossRef]
- Shao, N.; Kuang, H.Y.; Hao, M.; Gao, X.Y.; Lin, W.J.; Zou, W. Benefits of exenatide on obesity and non-alcoholic fatty liver disease with elevated liver enzymes in patients with type 2 diabetes. Diabetes/Metab. Res. Rev. 2014, 30, 521–529. [Google Scholar] [CrossRef]
- Klonoff, D.C.; Buse, J.B.; Nielsen, L.L.; Guan, X.; Bowlus, C.L.; Holcombe, J.H.; Wintle, M.E.; Maggs, D.G. Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Curr. Med. Res. Opin. 2008, 24, 275–286. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Repetto, E.; Guja, C.; Hardy, E.; Han, J.; Jabbour, S.A.; Ferrannini, E. Exenatide and dapagliflozin combination improves markers of liver steatosis and fibrosis in patients with type 2 diabetes. Diabetesobesity Metab. 2020, 22, 393–403. [Google Scholar] [CrossRef]
- Liu, L.; Yan, H.; Xia, M.; Zhao, L.; Lv, M.; Zhao, N.; Rao, S.; Yao, X.; Wu, W.; Pan, B.; et al. Efficacy of exenatide and insulin glargine on nonalcoholic fatty liver disease in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 2020, e3292. [Google Scholar] [CrossRef]
- Gluud, L.L.; Knop, F.K.; Vilsboll, T. Effects of lixisenatide on elevated liver transaminases: Systematic review with individual patient data meta-analysis of randomised controlled trials on patients with type 2 diabetes. BMJ Open 2014, 4, e005325. [Google Scholar] [CrossRef]
- Cui, J.; Philo, L.; Nguyen, P.; Hofflich, H.; Hernandez, C.; Bettencourt, R.; Richards, L.; Salotti, J.; Bhatt, A.; Hooker, J.; et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: A randomized controlled trial. J. Hepatol. 2016, 65, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Tamura, Y.; Kakehi, S.; Funayama, T.; Gastaldelli, A.; Takeno, K.; Kawaguchi, M.; Yamamoto, R.; Sato, F.; Ikeda, S.; et al. Effects of sitagliptin on ectopic fat contents and glucose metabolism in type 2 diabetic patients with fatty liver: A pilot study. J. Diabetes Investig. 2015, 6, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Joy, T.R.; McKenzie, C.A.; Tirona, R.G.; Summers, K.; Seney, S.; Chakrabarti, S.; Malhotra, N.; Beaton, M.D. Sitagliptin in patients with non-alcoholic steatohepatitis: A randomized, placebo-controlled trial. World J. Gastroenterol. 2017, 23, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Sumida, Y.; Tanaka, S.; Mori, K.; Taketani, H.; Ishiba, H.; Hara, T.; Okajima, A.; Umemura, A.; Nishikawa, T.; et al. Effect of sodium glucose cotransporter 2 inhibitor on liver function tests in Japanese patients with non-alcoholic fatty liver disease and type 2 diabetes mellitus. Hepatol. Res. 2017, 47, 1072–1078. [Google Scholar] [CrossRef]
- Kahl, S.; Gancheva, S.; Strassburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 2020, 43, 298–305. [Google Scholar] [CrossRef]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Farooqui, K.J.; Singh, M.K.; Wasir, J.S.; Bansal, B.; Kaur, P.; Jevalikar, G.; Gill, H.K.; et al. Effect of Empagliflozin on Liver Fat in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial (E-LIFT Trial). Diabetes Care 2018, 41, 1801–1808. [Google Scholar] [CrossRef]
- Lai, L.L.; Vethakkan, S.R.; Nik Mustapha, N.R.; Mahadeva, S.; Chan, W.K. Empagliflozin for the Treatment of Nonalcoholic Steatohepatitis in Patients with Type 2 Diabetes Mellitus. Dig. Dis. Sci. 2020, 65, 623–631. [Google Scholar] [CrossRef]
- Cusi, K.; Bril, F.; Barb, D.; Polidori, D.; Sha, S.; Ghosh, A.; Farrell, K.; Sunny, N.E.; Kalavalapalli, S.; Pettus, J.; et al. Effect of canagliflozin treatment on hepatic triglyceride content and glucose metabolism in patients with type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 812–821. [Google Scholar] [CrossRef]
- Inoue, M.; Hayashi, A.; Taguchi, T.; Arai, R.; Sasaki, S.; Takano, K.; Inoue, Y.; Shichiri, M. Effects of canagliflozin on body composition and hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease. J. Diabetes Investig. 2019, 10, 1004–1011. [Google Scholar] [CrossRef]
- Itani, T.; Ishihara, T. Efficacy of canagliflozin against nonalcoholic fatty liver disease: A prospective cohort study. Obes. Sci Pr. 2018, 4, 477–482. [Google Scholar] [CrossRef]
- Leiter, L.A.; Forst, T.; Polidori, D.; Balis, D.A.; Xie, J.; Sha, S. Effect of canagliflozin on liver function tests in patients with type 2 diabetes. Diabetes Metab. 2016, 42, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, Y.; Ye, Z.; Yang, H.; Cui, X.; Wang, Z.; Liu, L. Effects of Canagliflozin on Fatty Liver Indexes in Patients with Type 2 Diabetes: A Meta-analysis of Randomized Controlled Trials. J. Pharm. Pharm. Sci. 2018, 21, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Nishikawa, T.; Umemura, A.; Yamaguchi, K.; Moriguchi, M.; Yasui, K.; Kimura, M.; Iijima, H.; Hashimoto, T.; Sumida, Y.; et al. Efficacy and safety of canagliflozin in type 2 diabetes mellitus patients with biopsy-proven nonalcoholic steatohepatitis classified as stage 1-3 fibrosis. Diabetes Metab. Syndr. Obes. 2018, 11, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.H.; Gu, Y.; Yeung, M.Y.; Fong, C.H.Y.; Woo, Y.C.; Chow, W.S.; Tan, K.; Lam, K.S.L. Dapagliflozin and Empagliflozin Ameliorate Hepatic Dysfunction Among Chinese Subjects with Diabetes in Part Through Glycemic Improvement: A Single-Center, Retrospective, Observational Study. Diabetes 2018, 9, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Gross, J.L.; Pieters, A.; Bastien, A.; List, J.F. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 375, 2223–2233. [Google Scholar] [CrossRef]
- Tobita, H.; Sato, S.; Miyake, T.; Ishihara, S.; Kinoshita, Y. Effects of Dapagliflozin on Body Composition and Liver Tests in Patients with Nonalcoholic Steatohepatitis Associated with Type 2 Diabetes Mellitus: A Prospective, Open-label, Uncontrolled Study. Curr Res. Clin. Exp. 2017, 87, 13–19. [Google Scholar] [CrossRef]
- Eriksson, J.W.; Lundkvist, P.; Jansson, P.A.; Johansson, L.; Kvarnstrom, M.; Moris, L.; Miliotis, T.; Forsberg, G.B.; Riserus, U.; Lind, L.; et al. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: A double-blind randomised placebo-controlled study. Diabetologia 2018, 61, 1923–1934. [Google Scholar] [CrossRef]
- Liu, J.; Tarasenko, L.; Terra, S.G.; Huyck, S.; Wu, L.; Pong, A.; Calle, R.A.; Gallo, S.; Darekar, A.; Mancuso, J.P. Efficacy of ertugliflozin in monotherapy or combination therapy in patients with type 2 diabetes: A pooled analysis of placebo-controlled studies. Diab Vasc Dis. Res. 2019, 16, 415–423. [Google Scholar] [CrossRef]
- Han, E.; Lee, Y.H.; Lee, B.W.; Kang, E.S.; Cha, B.S. Ipragliflozin Additively Ameliorates Non-Alcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Controlled with Metformin and Pioglitazone: A 24-Week Randomized Controlled Trial. J. Clin. Med. 2020, 9, 259. [Google Scholar] [CrossRef]
