Targeting Hypoxia Sensitizes TNBC to Cisplatin and Promotes Inhibition of Both Bulk and Cancer Stem Cells



Abstract
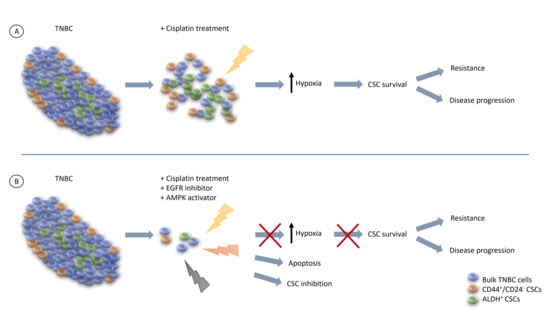
1. Introduction
2. Results
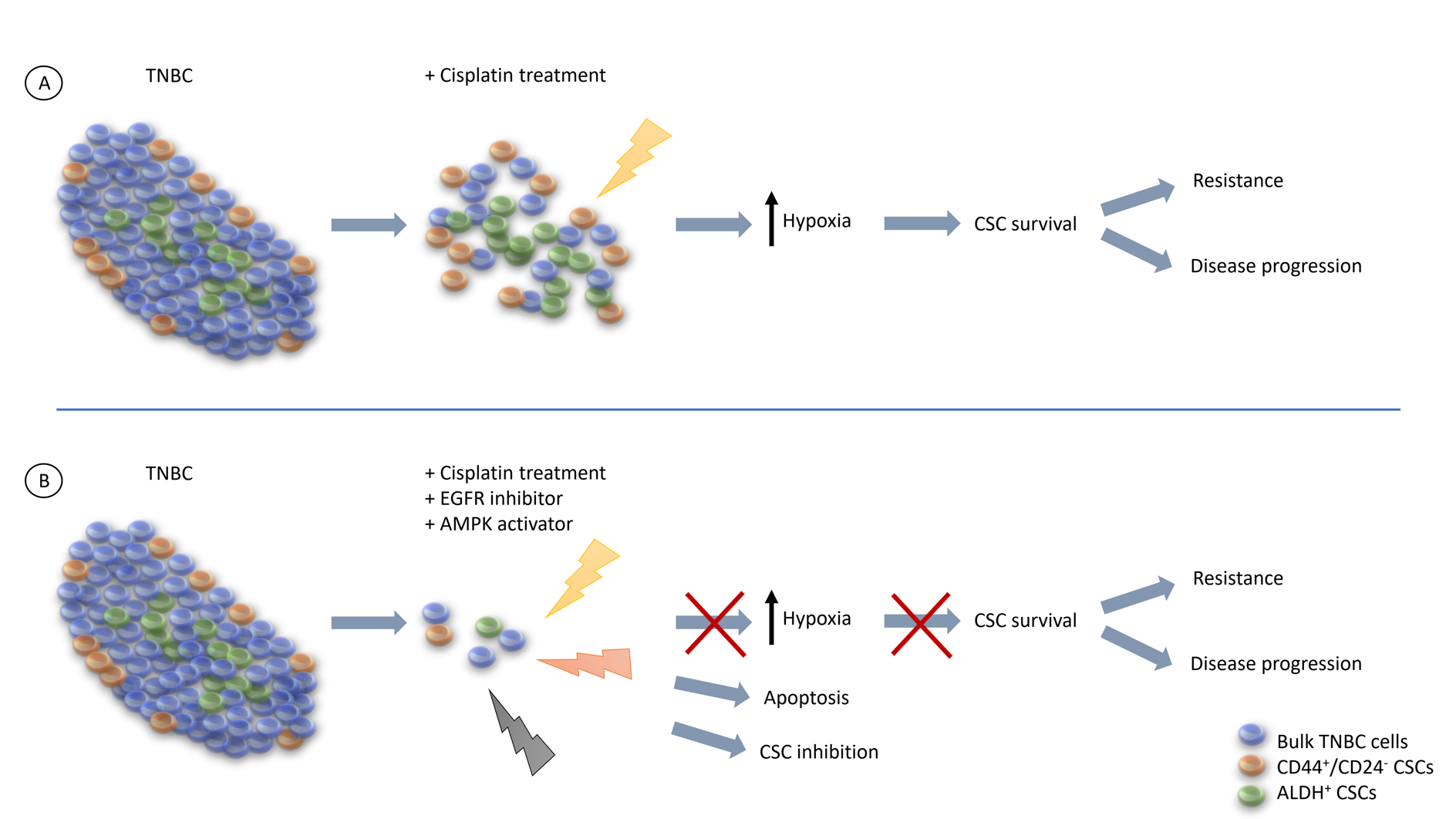
2.1. Upregulated Gene Expressions of EGFR and Hypoxia Associate with Cisplatin Resistance, Anti-Apoptosis, and Stemness
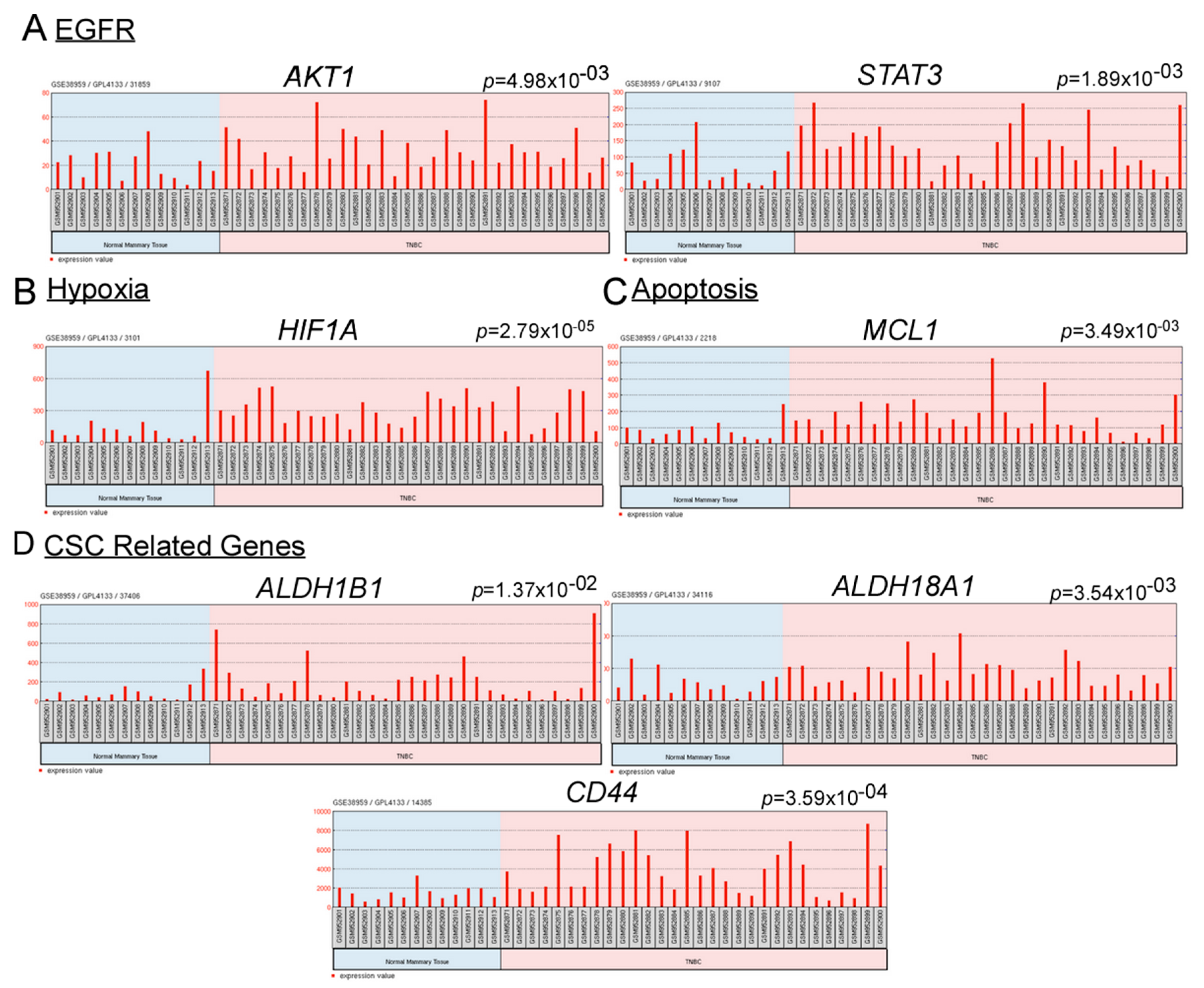
2.2. Combination of Hypoxia and EGFR Inhibitors with Cisplatin Effectively Suppress TNBC Bulk and CSC Populations
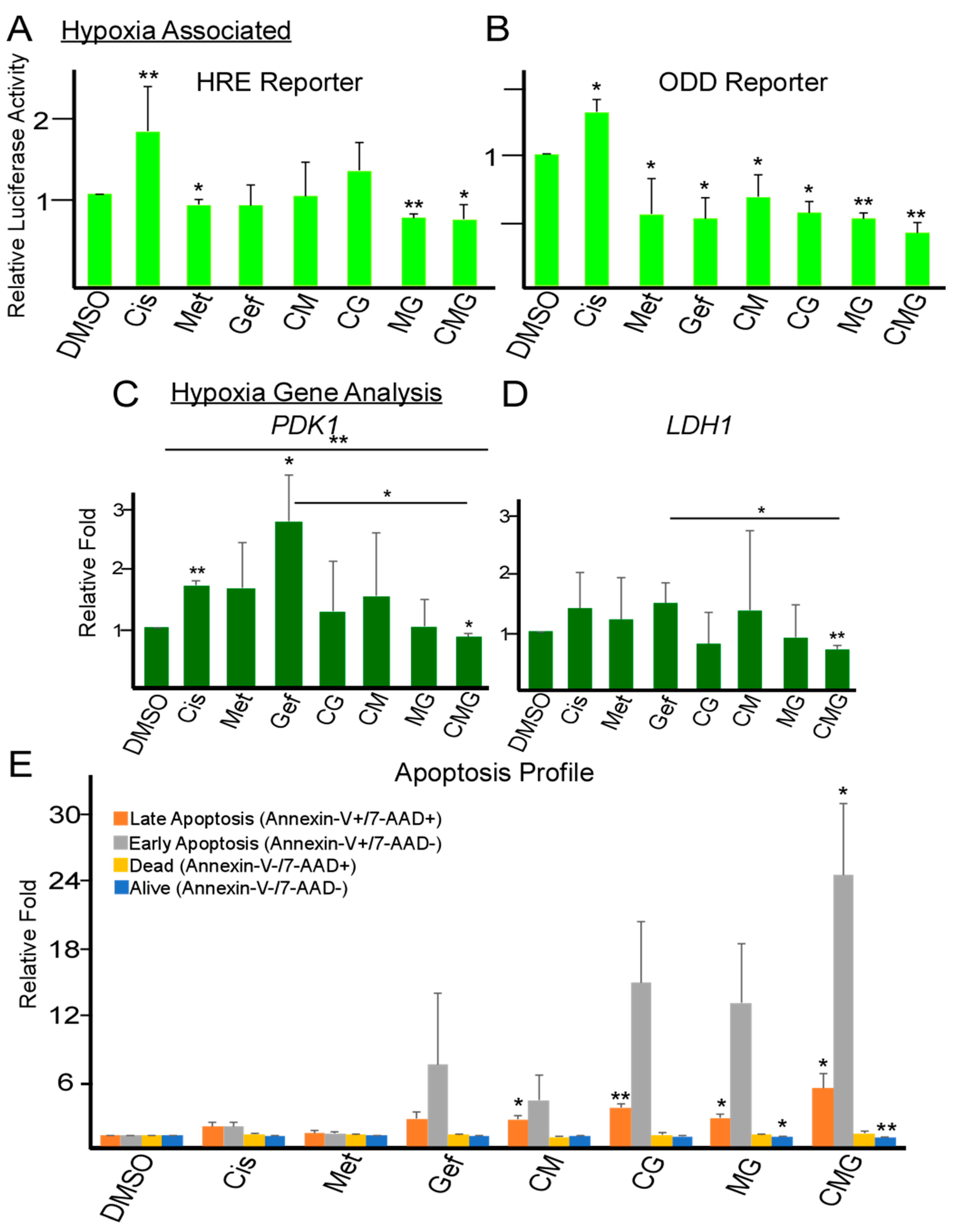
2.3. Combination of Metformin, Gefitinib and Cisplatin Effectively Repress Hypoxia and Promote Apoptosis in TNBC
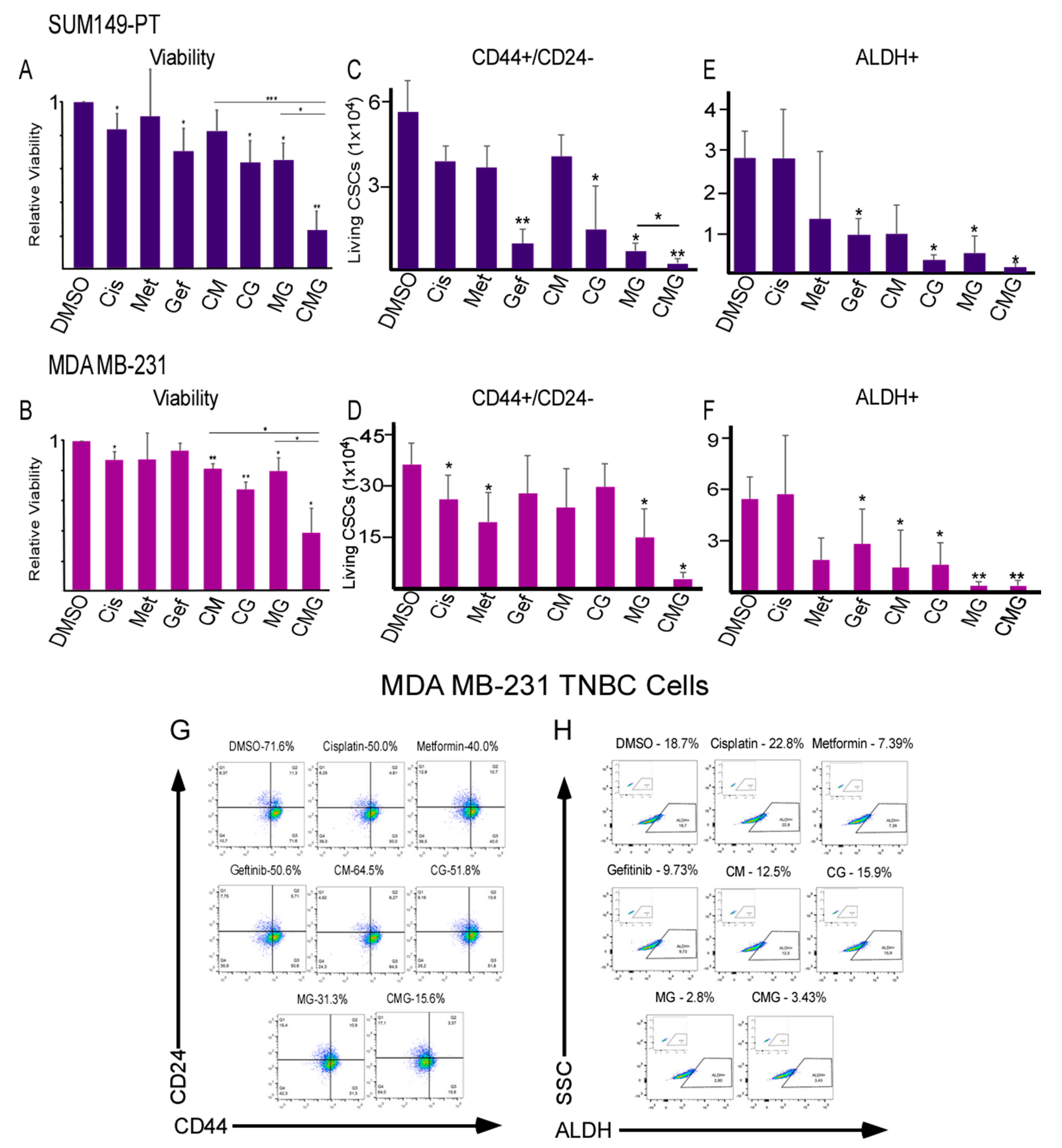
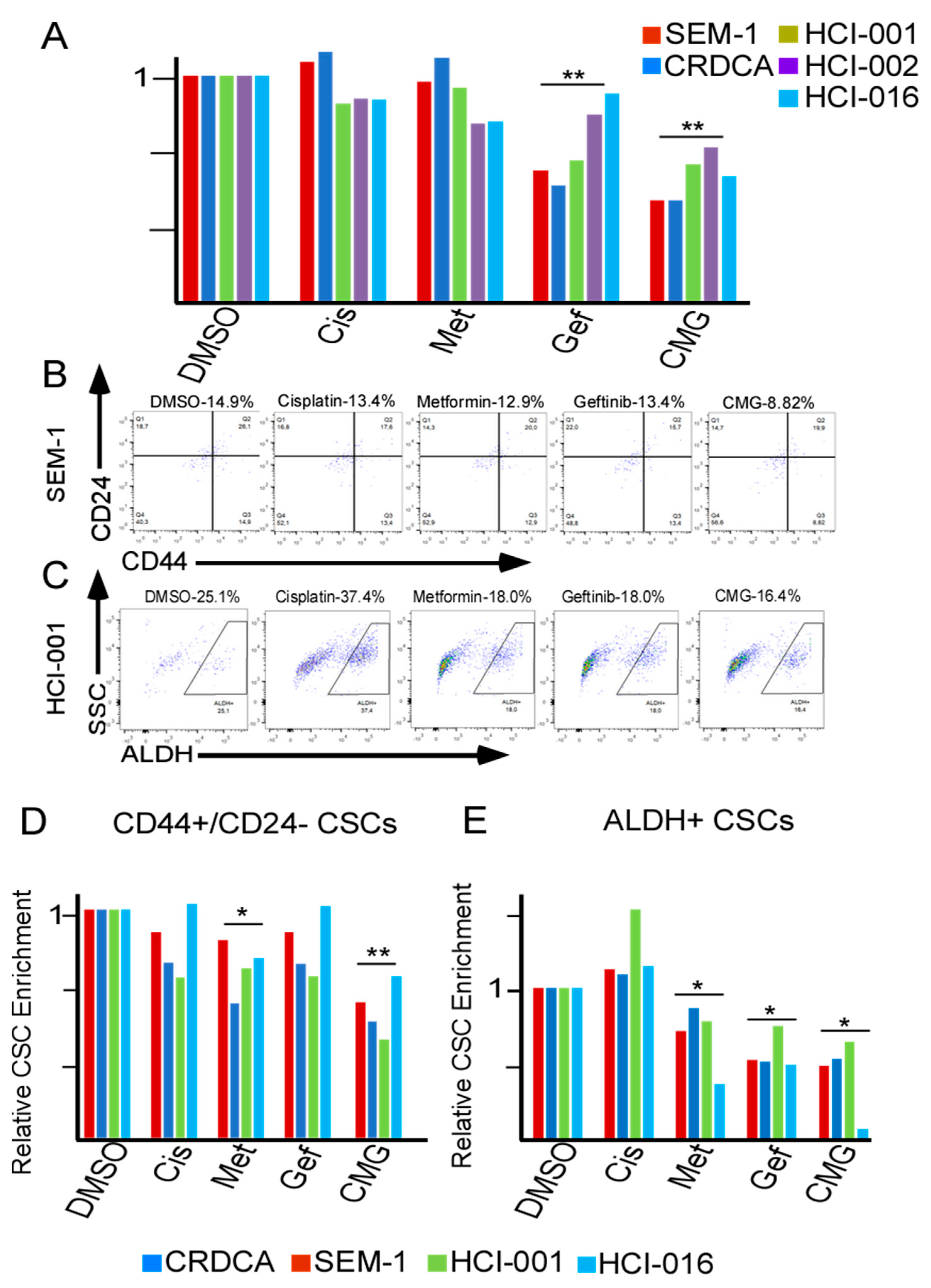
2.4. CMG Treatment Effectively Inhibits the Viability of TNBC Organotypic Cultures and Reduces Their CSC Subpopulations
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Organotypic Cultures of Patient TNBC Breast Tumor and Patient-Derived Xenograft Fragments
4.3. Flow Cytometry Analysis
4.4. Quantitative Real-Time PCR
4.5. Luciferase Reporter
4.6. Cell Viability Assays
4.7. Clinical Database Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | Epidermal growth factor receptor |
| CD44 | Cluster of differentiation 44 (hyaluronic acid receptor) |
| CD24 | Cluster of differentiation 24 |
| CSC | Cancer stem cell |
| ALDH | Aldehyde dehydrogenase |
| ESR1 | Estrogen receptor 1 (alpha) |
| PGR | Progesterone receptor |
| HER2 | Human epidermal growth factor receptor type 2 |
| PDX | Patient-derived xenograft |
| TNBC | Triple negative breast cancer |
| CMG | Cisplatin, Metformin and Gefitinib combination |
| MG | Metformin and gefitinib combination |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. Ca A Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Go, R.S.; Adjei, A.A. Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J. Clin. Oncol. 1999, 17, 409. [Google Scholar] [CrossRef]
- Hu, X.C.; Zhang, J.; Xu, B.H.; Cai, L.; Ragaz, J.; Wang, Z.H.; Wang, B.Y.; Teng, Y.E.; Tong, Z.S.; Pan, Y.Y.; et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 436–446. [Google Scholar] [CrossRef]