- Miyake, T.; Yoshida, S.; Furukawa, S.; Sakai, T.; Tada, F.; Senba, H.; Yamamoto, S.; Koizumi, Y.; Yoshida, O.; Hirooka, M.; et al. Ipragliflozin Ameliorates Liver Damage in Non-alcoholic Fatty Liver Disease. Open Med. 2018, 13, 402–409. [Google Scholar] [CrossRef]
- Ohki, T.; Isogawa, A.; Toda, N.; Tagawa, K. Effectiveness of Ipragliflozin, a Sodium-Glucose Co-transporter 2 Inhibitor, as a Second-line Treatment for Non-Alcoholic Fatty Liver Disease Patients with Type 2 Diabetes Mellitus Who Do Not Respond to Incretin-Based Therapies Including Glucagon-like Peptide-1 Analogs and Dipeptidyl Peptidase-4 Inhibitors. Clin. Drug Investig. 2016, 36, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Shimizu, S.; Inoue, K.; Saito, D.; Yanagisawa, M.; Inukai, K.; Akiyama, Y.; Morimoto, Y.; Noda, M.; Shimada, A. Comparison of Ipragliflozin and Pioglitazone Effects on Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes: A Randomized, 24-Week, Open-Label, Active-Controlled Trial. Diabetes Care 2017, 40, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Takase, T.; Nakamura, A.; Miyoshi, H.; Yamamoto, C.; Atsumi, T. Amelioration of fatty liver index in patients with type 2 diabetes on ipragliflozin: An association with glucose-lowering effects. Endocr. J. 2017, 64, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Kato, H.; Ishii, S.; Sasaki, Y.; Nakamura, Y.; Nakagawa, T.; Nagai, Y.; Tanaka, Y. Ipragliflozin, a sodium glucose co-transporter 2 inhibitor, reduces intrahepatic lipid content and abdominal visceral fat volume in patients with type 2 diabetes. Expert Opin. Pharmacother. 2017, 18, 1433–1438. [Google Scholar] [CrossRef]
- Bando, Y.; Ogawa, A.; Ishikura, K.; Kanehara, H.; Hisada, A.; Notumata, K.; Okafuji, K.; Toya, D. The effects of ipragliflozin on the liver-to-spleen attenuation ratio as assessed by computed tomography and on alanine transaminase levels in Japanese patients with type 2 diabetes mellitus. Diabetol. Int. 2017, 8, 218–227. [Google Scholar] [CrossRef]
- Ozaki, A.; Yoneda, M.; Kessoku, T.; Iwaki, M.; Kobayashi, T.; Honda, Y.; Ogawa, Y.; Imajo, K.; Sakai, E.; Taguri, M.; et al. Effect of tofogliflozin and pioglitazone on hepatic steatosis in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: A randomized, open-label pilot study (ToPiND study). Contemp. Clin. Trials Commun. 2020, 17, 100516. [Google Scholar] [CrossRef]
- Shibuya, T.; Fushimi, N.; Kawai, M.; Yoshida, Y.; Hachiya, H.; Ito, S.; Kawai, H.; Ohashi, N.; Mori, A. Luseogliflozin improves liver fat deposition compared to metformin in type 2 diabetes patients with non-alcoholic fatty liver disease: A prospective randomized controlled pilot study. Diabetes Obes. Metab. 2018, 20, 438–442. [Google Scholar] [CrossRef]
- Sumida, Y.; Murotani, K.; Saito, M.; Tamasawa, A.; Osonoi, Y.; Yoneda, M.; Osonoi, T. Effect of luseogliflozin on hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease: A prospective, single-arm trial (LEAD trial). Hepatol. Res. 2019, 49, 64–71. [Google Scholar] [CrossRef]
- Tang, A.; Rabasa-Lhoret, R.; Castel, H.; Wartelle-Bladou, C.; Gilbert, G.; Massicotte-Tisluck, K.; Chartrand, G.; Olivie, D.; Julien, A.S.; de Guise, J.; et al. Effects of Insulin Glargine and Liraglutide Therapy on Liver Fat as Measured by Magnetic Resonance in Patients With Type 2 Diabetes: A Randomized Trial. Diabetes Care 2015, 38, 1339–1346. [Google Scholar] [CrossRef]
- Yan, J.; Yao, B.; Kuang, H.; Yang, X.; Huang, Q.; Hong, T.; Li, Y.; Dou, J.; Yang, W.; Qin, G.; et al. Liraglutide, Sitagliptin, and Insulin Glargine Added to Metformin: The Effect on Body Weight and Intrahepatic Lipid in Patients With Type 2 Diabetes Mellitus and Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 2414–2426. [Google Scholar] [CrossRef]
- Buse, J.B.; Klonoff, D.C.; Nielsen, L.L.; Guan, X.; Bowlus, C.L.; Holcombe, J.H.; Maggs, D.G.; Wintle, M.E. Metabolic effects of two years of exenatide treatment on diabetes, obesity, and hepatic biomarkers in patients with type 2 diabetes: An interim analysis of data from the open-label, uncontrolled extension of three double-blind, placebo-controlled trials. Clin. Ther. 2007, 29, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Jendle, J.; Nauck, M.A.; Matthews, D.R.; Frid, A.; Hermansen, K.; During, M.; Zdravkovic, M.; Strauss, B.J.; Garber, A.J.; LEAD-2 and LEAD-3 Study Groups; et al. Weight loss with liraglutide, a once-daily human glucagon-like peptide-1 analogue for type 2 diabetes treatment as monotherapy or added to metformin, is primarily as a result of a reduction in fat tissue. Diabetesobesity Metab. 2009, 11, 1163–1172. [Google Scholar] [CrossRef]
- Kenny, P.R.; Brady, D.E.; Torres, D.M.; Ragozzino, L.; Chalasani, N.; Harrison, S.A. Exenatide in the treatment of diabetic patients with non-alcoholic steatohepatitis: A case series. Am. J. Gastroenterol. 2010, 105, 2707–2709. [Google Scholar] [CrossRef]
- Sathyanarayana, P.; Jogi, M.; Muthupillai, R.; Krishnamurthy, R.; Samson, S.L.; Bajaj, M. Effects of combined exenatide and pioglitazone therapy on hepatic fat content in type 2 diabetes. Obes. Silver Spring 2011, 19, 2310–2315. [Google Scholar] [CrossRef]
- Yilmaz, Y.; Yonal, O.; Deyneli, O.; Celikel, C.A.; Kalayci, C.; Duman, D.G. Effects of sitagliptin in diabetic patients with nonalcoholic steatohepatitis. Acta Gastro Enterol. Belg. 2012, 75, 240–244. [Google Scholar]
- Ohki, T.; Isogawa, A.; Iwamoto, M.; Ohsugi, M.; Yoshida, H.; Toda, N.; Tagawa, K.; Omata, M.; Koike, K. The effectiveness of liraglutide in nonalcoholic fatty liver disease patients with type 2 diabetes mellitus compared to sitagliptin and pioglitazone. Sci. World J. 2012, 2012, 496453. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, D.J.; Irwin, A.; Gardner, C.J.; Daousi, C.; Purewal, T.; Furlong, N.; Goenka, N.; Thomas, E.L.; Adams, V.L.; Pushpakom, S.P.; et al. Improved glycaemia correlates with liver fat reduction in obese, type 2 diabetes, patients given glucagon-like peptide-1 (GLP-1) receptor agonists. PLoS ONE 2012, 7, e50117. [Google Scholar] [CrossRef]
- Fan, H.; Pan, Q.; Xu, Y.; Yang, X. Exenatide improves type 2 diabetes concomitant with non-alcoholic fatty liver disease. Arq. Bras. De Endocrinol. E Metabol. 2013, 57, 702–708. [Google Scholar] [CrossRef]
- Suzuki, D.; Toyoda, M.; Kimura, M.; Miyauchi, M.; Yamamoto, N.; Sato, H.; Tanaka, E.; Kuriyama, Y.; Miyatake, H.; Abe, M.; et al. Effects of liraglutide, a human glucagon-like peptide-1 analogue, on body weight, body fat area and body fat-related markers in patients with type 2 diabetes mellitus. Intern. Med. 2013, 52, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Bergenstal, R.M.; Li, Y.; Porter, T.K.; Weaver, C.; Han, J. Exenatide once weekly improved glycaemic control, cardiometabolic risk factors and a composite index of an HbA1c <7%, without weight gain or hypoglycaemia, over 52 weeks. Diabetes Obes. Metab. 2013, 15, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Blaslov, K.; Zibar, K.; Bulum, T.; Duvnjak, L. Effect of exenatide therapy on hepatic fat quantity and hepatic biomarkers in type 2 diabetic patients. Clin. Res. Hepatol. Gastroenterol. 2014, 38, e61–e63. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Kitajima, Y.; Hyogo, H.; Takahashi, H.; Kojima, M.; Ono, M.; Araki, N.; Tanaka, K.; Yamaguchi, M.; Matsuda, Y.; et al. Pilot study of liraglutide effects in non-alcoholic steatohepatitis and non-alcoholic fatty liver disease with glucose intolerance in Japanese patients (LEAN-J). Hepatol. Res. 2015, 45, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Smits, M.M.; Tonneijck, L.; Muskiet, M.H.; Kramer, M.H.; Pouwels, P.J.; Pieters-van den Bos, I.C.; Hoekstra, T.; Diamant, M.; van Raalte, D.H.; Cahen, D.L. Twelve week liraglutide or sitagliptin does not affect hepatic fat in type 2 diabetes: A randomised placebo-controlled trial. Diabetologia 2016, 59, 2588–2593. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.M.; Cercueil, J.P.; Loffroy, R.; Denimal, D.; Bouillet, B.; Fourmont, C.; Chevallier, O.; Duvillard, L.; Verges, B. Effect of Liraglutide Therapy on Liver Fat Content in Patients with Inadequately Controlled Type 2 Diabetes: The Lira-NAFLD Study. J. Clin. Endocrinol. Metab. 2017, 102, 407–415. [Google Scholar] [CrossRef]
- Feng, W.; Gao, C.; Bi, Y.; Wu, M.; Li, P.; Shen, S.; Chen, W.; Yin, T.; Zhu, D. Randomized trial comparing the effects of gliclazide, liraglutide, and metformin on diabetes with non-alcoholic fatty liver disease. J. Diabetes 2017, 9, 800–809. [Google Scholar] [CrossRef]
- Khoo, J.; Hsiang, J.; Taneja, R.; Law, N.M.; Ang, T.L. Comparative effects of liraglutide 3 mg vs structured lifestyle modification on body weight, liver fat and liver function in obese patients with non-alcoholic fatty liver disease: A pilot randomized trial. Diabetesobesity Metab. 2017, 19, 1814–1817. [Google Scholar] [CrossRef]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef]
- Samson, S.L.; Gonzalez, E.V.; Yechoor, V.; Bajaj, M.; Oka, K.; Chan, L. Gene therapy for diabetes: Metabolic effects of helper-dependent adenoviral exendin 4 expression in a diet-induced obesity mouse model. Mol. Ther. 2008, 16, 1805–1812. [Google Scholar] [CrossRef]
- Samson, S.L.; Sathyanarayana, P.; Jogi, M.; Gonzalez, E.V.; Gutierrez, A.; Krishnamurthy, R.; Muthupillai, R.; Chan, L.; Bajaj, M. Exenatide decreases hepatic fibroblast growth factor 21 resistance in non-alcoholic fatty liver disease in a mouse model of obesity and in a randomised controlled trial. Diabetologia 2011, 54, 3093–3100. [Google Scholar] [CrossRef]
- Sharma, S.; Mells, J.E.; Fu, P.P.; Saxena, N.K.; Anania, F.A. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS ONE 2011, 6, e25269. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Houlihan, D.D.; Rowe, I.A.; Clausen, W.H.; Elbrond, B.; Gough, S.C.; Tomlinson, J.W.; Newsome, P.N. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: Individual patient data meta-analysis of the LEAD program. Aliment. Pharmacol. Ther. 2013, 37, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am. J. Physiol. 2012, 302, G762–G772. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lv, Q.; Li, S.; Wu, Y.; Li, L.; Li, J.; Zhang, F.; Sun, X.; Tong, N. Efficacy and safety of glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Prigeon, R.L.; Quddusi, S.; Paty, B.; D’Alessio, D.A. Suppression of glucose production by GLP-1 independent of islet hormones: A novel extrapancreatic effect. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E701–E707. [Google Scholar] [CrossRef] [PubMed]
- Seghieri, M.; Rebelos, E.; Gastaldelli, A.; Astiarraga, B.D.; Casolaro, A.; Barsotti, E.; Pocai, A.; Nauck, M.; Muscelli, E.; Ferrannini, E. Direct effect of GLP-1 infusion on endogenous glucose production in humans. Diabetologia 2013, 56, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.A.; Kolachala, V.L.; Jiang, R.; Abramowsky, C.; Romero, R.; Fifadara, N.; Anania, F.; Knechtle, S.; Kirk, A. The glucagon-like peptide-1 receptor agonist Exendin 4 has a protective role in ischemic injury of lean and steatotic liver by inhibiting cell death and stimulating lipolysis. Am. J. Pathol. 2012, 181, 1693–1701. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. Off. J. Int. Assoc. Study Liver 2011, 31, 1285–1297. [Google Scholar] [CrossRef]
- Cersosimo, E.; Gastaldelli, A.; Cervera, A.; Wajcberg, E.; Sriwijilkamol, A.; Fernandez, M.; Zuo, P.; Petz, R.; Triplitt, C.; Musi, N.; et al. Effect of exenatide on splanchnic and peripheral glucose metabolism in type 2 diabetic subjects. J. Clin. Endocrinol. Metab. 2011, 96, 1763–1770. [Google Scholar] [CrossRef]
- Vahl, T.P.; Tauchi, M.; Durler, T.S.; Elfers, E.E.; Fernandes, T.M.; Bitner, R.D.; Ellis, K.S.; Woods, S.C.; Seeley, R.J.; Herman, J.P.; et al. Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology 2007, 148, 4965–4973. [Google Scholar] [CrossRef]
- Pyke, C.; Heller, R.S.; Kirk, R.K.; Orskov, C.; Reedtz-Runge, S.; Kaastrup, P.; Hvelplund, A.; Bardram, L.; Calatayud, D.; Knudsen, L.B. GLP-1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology 2014, 155, 1280–1290. [Google Scholar] [CrossRef]
- Wei, Y.; Mojsov, S. Tissue-specific expression of the human receptor for glucagon-like peptide-I: Brain, heart and pancreatic forms have the same deduced amino acid sequences. Febs Lett. 1995, 358, 219–224. [Google Scholar] [CrossRef]
- Seo, D.; Faintuch, B.L.; Aparecida de Oliveira, E.; Faintuch, J. Pancreas and liver uptake of new radiolabeled incretins (GLP-1 and Exendin-4) in models of diet-induced and diet-restricted obesity. Nucl. Med. Biol. 2017, 49, 57–64. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Okerson, T.; Viswanathan, P.; Guan, X.; Holcombe, J.H.; MacConell, L. Effects of exenatide versus sitagliptin on postprandial glucose, insulin and glucagon secretion, gastric emptying, and caloric intake: A randomized, cross-over study. Curr. Med. Res. Opin. 2008, 24, 2943–2952. [Google Scholar] [CrossRef] [PubMed]
- Matikainen, N.; Manttari, S.; Schweizer, A.; Ulvestad, A.; Mills, D.; Dunning, B.E.; Foley, J.E.; Taskinen, M.R. Vildagliptin therapy reduces postprandial intestinal triglyceride-rich lipoprotein particles in patients with type 2 diabetes. Diabetologia 2006, 49, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Camastra, S.; Astiarraga, B.; Tura, A.; Frascerra, S.; Ciociaro, D.; Mari, A.; Gastaldelli, A.; Ferrannini, E. Effect of exenatide on postprandial glucose fluxes, lipolysis, and ss-cell function in non-diabetic, morbidly obese patients. Diabetes Obes. Metab. 2017, 19, 412–420. [Google Scholar] [CrossRef]