- Rodler, E.T.; Kurland, B.F.; Griffin, M.; Gralow, J.R.; Porter, P.; Yeh, R.F.; Gadi, V.K.; Guenthoer, J.; Beumer, J.H.; Korde, L.; et al. Phase I study of veliparib (ABT-888) combined with cisplatin and vinorelbine in advanced triple-negative breast cancer and/or BRCA mutation–associated breast cancer. Clin. Cancer Res. 2016, 22, 2855–2864. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Metzger-Filho, O.; Sarmento, R.; Bines, J. Neoadjuvant Treatment of Stage IIB/III Triple Negative Breast Cancer with Cyclophosphamide, Doxorubicin, and Cisplatin (CAP Regimen): A Single Arm, Single Center Phase II Study (GBECAM 2008/02). Front. Oncol. 2018, 7, 329. [Google Scholar] [CrossRef]
- Su, Y.W.; Hung, C.Y.; Lam, H.B.; Chang, Y.C.; Yang, P.S. A single institution experience of incorporation of cisplatin into adjuvant chemotherapy for patients with triple-negative breast cancer of unknown BRCA mutation status. Clin. Med. Insights Oncol. 2018, 12, 1179554918794672. [Google Scholar] [CrossRef]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol 2010, 28, 1145. [Google Scholar] [CrossRef]
- Zhang, F.; Duan, S.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O.; Chen, Y. Cisplatin treatment increases stemness through upregulation of hypoxia-inducible factors by interleukin-6 in non-small cell lung cancer. Cancer Sci. 2016, 107, 746–754. [Google Scholar] [CrossRef]
- Jia, D.; Tan, Y.; Liu, H.; Ooi, S.; Li, L.; Wright, K.; Bennett, S.; Addison, C.L.; Wang, L.J.O. Cardamonin reduces chemotherapy-enriched breast cancer stem-like cells in vitro and in vivo. Oncotarget 2016, 7, 771. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; Li, L.; Jia, D.; Ooi, S.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; Nessim, C.; Yao, Z.; et al. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol. Oncol. 2018, 12, 423–440. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Lee, H.H.; Chang, S.S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR signaling enhances aerobic glycolysis in triple-negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609. [Google Scholar]