- Ben-Shlomo, S.; Zvibel, I.; Shnell, M.; Shlomai, A.; Chepurko, E.; Halpern, Z.; Barzilai, N.; Oren, R.; Fishman, S. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol. 2011, 54, 1214–1223. [Google Scholar] [CrossRef]
- Parlevliet, E.T.; Wang, Y.; Geerling, J.J.; Schroder-Van der Elst, J.P.; Picha, K.; O’Neil, K.; Stojanovic-Susulic, V.; Ort, T.; Havekes, L.M.; Romijn, J.A.; et al. GLP-1 receptor activation inhibits VLDL production and reverses hepatic steatosis by decreasing hepatic lipogenesis in high-fat-fed APOE*3-Leiden mice. PLoS ONE 2012, 7, e49152. [Google Scholar] [CrossRef]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef]
- Lee, Y.S.; Park, M.S.; Choung, J.S.; Kim, S.S.; Oh, H.H.; Choi, C.S.; Ha, S.Y.; Kang, Y.; Kim, Y.; Jun, H.S. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia 2012, 55, 2456–2468. [Google Scholar] [CrossRef]
- Shirakawa, J.; Fujii, H.; Ohnuma, K.; Sato, K.; Ito, Y.; Kaji, M.; Sakamoto, E.; Koganei, M.; Sasaki, H.; Nagashima, Y.; et al. Diet-induced adipose tissue inflammation and liver steatosis are prevented by DPP-4 inhibition in diabetic mice. Diabetes 2011, 60, 1246–1257. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, M.; Ren, H.; Hu, H.; Boden, G.; Li, L.; Yang, G. GLP-1 analogue prevents NAFLD in ApoE KO mice with diet and Acrp30 knockdown by inhibiting c-JNK. Liver Int. Off. J. Int. Assoc. Study Liver 2013, 33, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.A.; Koska, J.; Mullin, M.P.; Syoufi, I.; Schwenke, D.C.; Reaven, P.D. Exenatide suppresses postprandial elevations in lipids and lipoproteins in individuals with impaired glucose tolerance and recent onset type 2 diabetes mellitus. Atherosclerosis 2010, 212, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Hermansen, K.; Baekdal, T.A.; During, M.; Pietraszek, A.; Mortensen, L.S.; Jorgensen, H.; Flint, A. Liraglutide suppresses postprandial triglyceride and apolipoprotein B48 elevations after a fat-rich meal in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled, cross-over trial. Diabetesobesity Metab. 2013, 15, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.B.; Shojaee-Moradie, F.; Sharaf, S.E.; Jackson, N.C.; Fielding, B.; Hovorka, R.; Mendis, J.; Russell-Jones, D.; Umpleby, A.M. Lixisenatide Reduces Chylomicron Triacylglycerol by Increased Clearance. J. Clin. Endocrinol. Metab. 2019, 104, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Kato, M.; Tanaka, K.; Tanaka, M.; Kohjima, M.; Nakamura, K.; Enjoji, M.; Nakamuta, M.; Kotoh, K.; Takayanagi, R. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol. Med. Rep. 2012, 5, 729–733. [Google Scholar] [CrossRef]
- Firneisz, G.; Varga, T.; Lengyel, G.; Feher, J.; Ghyczy, D.; Wichmann, B.; Selmeci, L.; Tulassay, Z.; Racz, K.; Somogyi, A. Serum dipeptidyl peptidase-4 activity in insulin resistant patients with non-alcoholic fatty liver disease: A novel liver disease biomarker. PLoS ONE 2010, 5, e12226. [Google Scholar] [CrossRef]
- Balaban, Y.H.; Korkusuz, P.; Simsek, H.; Gokcan, H.; Gedikoglu, G.; Pinar, A.; Hascelik, G.; Asan, E.; Hamaloglu, E.; Tatar, G. Dipeptidyl peptidase IV (DDP IV) in NASH patients. Ann. Hepatol. 2007, 6, 242–250. [Google Scholar] [CrossRef]
- Olivares, M.; Schuppel, V.; Hassan, A.M.; Beaumont, M.; Neyrinck, A.M.; Bindels, L.B.; Benitez-Paez, A.; Sanz, Y.; Haller, D.; Holzer, P.; et al. The Potential Role of the Dipeptidyl Peptidase-4-Like Activity From the Gut Microbiota on the Host Health. Front. Microbiol. 2018, 9, 1900. [Google Scholar] [CrossRef]
- Mulvihill, E.E.; Varin, E.M.; Gladanac, B.; Campbell, J.E.; Ussher, J.R.; Baggio, L.L.; Yusta, B.; Ayala, J.; Burmeister, M.A.; Matthews, D.; et al. Cellular Sites and Mechanisms Linking Reduction of Dipeptidyl Peptidase-4 Activity to Control of Incretin Hormone Action and Glucose Homeostasis. Cell Metab. 2017, 25, 152–165. [Google Scholar] [CrossRef]
- Sanchez-Garrido, M.A.; Brandt, S.J.; Clemmensen, C.; Muller, T.D.; DiMarchi, R.D.; Tschop, M.H. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia 2017, 60, 1851–1861. [Google Scholar] [CrossRef]
- Seghieri, M.; Christensen, A.S.; Andersen, A.; Solini, A.; Knop, F.K.; Vilsboll, T. Future Perspectives on GLP-1 Receptor Agonists and GLP-1/glucagon Receptor Co-agonists in the Treatment of NAFLD. Front. Endocrinology 2018, 9, 649. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.J.; Muller, T.D.; DiMarchi, R.D.; Tschop, M.H.; Stemmer, K. Peptide-based multi-agonists: A new paradigm in metabolic pharmacology. J. Intern. Med. 2018, 284, 581–602. [Google Scholar] [CrossRef] [PubMed]
- Asmar, M.; Simonsen, L.; Madsbad, S.; Stallknecht, B.; Holst, J.J.; Bulow, J. Glucose-dependent insulinotropic polypeptide may enhance fatty acid re-esterification in subcutaneous abdominal adipose tissue in lean humans. Diabetes 2010, 59, 2160–2163. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.G.; Snead, W.L.; Campbell, P.J. Regulation of free fatty acid metabolism by glucagon. J. Clin. Endocrinol. Metab. 1993, 77, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E.; Lorenzi, M.; Bier, D.M.; Tsalikian, E.; Schneider, V.; Karam, J.H.; Forsham, P.H. Effects of physiologic levels of glucagon and growth hormone on human carbohydrate and lipid metabolism. Studies involving administration of exogenous hormone during suppression of endogenous hormone secretion with somatostatin. J. Clin. Investig. 1976, 57, 875–884. [Google Scholar] [CrossRef]
- Tillner, J.; Posch, M.G.; Wagner, F.; Teichert, L.; Hijazi, Y.; Einig, C.; Keil, S.; Haack, T.; Wagner, M.; Bossart, M.; et al. A novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: Results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetesobesity Amp Metab. 2019, 21, 120–128. [Google Scholar] [CrossRef]
- You, S.; Mcdonald, M.; Case, M.; Steiner, D.; Tat, T.; Jenkinson, C.; Pick, R.; Hart, J.; Moreno, V.; Parise, J.; et al. Long-Acting GLP-1 and Glucagon Receptor Dual Agonists for the Treatment of Type 2 Diabetes. Diabetes 2016, 65 (Suppl. 1), A221–A360. [Google Scholar] [CrossRef][Green Version]
- Hershkovitz, O.; Bar-IIan, A.; Hart, G.; Fima, E. The long-acting dual GLP-1/Glucagon Agonist, MOD-6030 improves glycaemic control and induces a prolonged weight loss in diet induced obesity mice following a once weekly administration. In Proceedings of the Endocrine Society’s 95th Annual Meeting and Expo, San Francisco, CA, USA, 15–18 June 2013. [Google Scholar]
- Day, J.W.; Ottaway, N.; Patterson, J.T.; Gelfanov, V.; Smiley, D.; Gidda, J.; Findeisen, H.; Bruemmer, D.; Drucker, D.J.; Chaudhary, N.; et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5, 749–757. [Google Scholar] [CrossRef]
- Day, J.W.; Gelfanov, V.; Smiley, D.; Carrington, P.E.; Eiermann, G.; Chicchi, G.; Erion, M.D.; Gidda, J.; Thornberry, N.A.; Tschop, M.H.; et al. Optimization of co-agonism at GLP-1 and glucagon receptors to safely maximize weight reduction in DIO-rodents. Biopolymers 2012, 98, 443–450. [Google Scholar] [CrossRef]
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; Du, X.; Petrov, A.; Lassman, M.E.; Jiang, G.; et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58, 2258–2266. [Google Scholar] [CrossRef]
- Valdecantos, M.P.; Pardo, V.; Ruiz, L.; Castro-Sanchez, L.; Lanzon, B.; Fernandez-Millan, E.; Garcia-Monzon, C.; Arroba, A.I.; Gonzalez-Rodriguez, A.; Escriva, F.; et al. A novel glucagon-like peptide 1/glucagon receptor dual agonist improves steatohepatitis and liver regeneration in mice. Hepatology 2017, 65, 950–968. [Google Scholar] [CrossRef]
- Clemmensen, C.; Chabenne, J.; Finan, B.; Sullivan, L.; Fischer, K.; Kuchler, D.; Sehrer, L.; Ograjsek, T.; Hofmann, S.M.; Schriever, S.C.; et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes 2014, 63, 1422–1427. [Google Scholar] [CrossRef]
- Patel, V.; Joharapurkar, A.; Kshirsagar, S.; Sutariya, B.; Patel, M.; Patel, H.; Pandey, D.; Patel, D.; Ranvir, R.; Kadam, S.; et al. Coagonist of GLP-1 and Glucagon Receptor Ameliorates Development of Non-Alcoholic Fatty Liver Disease. Cardiovasc. Hematol. Agents Med. Chem. 2018, 16, 35–43. [Google Scholar] [CrossRef]
- Henderson, S.J.; Konkar, A.; Hornigold, D.C.; Trevaskis, J.L.; Jackson, R.; Fritsch Fredin, M.; Jansson-Lofmark, R.; Naylor, J.; Rossi, A.; Bednarek, M.A.; et al. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetesobesity Metab. 2016, 18, 1176–1190. [Google Scholar] [CrossRef]
- Schmitt, C.; Portron, A.; Jadidi, S.; Sarkar, N.; DiMarchi, R. Pharmacodynamics, pharmacokinetics and safety of multiple ascending doses of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 in people with type 2 diabetes mellitus. Diabetesobesity Metab. 2017, 19, 1436–1445. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Smiley, D.L.; Ma, T.; Clemmensen, C.; Chabenne, J.; Zhang, L.; Habegger, K.M.; Fischer, K.; et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 2015, 21, 27–36. [Google Scholar] [CrossRef]
- Jall, S.; Sachs, S.; Clemmensen, C.; Finan, B.; Neff, F.; DiMarchi, R.D.; Tschop, M.H.; Muller, T.D.; Hofmann, S.M. Monomeric GLP-1/GIP/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol. Metab. 2017, 6, 440–446. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Neish, A.S. Microbes in gastrointestinal health and disease. Gastroenterology 2009, 136, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Backhed, F. The gut microbiota--masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Starkel, P.; Windey, K.; Tremaroli, V.; Backhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Idilman, R.; Mabudian, L.; Hood, M.; Fagan, A.; Turan, D.; White, M.B.; Karakaya, F.; Wang, J.; Atalay, R.; et al. Diet affects gut microbiota and modulates hospitalization risk differentially in an international cirrhosis cohort. Hepatology 2018, 68, 234–247. [Google Scholar] [CrossRef]
- Davis, B.C.; Bajaj, J.S. The Human Gut Microbiome in Liver Diseases. Semin. Liver Dis. 2017, 37, 128–140. [Google Scholar] [CrossRef]
- Llorente, C.; Jepsen, P.; Inamine, T.; Wang, L.; Bluemel, S.; Wang, H.J.; Loomba, R.; Bajaj, J.S.; Schubert, M.L.; Sikaroodi, M.; et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat. Commun. 2017, 8, 837. [Google Scholar] [CrossRef]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Rychlicki, C.; Agostinelli, L.; Giordano, D.M.; Gaggini, M.; Fraumene, C.; Saponaro, C.; Manghina, V.; Sartini, L.; Mingarelli, E.; et al. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Sci. Rep. 2017, 7, 12200. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Rychlicki, C.; Agostinelli, L.; Saccomanno, S.; Candelaresi, C.; Trozzi, L.; Mingarelli, E.; Facinelli, B.; Magi, G.; Palmieri, C.; et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 2014, 59, 1738–1749. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated With Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandona, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernandez-Real, J.M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Duan, Y.; Yang, L.; Schnabl, B. Small metabolites, possible big changes: A microbiota-centered view of non-alcoholic fatty liver disease. Gut 2019, 68, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Bashiardes, S.; Shapiro, H.; Rozin, S.; Shibolet, O.; Elinav, E. Non-alcoholic fatty liver and the gut microbiota. Mol. Metab. 2016, 5, 782–794. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Sandler, N.G.; Koh, C.; Roque, A.; Eccleston, J.L.; Siegel, R.B.; Demino, M.; Kleiner, D.E.; Deeks, S.G.; Liang, T.J.; Heller, T.; et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology 2011, 141, 1220–1230.e3. [Google Scholar] [CrossRef]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Trauner, M.; Fuchs, C.D.; Halilbasic, E.; Paumgartner, G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology 2017, 65, 1393–1404. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.; Siddiqui, M.S.; Boyett, S.; Min, H.K.; et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Liu, P.; Wei, R.; Chen, W.; Rajani, C.; Hernandez, B.Y.; Alegado, R.; Dong, B.; Li, D.; et al. Distinctly altered gut microbiota in the progression of liver disease. Oncotarget 2016, 7, 19355–19366. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Traussnigg, S.A.; Trauner, M. Nuclear Receptor Modulation for the Treatment of Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2016, 36, 69–86. [Google Scholar] [CrossRef]
- Fickert, P.; Fuchsbichler, A.; Moustafa, T.; Wagner, M.; Zollner, G.; Halilbasic, E.; Stoger, U.; Arrese, M.; Pizarro, M.; Solis, N.; et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am. J. Pathol. 2009, 175, 2392–2405. [Google Scholar] [CrossRef]
- Intercept Pharmaceuticals. Intercept Receives Complete Response Letter from FDA for Obeticholic Acid for the Treatment of Fibrosis Due to NASH; Intercept Pharmaceuticals: New York, NY, USA, 2020. [Google Scholar]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef]
- Degirolamo, C.; Modica, S.; Vacca, M.; Di Tullio, G.; Morgano, A.; D’Orazio, A.; Kannisto, K.; Parini, P.; Moschetta, A. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology 2015, 61, 161–170. [Google Scholar] [CrossRef]
- Fang, S.; Suh, J.M.; Reilly, S.M.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef]