- Rho, J.K.; Choi, Y.J.; Lee, J.K.; Ryoo, B.Y.; Na, I.I.; Yang, S.H.; Kim, C.H.; Yoo, Y.D.; Lee, J.C. Gefitinib circumvents hypoxia-induced drug resistance by the modulation of HIF-1α. Oncol. Rep. 2009, 21, 801–807. [Google Scholar]
- Liu, S.; Yang, H.; Ge, X.; Su, L.; Zhang, A.; Liang, L. Drug resistance analysis of gefitinib-targeted therapy in non-small cell lung cancer. Oncol. Lett. 2016, 12, 3941–3943. [Google Scholar] [CrossRef]
- Minakata, K.; Takahashi, F.; Nara, T.; Hashimoto, M.; Tajima, K.; Murakami, A.; Nurwidya, F.; Yae, S.; Koizumi, F.; Moriyama, H.; et al. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Sci. 2012, 103, 1946–1954. [Google Scholar] [CrossRef]
- Huang, W.C.; Chen, Y.J.; Li, L.Y.; Wei, Y.L.; Hsu, S.C.; Tsai, S.L.; Chiu, P.C.; Huang, W.P.; Wang, Y.N.; Chen, C.H.; et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J. Biol. Chem. 2011, 286, 20558–20568. [Google Scholar] [CrossRef]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M.; Shi, B.; Fu, X.; Chen, Y.; Chen, L.; et al. Metformin suppresses hypoxia-induced stabilization of HIF-1α through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873. [Google Scholar] [CrossRef]
- Yin, X.; Wei, Z.; Song, C.; Tang, C.; Xu, W.; Wang, Y.; Xie, J.; Lin, Z.; Han, W. Metformin sensitizes hypoxia-induced gefitinib treatment resistance of HNSCC via cell cycle regulation and EMT reversal. Cancer Manag. Res. 2018, 10, 5785. [Google Scholar] [CrossRef]
- Fujita, H.; Hirose, K.; Sato, M.; Fujioka, I.; Fujita, T.; Aoki, M.; Takai, Y. Metformin attenuates hypoxia-induced resistance to cisplatin in the HepG2 cell line. Oncol. Lett. 2019, 17, 2431–2440. [Google Scholar] [CrossRef]
- Shi, P.; Liu, W.; Wang, H.; Li, F.; Zhang, H.; Wu, Y.; Kong, Y.; Zhou, Z.; Wang, C.; Chen, W.; et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 2017, 3, 1–13. [Google Scholar] [CrossRef]
- Soleymani Abyaneh, H.; Gupta, N.; Radziwon-Balicka, A.; Jurasz, P.; Seubert, J.; Lai, R.; Lavasanifar, A.J.C. STAT3 but not HIF-1α is important in mediating Hypoxia-Induced chemoresistance in MDA-MB-231, a triple negative breast cancer cell line. Cancers 2017, 9, 137. [Google Scholar] [CrossRef]
- Kim, E.M.; Mueller, K.; Gartner, E.; Boerner, J. Dasatinib is synergistic with cetuximab and cisplatin in triple-negative breast cancer cells. J. Surg. Res. 2013, 185, 231–239. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Shiraishi, A.; Tachi, K.; Essid, N.; Tsuboi, I.; Nagano, M.; Kato, T.; Yamashita, T.; Bando, H.; Hara, H.; Ohneda, O. Hypoxia promotes the phenotypic change of aldehyde dehydrogenase activity of breast cancer stem cells. Cancer Sci. 2017, 108, 362–372. [Google Scholar] [CrossRef]
- Crowder, S.W.; Balikov, D.A.; Hwang, Y.S.; Sung, H.J. Cancer stem cells under hypoxia as a chemoresistance factor in the breast and brain. Curr. Pathobiol. Rep. 2014, 2, 33–40. [Google Scholar] [CrossRef]
- Emerling, B.M.; Weinberg, F.; Liu, J.L.; Mak, T.W.; Chandel, N.S. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). Proc. Natl. Acad. Sci. USA 2008, 105, 2622–2627. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.D.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Safran, M.; Kim, W.Y.; O’Connell, F.; Flippin, L.; Günzler, V.; Horner, J.W.; DePinho, R.A.; Kaelin, W.G. Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: Assessment of an oral agent that stimulates erythropoietin production. Proc. Natl. Acad. Sci. USA 2006, 103, 105–110. [Google Scholar] [CrossRef]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M.R.; et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef]
- McGinn, O.; Gupta, V.K.; Dauer, P.; Arora, N.; Sharma, N.; Nomura, A.; Dudeja, V.; Saluja, A.; Banerjee, S. Inhibition of hypoxic response decreases stemness and reduces tumorigenic signaling due to impaired assembly of HIF1 transcription complex in pancreatic cancer. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Group, I.A.L.C.T.C. Cisplatin-based adjuvant chemotherapy in patients with completely resected non–small-cell lung cancer. New Engl. J. Med. 2004, 350, 351–360. [Google Scholar]