- Jiang, C.; Xie, C.; Lv, Y.; Li, J.; Krausz, K.W.; Shi, J.; Brocker, C.N.; Desai, D.; Amin, S.G.; Bisson, W.H.; et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 2015, 6, 10166. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Mangelsdorf, D.J. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Nicholes, K.; Guillet, S.; Tomlinson, E.; Hillan, K.; Wright, B.; Frantz, G.D.; Pham, T.A.; Dillard-Telm, L.; Tsai, S.P.; Stephan, J.P.; et al. A mouse model of hepatocellular carcinoma: Ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am. J. Pathol. 2002, 160, 2295–2307. [Google Scholar] [CrossRef]
- Miura, S.; Mitsuhashi, N.; Shimizu, H.; Kimura, F.; Yoshidome, H.; Otsuka, M.; Kato, A.; Shida, T.; Okamura, D.; Miyazaki, M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 2012, 12, 56. [Google Scholar] [CrossRef]
- Zhou, M.; Yang, H.; Learned, R.M.; Tian, H.; Ling, L. Non-cell-autonomous activation of IL-6/STAT3 signaling mediates FGF19-driven hepatocarcinogenesis. Nat. Commun. 2017, 8, 15433. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, X.; Phung, V.; Lindhout, D.A.; Mondal, K.; Hsu, J.Y.; Yang, H.; Humphrey, M.; Ding, X.; Arora, T.; et al. Separating Tumorigenicity from Bile Acid Regulatory Activity for Endocrine Hormone FGF19. Cancer Res. 2014, 74, 3306–3316. [Google Scholar] [CrossRef]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; DePaoli, A.M.; Tian, H.; Ling, L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 1024–1042. [Google Scholar] [CrossRef]
- Zhou, M.; Luo, J.; Chen, M.; Yang, H.; Learned, R.M.; DePaoli, A.M.; Tian, H.; Ling, L. Mouse species-specific control of hepatocarcinogenesis and metabolism by FGF19/FGF15. J. Hepatol. 2017, 66, 1182–1192. [Google Scholar] [CrossRef]
- Pearce, S.C.; Al-Jawadi, A.; Kishida, K.; Yu, S.; Hu, M.; Fritzky, L.F.; Edelblum, K.L.; Gao, N.; Ferraris, R.P. Marked differences in tight junction composition and macromolecular permeability among different intestinal cell types. BMC Biol. 2018, 16, 19. [Google Scholar] [CrossRef]
- Fasano, A. Intestinal permeability and its regulation by zonulin: Diagnostic and therapeutic implications. Clin. Gastroenterol. Hepatol. 2012, 10, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A. Regulation of intercellular tight junctions by zonula occludens toxin and its eukaryotic analogue zonulin. Ann. N. Y. Acad. Sci. 2000, 915, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Uzzau, S.; Goldblum, S.E.; Fasano, A. Human zonulin, a potential modulator of intestinal tight junctions. J. Cell Sci. 2000, 113 Pt 24, 4435–4440. [Google Scholar]
- Sturgeon, C.; Lan, J.; Fasano, A. Zonulin transgenic mice show altered gut permeability and increased morbidity/mortality in the DSS colitis model. Ann. N. Y. Acad. Sci. 2017, 1397, 130–142. [Google Scholar] [CrossRef]
- Scheppach, W. Effects of short chain fatty acids on gut morphology and function. Gut 1994, 35, S35–S38. [Google Scholar] [CrossRef] [PubMed]
- Wirth, R.; Bodi, N.; Maroti, G.; Bagyanszki, M.; Talapka, P.; Fekete, E.; Bagi, Z.; Kovacs, K.L. Regionally distinct alterations in the composition of the gut microbiota in rats with streptozotocin-induced diabetes. PLoS ONE 2014, 9, e110440. [Google Scholar] [CrossRef] [PubMed]
- Graziani, C.; Talocco, C.; De Sire, R.; Petito, V.; Lopetuso, L.R.; Gervasoni, J.; Persichilli, S.; Franceschi, F.; Ojetti, V.; Gasbarrini, A.; et al. Intestinal permeability in physiological and pathological conditions: Major determinants and assessment modalities. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 795–810. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef]
- Gao, J.H.; Wen, S.L.; Tong, H.; Wang, C.H.; Yang, W.J.; Tang, S.H.; Yan, Z.P.; Tai, Y.; Ye, C.; Liu, R.; et al. Inhibition of cyclooxygenase-2 alleviates liver cirrhosis via improvement of the dysfunctional gut-liver axis in rats. Am. J. Physiol. 2016, 310, G962–G972. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Miele, L.; Principessa, L.; Ferretti, F.; Villa, M.P.; Negro, V.; Grieco, A.; Alisi, A.; Nobili, V. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Stud. Liver 2014, 46, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2019, 10. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E.; et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol. 2011, 300, G433–G441. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef]
- Fuchs, M.; Sanyal, A.J. Lipotoxicity in NASH. J. Hepatol. 2012, 56, 291–293. [Google Scholar] [CrossRef]
- Yu, L.X.; Yan, H.X.; Liu, Q.; Yang, W.; Wu, H.P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.Q.; Zou, S.S.; et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef]
- Machida, K.; Tsukamoto, H.; Mkrtchyan, H.; Duan, L.; Dynnyk, A.; Liu, H.M.; Asahina, K.; Govindarajan, S.; Ray, R.; Ou, J.H.; et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc. Natl. Acad. Sci. USA 2009, 106, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Pena, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Damms-Machado, A.; Louis, S.; Schnitzer, A.; Volynets, V.; Rings, A.; Basrai, M.; Bischoff, S.C. Gut permeability is related to body weight, fatty liver disease, and insulin resistance in obese individuals undergoing weight reduction. Am. J. Clin. Nutr. 2017, 105, 127–135. [Google Scholar] [CrossRef]
- Volynets, V.; Kuper, M.A.; Strahl, S.; Maier, I.B.; Spruss, A.; Wagnerberger, S.; Konigsrainer, A.; Bischoff, S.C.; Bergheim, I. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2012, 57, 1932–1941. [Google Scholar] [CrossRef]
- Bardella, M.T.; Valenti, L.; Pagliari, C.; Peracchi, M.; Fare, M.; Fracanzani, A.L.; Fargion, S. Searching for coeliac disease in patients with non-alcoholic fatty liver disease. Dig. Liver Dis. 2004, 36, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Reilly, N.R.; Lebwohl, B.; Hultcrantz, R.; Green, P.H.; Ludvigsson, J.F. Increased risk of non-alcoholic fatty liver disease after diagnosis of celiac disease. J. Hepatol. 2015, 62, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Duarte, N.; Coelho, I.C.; Patarrao, R.S.; Almeida, J.I.; Penha-Goncalves, C.; Macedo, M.P. How Inflammation Impinges on NAFLD: A Role for Kupffer Cells. Biomed. Res. Int. 2015, 2015, 984578. [Google Scholar] [CrossRef]
- Meli, R.; Mattace Raso, G.; Calignano, A. Role of Innate Immune Response in Non-Alcoholic Fatty Liver Disease: Metabolic Complications and Therapeutic Tools. Front. Immunol. 2014, 5, 177. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [CrossRef]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.L.; Gaudin, F.; Naveau, S.; Prevot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Duarte, N.; Coelho, I.; Holovanchuk, D.; Ines Almeida, J.; Penha-Goncalves, C.; Paula Macedo, M. Dipeptidyl Peptidase-4 Is a Pro-Recovery Mediator During Acute Hepatotoxic Damage and Mirrors Severe Shifts in Kupffer Cells. Hepatol. Commun. 2018, 2, 1080–1094. [Google Scholar] [CrossRef]
- Cheng, C.; Tan, J.; Qian, W.; Zhang, L.; Hou, X. Gut inflammation exacerbates hepatic injury in the high-fat diet induced NAFLD mouse: Attention to the gut-vascular barrier dysfunction. Life Sci. 2018, 209, 157–166. [Google Scholar] [CrossRef]
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velasquez-Mejia, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin Is Associated With Higher Relative Abundance of Mucin-Degrading Akkermansia muciniphila and Several Short-Chain Fatty Acid-Producing Microbiota in the Gut. Diabetes Care 2017, 40, 54–62. [Google Scholar] [CrossRef]
- Xue, Y.; Zhang, H.; Sun, X.; Zhu, M.J. Metformin Improves Ileal Epithelial Barrier Function in Interleukin-10 Deficient Mice. PLoS ONE 2016, 11, e0168670. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Ren, L.W.; Zhan, P.; Yang, H.Y.; Chai, D.D.; Yu, Z.W. Metformin exerts glucose-lowering action in high-fat fed mice via attenuating endotoxemia and enhancing insulin signaling. Acta Pharm. Sin. 2016, 37, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Feng, B.; Li, P.; Tang, Z.; Wang, L. Microflora Disturbance during Progression of Glucose Intolerance and Effect of Sitagliptin: An Animal Study. J. Diabetes Res. 2016, 2016, 10. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiao, X.; Li, M.; Yu, M.; Ping, F.; Zheng, J.; Wang, T.; Wang, X. Vildagliptin increases butyrate-producing bacteria in the gut of diabetic rats. PLoS ONE 2017, 12, e0184735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiao, X.; Zheng, J.; Li, M.; Yu, M.; Ping, F.; Wang, T.; Wang, X. Featured article: Structure moderation of gut microbiota in liraglutide-treated diabetic male rats. Exp. Biol. Med. 2018, 243, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Battson, M.L.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Weir, T.L.; Gentile, C.L. SGLT2 inhibition via dapagliflozin improves generalized vascular dysfunction and alters the gut microbiota in type 2 diabetic mice. Cardiovasc. Diabetol. 2018, 17, 62. [Google Scholar] [CrossRef]
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588. [Google Scholar] [CrossRef]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Suez, J.; Zmora, N.; Zilberman-Schapira, G.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Zur, M.; Regev-Lehavi, D.; Ben-Zeev Brik, R.; Federici, S.; et al. Post-Antibiotic Gut Mucosal Microbiome Reconstitution Is Impaired by Probiotics and Improved by Autologous FMT. Cell 2018, 174, 1406–1423. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef] [PubMed]




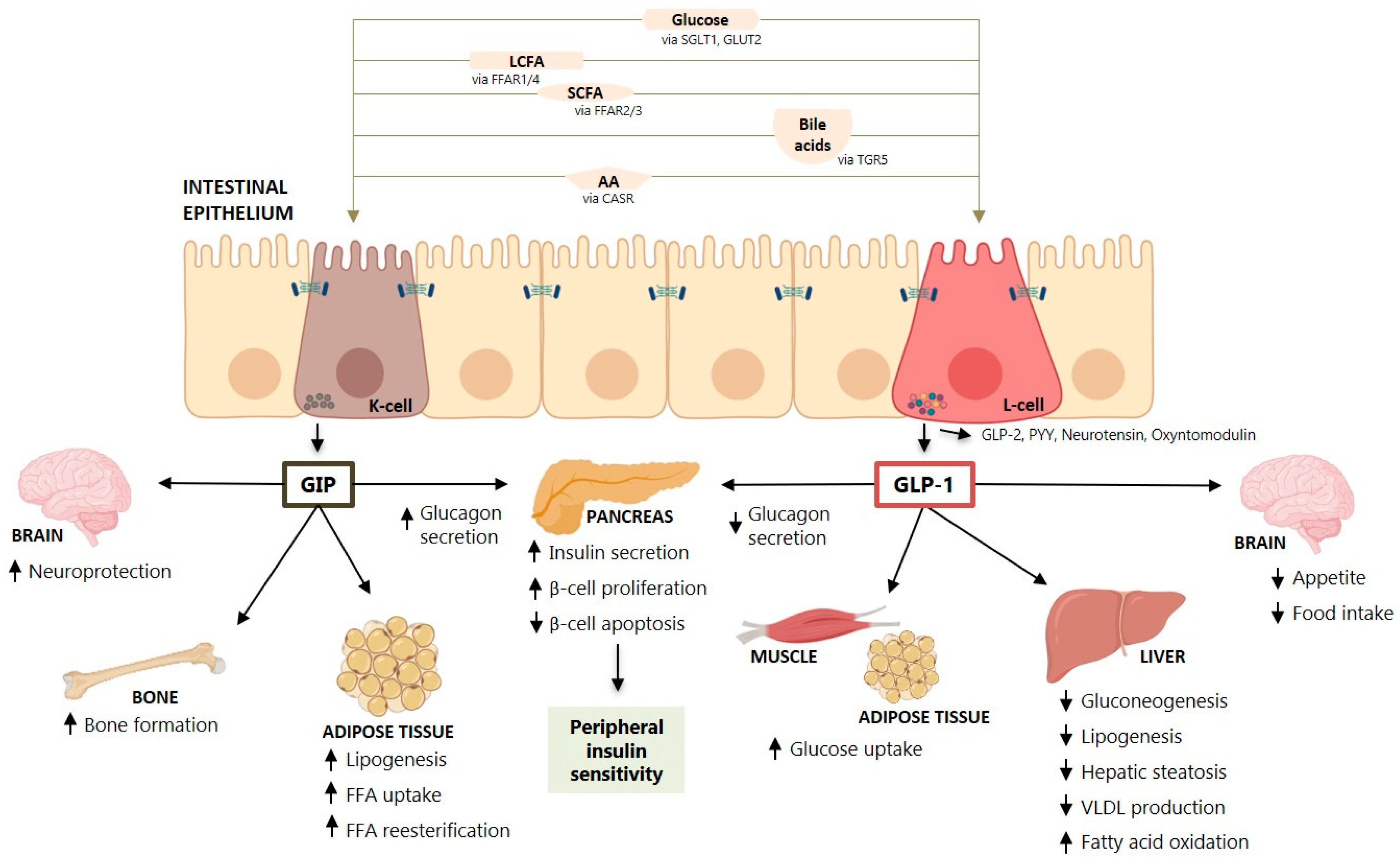{kind=link}
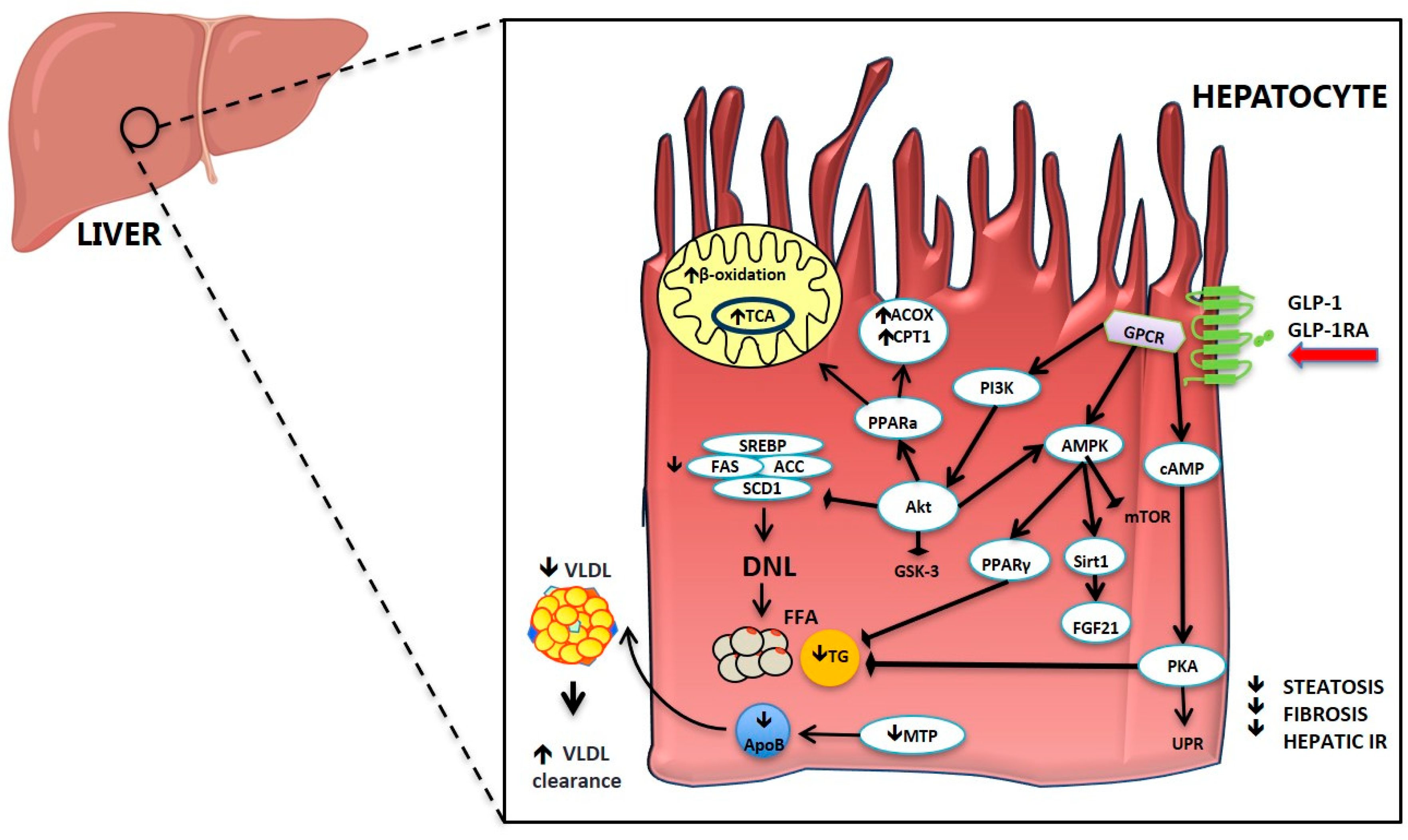{kind=link}
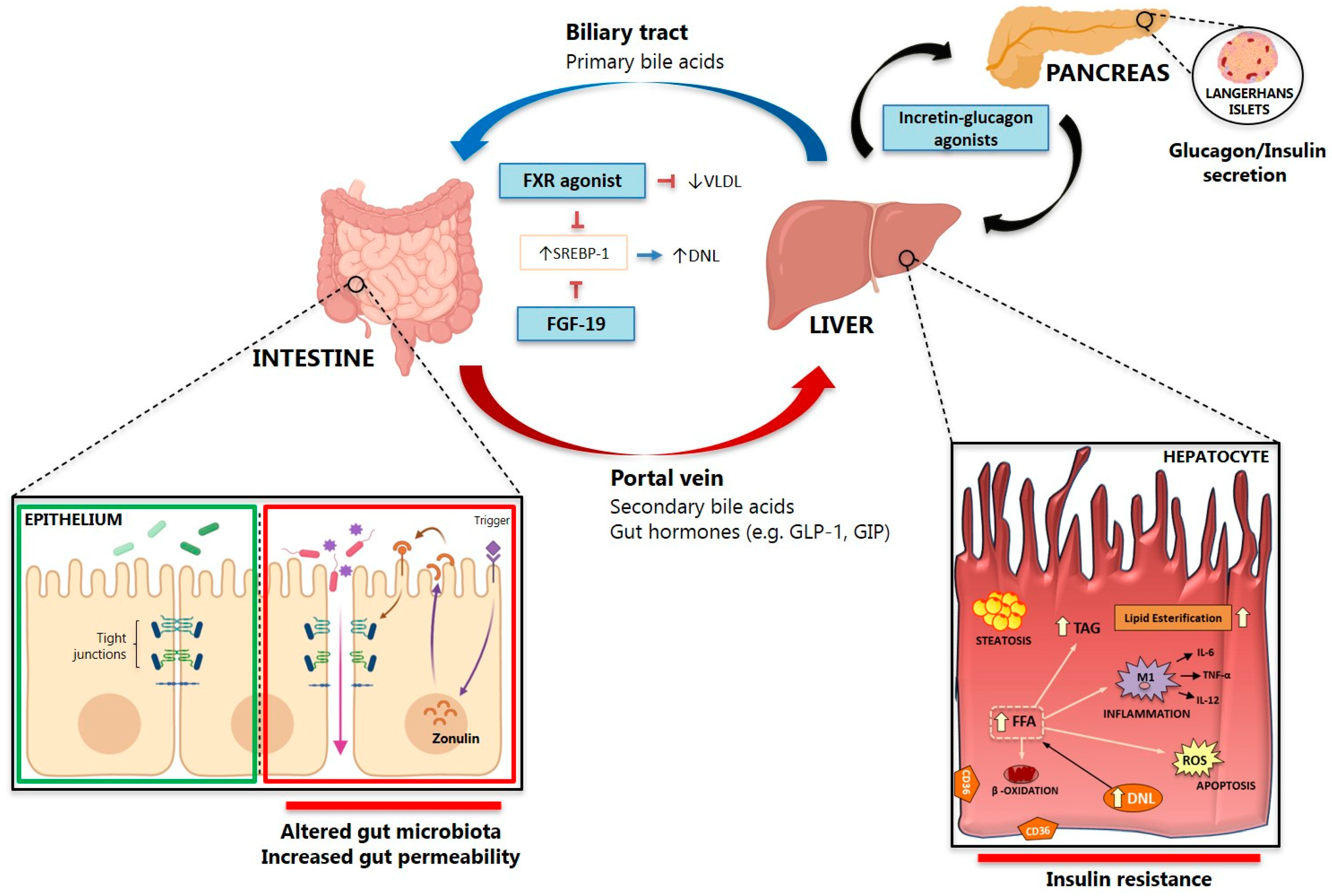{kind=link}
| Drug Name | Mechanism of Action | Admini-Stration | Stage of Development | Effect on Steatosis | Effect on Fibrosis | Effect on LFTs | Effect on Glucose Metabolism | Effect on Insulin Resistance | References |
|---|---|---|---|---|---|---|---|---|---|
| HM15211 | GLP-1/GIP/glucagon | SC | Phase I | Yes * | Yes * | Yes * | NA | NA | [40,41,42] |
| NGM313 MK3655 | Activator of FGFR1c/KL | SC (once monthly) | Phase I | Yes | NA | NA | Yes | Yes | [43,44] |
| RO5093151 | 11β-HSD1 inhibitor | PO | Phase I/II | Yes | NA | Yes | Yes | Yes | [45,46] |
| MK-0533 | PPAR γ/SPPARM | PO | Phase II | NA | NA | NA | Yes | Yes | [47] |
| Pemafibrate | PPAR α/SPPARM | PO | Phase II | NA | NA | Yes | NA | Yes | [48] |
| Saroglitazar | PPAR α/γ | PO | Phase IIa | Yes | Yes | Yes | Yes | Yes | [49,50,51] |
| Lanifibranor | PPAR α/γ/δ | PO | Phase IIa | Yes | Yes | Yes | Yes | Yes | [52,53] |
| Tropifexor | FXR agonist | PO | Phase IIb | Yes | Yes | Yes | NA | NA | [54,55] |
| Cilofexor | FXR agonist | PO | Phase II | Yes | No | Yes | NA | NA | [56,57] |
| NGM282 | FGF19 analogue | SC | Phase II | Yes | Yes | Yes | No | No | [58,59,60,61,62,63] |
| Cotadutide (MEDI0382) | GLP-1/glucagon | SC | Phase II | Yes | NA | Yes | Yes | Yes | [64,65] |
| ZP2929/BI 456906 | GLP-1/glucagon | SC | Phase II | Yes * | Yes * | NA | Yes * | Yes | [66,67] |
| Licogliflozin | Dual SGLT1/2 inhibitor | PO | Phase IIa | Yes | NA | Yes | Yes | Related to weight loss | [68,69] |
| BIO89-100 | PEG-FGF21 analogue | SC | Phase Ib/IIa | Yes | NA | Yes | Yes | Yes | [70] |
| PF-06835919 | Ketohexokinase (KHK) Inhibitor | PO | Phase II | Yes | NA | Yes | NA | Yes | [71] |
| TVB-2640 | Fatty acid synthase (FAS) inhibitor | PO | Phase II | Yes | NA | Yes | No | No | [72] |
| GS-0976 (Firsocostat) | Acetyl-CoA carboxylase (ACC) inhibitor | PO | Phase II | Yes | Yes | Yes | No | No | [73] |
| Resmetirom (MGL-3196) | Hepatic thyroid hormone receptor-β agonist | PO | Phase II/III | Yes | Yes | Yes | NA | NA | [74] |
| MSDC-0602K | Mitochondrial pyruvate carrier (MPC) | PO | Phase IIb | Yes | NA | Yes | Yes | Yes | [75,76] |
| Lobeglitazone | PPAR γ | PO | Phase III | Yes | No | NA | NA | Yes | [77] |
| INT-131 besylate | PPAR γ/SPPARM | PO | Phase III | Yes | No | NA | Yes | Yes | [78] |
| Elafibranor | PPAR α/δ | PO | Phase III | Yes | Yes | Yes | Yes | Yes | [79,80] |
| Obeticholic acid | FXR agonist | PO | Phase III | Yes, mild | Yes | Yes | No | NA | [81,82,83] |
| Tirzepatide (LY3298176) | GLP-1/GIP | SC | Phase III | Yes | Yes | Yes | Yes | Yes | [84,85,86] |
| NNC0090-2746/RG7697 | GLP-1/GIP | SC | Phase III | Yes | Yes | Yes | Yes | Yes | [87] |
| Pegbelfermin | Long-acting FGF21 analogue | SC | Phase III | Yes | Yes | Yes | No | No | [88,89] |
| Aramchol | Stearoyl-coenzyme-A-desaturase-1 (SCD1) inhibitor | PO | Phase III/IV | Yes | Yes | Yes | Yes | No | [90,91] |
| Pioglitazone | PPAR γ | PO | Phase IV | Marked | Yes | Yes | Yes | Yes | [92,93,94,95] |
| Fenofibrate | PPAR α | PO | Phase IV | Minimal | No | Yes | No | No | [96,97,98] |
| Bezafibrate | PPAR α | PO | Phase IV | NA | NA | Yes | No | No | [99,100,101] |
| Liraglutide | GLP-1RA | SC | Phase IV | Yes | Yes | Yes | Yes | Related to weight loss | [102,103,104] |
| Semaglutide | GLP-1RA | SC/PO | Phase IV | Yes | Yes | Yes | Yes | Related to weight loss | [105] |
| Dulaglutide | GLP-1RA | SC | Phase IV | Yes | Yes | Yes | Yes | Related to weight loss | [106,107] |
| Exenatide | GLP-1RA | SC | Phase IV | Yes | Yes | Yes | Yes | Related to weight loss | [24,108,109,110,111,112] |
| Lixisenatide | GLP-1RA | SC | Phase IV | NA | NA | Yes | Yes | Related to weight loss | [113] |
| Sitagliptin | DPP-4 inhibitor | PO | Phase IV | No | No | No | Yes | No | [114,115,116,117] |
| Empagliflozin | SGLT2 inhibitor | PO | Phase IV | Yes | Yes | Yes | Yes | Related to weight loss | [118,119,120] |