- Collaboration, N.C.f.L.A.C.C.M. Neoadjuvant chemotherapy for locally advanced cervical cancer: A systematic review and meta-analysis of individual patient data from 21 randomised trials. Eur. J. Cancer 2003, 39, 2470–2486. [Google Scholar]
- McClay, E.F.; Howell, S.B. A review: Intraperitoneal cisplatin in the management of patients with ovarian cancer. Gynecol. Oncol. 1990, 36, 1–6. [Google Scholar] [CrossRef]
- Andronescu, E.; Ficai, A.; Albu, M.G.; Mitran, V.; Sonmez, M.; Ficai, D.; Ion, R.; Cimpean, A. Collagen-hydroxyapatite/cisplatin drug delivery systems for locoregional treatment of bone cancer. Technol. Cancer Res. Treat. 2013, 12, 275–284. [Google Scholar] [CrossRef]
- Walsh, T.N.; Noonan, N.; Hollywood, D.; Kelly, A.; Keeling, N.; Hennessy, T.P. A comparison of multimodal therapy and surgery for esophageal adenocarcinoma. New Engl. J. Med. 1996, 335, 462–467. [Google Scholar] [CrossRef]
- Zhang, J.; Lin, Y.; Sun, X.; Wang, B.; Wang, Z.; Luo, J.; Wang, L.; Zhang, S.; Cao, J.; Tao, Z.; et al. Biomarker assessment of the CBCSG006 trial: A randomized phase III trial of cisplatin plus gemcitabine compared with paclitaxel plus gemcitabine as first-line therapy for patients with metastatic triple-negative breast cancer. Ann. Oncol. 2018, 29, 1741–1747. [Google Scholar] [CrossRef]
- Baselga, J.; Gómez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pêgo, A.; Chan, A.; et al. Randomized phase II study of the anti–epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586. [Google Scholar] [CrossRef]
- Jovanović, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus in patients with stage II/III Triple-Negative Breast Cancer (TNBC): Responses and long-term outcome correlated with increased frequency of DNA damage response gene mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar]
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A multicenter phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J. Clin. Oncol. 2015, 33, 1902. [Google Scholar] [CrossRef]
- Baek, D.W.; Park, J.Y.; Lee, S.J.; Chae, Y.S. Impressive effect of cisplatin monotherapy on a patient with heavily pretreated triple-negative breast cancer with poor performance. Yeungnam Univ. J. Med. 2020, 37, 230–235. [Google Scholar] [CrossRef]