| Canagliflozin | SGLT2 inhibitor | PO | Phase IV | Yes | NA | Yes | Yes | Related to weight loss | [121,122,123,124,125,126] |
| Dapagliflozin | SGLT2 inhibitor | PO | Phase IV | Yes | NA | Yes | Yes | Related to weight loss | [111,127,128,129,130] |
| Ertugliflozin | SGLT2 inhibitor | PO | Phase IV | NA | NA | Yes | Yes | Related to weight loss | [131] |
| Ipragliflozin | SGLT2 inhibitor | PO | Approved by PMDA | Yes | Tendency to decrease | Yes | Yes | Related to weight loss | [117,132,133,134,135,136,137,138] |
| Tofogliflozin | SGLT2 inhibitor | PO | Approved by PMDA | Yes | NA | Yes | Yes | Related to weight loss | [139] |
| Luseogliflozin | SGLT2 inhibitor | PO | Approved by PMDA | Yes | NA | Yes | Yes | Related to weight loss | [140,141] |
| Glargine | Insulin | SC | Phase IV | Yes | NA | No | Yes | - | [112,142,143] |
| References | Subjects | Weeks of Treatment | Treatment Agent | Histology (Yes/No) | Fibrosis | Steatosis | Visceral Fat | ALT | BMI | HbA1c |
|---|---|---|---|---|---|---|---|---|---|---|
| Buse et al. (2007) [144] | T2D (n = 283) | 30 + 2 years follow up | Exenatide | No | - | - | - | ↓ (if elevated at baseline) | ↓ | ↓ |
| Klonoff et al. (2008) [110] | T2D (n = 217) | 30 + 3 years follow up | Exenatide | No | - | - | - | ↓ (if elevated at baseline) | ↓ | ↓ |
| Jendle et al. (2009) [145] | T2D (n = 131) | 26 | Liraglutide + Metformin | No | - | ↓ (CT) | ↓ (CT) | ↓ | ↓ | ↓ |
| Kenny et al. (2010) [146] | T2D and biopsy-proven NAFLD (n = 8) | 28 | Exenatide | Yes | ↓ | ↓ | - | ↓ | ↓ | ↓ |
| Sathyanarayana et al. (2011) [147] | T2D (n = 21) | 52 | Exenatide + Pioglitazone | No | - | ↓ (MRS) | - | ↓ | ↓ | ↓ |
| Yilmaz et al. (2012) [148] | T2D+ NASH (n = 15) | 52 | Sitagliptin | Yes | = | = | - | ↓ | ↓ | = |
| Ohki et al. (2012) [149] | T2D and NAFLD (n = 26) | NA | Liraglutide | No | ↓ (APRI) | - | - | ↓ | ↓ | ↓ |
| Ohki et al. (2012) [149] | T2D and NAFLD (n = 36) | NA | Sitagliptin | No | = (APRI) | - | - | ↓ | = | ↓ |
| Cuthberson et al. (2012) [150] | Obese, T2D and NAFLD (n = 25, 19/6) | 26 | Exenatide or Liraglutide | No | - | ↓ (MRS) | ↓ (MRI) | ↓ | ↓ | ↓ |
| Fan et al. (2013) [151] | T2D and NAFLD (n = 117) | 12 | Exenatide or Metformin | No | - | - | - | ↓ | ↓ | = |
| Suzuki et al. (2013) [152] | T2D (n = 46) | 26 | Liraglutide + Pioglitazone | No | - | ↓ (CT) | ↓ (CT) | - | ↓ | = |
| Bergenstal et al. (2013) [153] | T2D (n = 534) | 52 | Exenatide | No | - | - | - | ↓ | ↓ | ↓ |
| Blaslov et al. (2014) [154] | T2D (n = 125) | 26 | Exenatide alone or add-on metformin or/and sulphonylurea | No | - | ↓ (FLI) | - | ↓ | ↓ | ↓ |
| Shao et al. (2014) [109] | Obese, NAFLD with elevated liver enzymes and T2D (n = 60) | 12 | Exenatide + Insulin | No | - | ↓ (US) | - | ↓ | ↓ | ↓ |
| Watanabe et al. (2015) [115] | T2D and NAFLD (n = 7) | 12 | Sitagliptin | No | - | ↓ (MRS) | = (MRI) | = | = | ↓ |
| Tang et al. (2015) [142] | T2D (n = 35) | 12 | Liraglutide or Insulin glargine | No | - | = (MRS) | - | = | ↓ | ↓ |
| Eguchi et al.* (2015) [155] | Diabetic NAFLD/NASH (n = 19) | 24 + 24 | Lifestyle + Liraglutide | No | ↓ (FIB-4) | ↓ (CT) | ↓ (CT) | ↓ | ↓ | ↓ |
| Eguchi et al.* (2015) [155] | T2D+ NAFLD/NASH (n = 10) | 24 + 24 + 96 | Lifestyle + Liraglutide + Follow-up Liraglutide | Yes | ↓ | ↓ | - | ↓ | ↓ | ↓ |
| Smits et al. (2016) [156] | Overweight T2D (n = 52) | 12 | Liraglutide or Sitagliptin | No | = (APRI/FIB-4/NFS) | = (MRS) | - | ↓ | = | ↓ |
| Cui et al. (2016) [114] | Prediabetic NAFLD (n = 50) | 24 | Sitagliptin | No | = (MRE) | = (MRI) | - | = | = | = |
| Armstrong et al. (2016) [103] | Biopsy-proven NASH (n = 23) | 48 | Liraglutide | Yes | ↓ | ↓ | - | ↓ | ↓ | ↓ |
| Joy et al. (2017) [116] | Biopsy-proven NASH (n = 12) | 26 | Sitagliptin | Yes | = (NAS) | = (MRI) | = (MRI) | = | = | ↓ |
| Petit et al. (2017) [157] | T2D (n = 68) | 26 | Liraglutide | No | - | ↓ (MRS) | - | ↓ | ↓ | ↓ |
| Feng et al. (2017) [158] | T2D and NAFLD (n = 87) | 24 | Liraglutide, Metformin or Gliclazide | No | - | ↓ (US) | - | ↓ | ↓ | ↓ |
| Khoo et al. (2017) [159] | Obese NAFLD (n = 24) | 26 | Liraglutide versus Lifestyle | No | - | ↓ (MRI) | - | ↓ | ↓ | - |
| Seko et al. (2017) [107] | T2D and biopsy-proven NAFLD (n = 15) | 12 | Dulaglutide | Yes | ↓ | ↓ | - | ↓ | ↓ | ↓ |
| Cusi et al. (2018) [106] | T2D (n = 971) | 26 | Dulaglutide | No | - | - | - | ↓ | - | ↓ |
| Yan et al. (2019) [143] | T2D and NAFLD (n = 18) | 26 | Liraglutide + Metformin | No | - | ↓ (MRI) | ↓ (MRI) | - | ↓ | ↓ |
| Gastaldelli et al. (2019) [111] | T2D (n = 228) | 28/52 | Exenatide + Dapagliflozin | No | - | - | - | ↓ | - | ↓ |
| Co-agonism | Drug | Company | Stage of Development | References |
|---|---|---|---|---|
| GLP-1/glucagon | Cotadutide (MEDI0382) | Medimmune | Phase II | [64,65] |
| HM12525A; JNJ-64565111 | Hanmi Pharmaceuticals | Phase II | [67] | |
| MK-8521 | Merck | Phase II | [67] | |
| SAR425899 | Sanofi | Phase II | [199] | |
| ZP2929; BI 456906 | Zealand Pharma | Phase II | [66,67] | |
| JNJ-54728518 | Janssen Pharmaceuticals | Phase I | [200] | |
| NN9277; NNC 9204-1177 | Novo Nordisk | Phase I | - | |
| MOD-6030 | Prolor/OPKO Biologics | Phase I | [201] | |
| VPD-107 | Spitfire Pharma | Pre-clinical | - | |
| GLP-1/GIP | Tirzepatide (LY3298176) | Eli Lilly | Phase III | [84,85,86] |
| NNC0090-2746; NN970 9; MAR709; RG7697 | Novo Nordisk/Marcadia | Phase III-stopped | [87] | |
| CPD86 | Eli Lilly | Pre-clinical | - | |
| ZP-I-98 | Zealand Pharma | Pre-clinical | - | |
| ZP-DI-70 | Zealand Pharma | Pre-clinical | - | |
| GLP-1/GIP/glucagon | HM15211 | Hanmi Pharmaceuticals | Phase I | [40,41,42] |
| NN9423; NNC 92041706; Tri-agonist 1706 | Novo Nordisk | Phase I | [67] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svegliati-Baroni, G.; Patrício, B.; Lioci, G.; Macedo, M.P.; Gastaldelli, A. Gut-Pancreas-Liver Axis as a Target for Treatment of NAFLD/NASH. Int. J. Mol. Sci. 2020, 21, 5820. https://doi.org/10.3390/ijms21165820
Svegliati-Baroni G, Patrício B, Lioci G, Macedo MP, Gastaldelli A. Gut-Pancreas-Liver Axis as a Target for Treatment of NAFLD/NASH. International Journal of Molecular Sciences. 2020; 21(16):5820. https://doi.org/10.3390/ijms21165820
Chicago/Turabian StyleSvegliati-Baroni, Gianluca, Bárbara Patrício, Gessica Lioci, Maria Paula Macedo, and Amalia Gastaldelli. 2020. "Gut-Pancreas-Liver Axis as a Target for Treatment of NAFLD/NASH" International Journal of Molecular Sciences 21, no. 16: 5820. https://doi.org/10.3390/ijms21165820
APA StyleSvegliati-Baroni, G., Patrício, B., Lioci, G., Macedo, M. P., & Gastaldelli, A. (2020). Gut-Pancreas-Liver Axis as a Target for Treatment of NAFLD/NASH. International Journal of Molecular Sciences, 21(16), 5820. https://doi.org/10.3390/ijms21165820