- Fan, Y.; Xu, B.; Yuan, P.; Ma, F.; Wang, J.; Ding, X.; Zhang, P.; Li, Q.; Cai, R. Docetaxel–cisplatin might be superior to docetaxel–capecitabine in the first-line treatment of metastatic triple-negative breast cancer. Ann. Oncol. 2012, 24, 1219–1225. [Google Scholar] [CrossRef]
- Ryan, P.; Tung, N.; Isakoff, S.; Golshan, M.; Richardson, A.; Corben, A.; Smith, B.; Gelman, R.; Winer, E.; Garber, J. Neoadjuvant cisplatin and bevacizumab in triple negative breast cancer (TNBC): Safety and efficacy. J. Clin. Oncol. 2009, 27, 551. [Google Scholar]
- Liu, C.C.; Lin, J.H.; Hsu, T.W.; Su, K.; Li, A.F.Y.; Hsu, H.S.; Hung, S.C. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int. J. Cancer 2015, 136, 547–559. [Google Scholar]
- Qin, J.J.; Yan, L.; Zhang, J.; Zhang, W.D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 195. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; Den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Kuo, W.Y.; Hwu, L.; Wu, C.Y.; Lee, J.S.; Chang, C.W.; Liu, R.S. STAT3/NF-κB-Regulated Lentiviral TK/GCV Suicide Gene Therapy for Cisplatin-Resistant Triple-Negative Breast Cancer. Theranostics 2017, 7, 647. [Google Scholar] [CrossRef] [PubMed]
- Morais, C.; Gobe, G.; Johnson, D.W.; Healy, H. Inhibition of nuclear factor kappa B transcription activity drives a synergistic effect of pyrrolidine dithiocarbamate and cisplatin for treatment of renal cell carcinoma. Apoptosis 2010, 15, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Ma, Y.; Zhang, Z.; Zhao, J.; Kobayashi, H.; Zhang, L.; Fu, L. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol. Rep. 2010, 23, 671–676. [Google Scholar] [PubMed]
- Shin, D.H.; Choi, Y.J.; Park, J.W. SIRT1 and AMPK mediate hypoxia-induced resistance of non–small cell lung cancers to cisplatin and doxorubicin. Cancer Res. 2014, 74, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Leithner, K.; Wohlkoenig, C.; Quehenberger, F.; Bertsch, A.; Olschewski, A.; Olschewski, H.; Hrzenjak, A. Panobinostat reduces hypoxia-induced cisplatin resistance of non-small cell lung carcinoma cells via HIF-1α destabilization. Mol. Cancer 2015, 14, 1. [Google Scholar] [CrossRef]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47+/CD73+/PDL1+ immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 201718197. [Google Scholar] [CrossRef]
- Ye, J.; Chen, K.; Qi, L.; Li, R.; Tang, H.; Zhou, C.; Zhai, W. Metformin suppresses hypoxia-induced migration via the HIF-1α/VEGF pathway in gallbladder cancer in vitro and in vivo. Oncol. Rep. 2018, 40, 3501–3510. [Google Scholar] [CrossRef]
- Tadakawa, M.; Takeda, T.; Li, B.; Tsuiji, K.; Yaegashi, N. The anti-diabetic drug metformin inhibits vascular endothelial growth factor expression via the mammalian target of rapamycin complex 1/hypoxia-inducible factor-1α signaling pathway in ELT-3 cells. Mol. Cell. Endocrinol. 2015, 399, 1–8. [Google Scholar] [CrossRef]
- Sahra, I.B.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.-F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef]
- Honjo, S.; Ajani, J.A.; Scott, A.W.; Chen, Q.; Skinner, H.D.; Stroehlein, J.; Johnson, R.L.; Song, S. Metformin sensitizes chemotherapy by targeting cancer stem cells and the mTOR pathway in esophageal cancer. Int. J. Oncol. 2014, 45, 567–574. [Google Scholar] [CrossRef]
- Solomon, B.; Binns, D.; Roselt, P.; Weibe, L.I.; McArthur, G.A.; Cullinane, C.; Hicks, R.J. Modulation of intratumoral hypoxia by the epidermal growth factor receptor inhibitor gefitinib detected using small animal PET imaging. Mol. Cancer Ther. 2005, 4, 1417–1422. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murakami, A.; Takahashi, F.; Nurwidya, F.; Kobayashi, I.; Minakata, K.; Hashimoto, M.; Nara, T.; Kato, M.; Tajima, K.; Shimada, N.; et al. Hypoxia increases gefitinib-resistant lung cancer stem cells through the activation of insulin-like growth factor 1 receptor. PLoS ONE 2014, 9, e86459. [Google Scholar] [CrossRef] [PubMed]
- PDXNet. PDX Delopment and Trial Centers Research Network. Available online: https://brandi-davis-7wsf.squarespace.com/hcibcm. (accessed on 20 June 2020).
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, P.; Mathew, B.S.; Das, S.; Isaiah, R.; John, S.; Prabha, R.; Fleming, D.H. Cisplatin concentrations in long and short duration infusion: Implications for the optimal time of radiation delivery. J. Clin. Diagn. Res. 2016, 10, XC01–XC04. [Google Scholar] [CrossRef]
- McCreight, L.J.; Stage, T.B.; Connelly, P.; Lonergan, M.; Nielsen, F.; Prehn, C.; Adamski, J.; Brøsen, K.; Pearson, E.R. Pharmacokinetics of metformin in patients with gastrointestinal intolerance. Diabetes Obes. Metab. 2018, 20, 1593–1601. [Google Scholar] [CrossRef]
- McKillop, D.; Partridge, E.A.; Kemp, J.V.; Spence, M.P.; Kendrew, J.; Barnett, S.; Wood, P.G.; Giles, P.B.; Patterson, A.B.; Bichat, F.; et al. Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Mol. Cancer Ther. 2005, 4, 641–649. [Google Scholar] [CrossRef]
- Borodovsky, A.; McQuiston, T.J.; Stetson, D.; Ahmed, A.; Whitston, D.; Zhang, J.; Grondine, M.; Lawson, D.; Challberg, S.S.; Zinda, M.; et al. Generation of stable PDX derived cell lines using conditional reprogramming. Mol. Cancer 2017, 16, 177. [Google Scholar] [CrossRef]
- Yuan, H.; Myers, S.; Wang, J.; Zhou, D.; Woo, J.A.; Kallakury, B.; Ju, A.; Bazylewicz, M.; Carter, Y.M.; Albanese, C.; et al. Use of reprogrammed cells to identify therapy for respiratory papillomatosis. New Engl. J. Med. 2012, 367, 1220–1227. [Google Scholar] [CrossRef]
- Fong, E.L.S.; Toh, T.B.; Lin, Q.X.X.; Liu, Z.; Hooi, L.; Rashid, M.B.M.A.; Benoukraf, T.; Chow, E.K.H.; Huynh, T.H.; Yu, H. Generation of matched patient-derived xenograft in vitro-in vivo models using 3D macroporous hydrogels for the study of liver cancer. Biomaterials 2018, 159, 229–240. [Google Scholar] [CrossRef]
- Roife, D.; Dai, B.; Kang, Y.a.; Perez, M.V.R.; Pratt, M.; Li, X.; Fleming, J.B. Ex vivo testing of patient-derived xenografts mirrors the clinical outcome of patients with pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2016, 22, 6021–6030. [Google Scholar] [CrossRef]
- Sulaiman, A.; Sulaiman, B.; Khouri, L.; McGarry, S.; Nessim, C.; Arnaout, A.; Li, X.; Addison, C.; Dimitroulakos, J.; Wang, L. Both bulk and cancer stem cell subpopulations in triple-negative breast cancer are susceptible to Wnt, HDAC, and ERα coinhibition. Febs Lett. 2016, 590, 4606–4616. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; McGarry, S.; Lam, K.M.; El-Sahli, S.; Chambers, J.; Kaczmarek, S.; Li, L.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; et al. Co-inhibition of mTORC1, HDAC and ESR1α retards the growth of triple-negative breast cancer and suppresses cancer stem cells. Cell Death Dis. 2018, 9, 815. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Prywes, R. Serum-induced expression of the cdc25AGene by relief of E2F-mediated repression. Mol. Cell. Biol. 1999, 19, 4695–4702. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Balch, C.; Montgomery, J.S.; Jeong, M.; Chung, J.H.; Yan, P.; Huang, T.H.; Kim, S.; Nephew, K.P. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. Bmc Med. Genom. 2009, 2, 34. [Google Scholar] [CrossRef]
- Komatsu, M.; Yoshimaru, T.; Matsuo, T.; Kiyotani, K.; Miyoshi, Y.; Tanahashi, T.; Rokutan, K.; Yamaguchi, R.; Saito, A.; Imoto, S.; et al. Molecular features of triple negative breast cancer cells by genome-wide gene expression profiling analysis. Int. J. Oncol. 2013, 42, 478–506. [Google Scholar] [CrossRef]
- Maubant, S.; Tesson, B.; Maire, V.; Ye, M.; Rigaill, G.; Gentien, D.; Cruzalegui, F.; Tucker, G.C.; Roman-Roman, S.; Dubois, T. Transcriptome analysis of Wnt3a-treated triple-negative breast cancer cells. PLoS ONE 2015, 10, e0122333. [Google Scholar] [CrossRef]
- Maire, V.; Baldeyron, C.; Richardson, M.; Tesson, B.; Vincent-Salomon, A.; Gravier, E.; Marty-Prouvost, B.; De Koning, L.; Rigaill, G.; Dumont, A.; et al. TTK/hMPS1 is an attractive therapeutic target for triple-negative breast cancer. PLoS ONE 2013, 8, e63712. [Google Scholar] [CrossRef]
- Maire, V.; Némati, F.; Richardson, M.; Vincent-Salomon, A.; Tesson, B.; Rigaill, G.; Gravier, E.; Marty-Prouvost, B.; De Koning, L.; Lang, G.; et al. Polo-like kinase 1: A potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res. 2013, 73, 813–823. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward | Reverse |
|---|---|---|
| 18S | AACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| GAPDH | ACAGTCAGCCGCATCTTCTT | GACAAGCTTCCCGTTCTCAG |
| PDK1 | CAACAGAGGTGTTTACCCCC | ATTTTCCTCAAAGGAACGCC |
| LDH1 | GGCCTGTGCCATCAGTATCT | GGAGATCCATCATCTCTCCC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulaiman, A.; McGarry, S.; Chambers, J.; Al-Kadi, E.; Phan, A.; Li, L.; Mediratta, K.; Dimitroulakos, J.; Addison, C.; Li, X.; et al. Targeting Hypoxia Sensitizes TNBC to Cisplatin and Promotes Inhibition of Both Bulk and Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21, 5788. https://doi.org/10.3390/ijms21165788
Sulaiman A, McGarry S, Chambers J, Al-Kadi E, Phan A, Li L, Mediratta K, Dimitroulakos J, Addison C, Li X, et al. Targeting Hypoxia Sensitizes TNBC to Cisplatin and Promotes Inhibition of Both Bulk and Cancer Stem Cells. International Journal of Molecular Sciences. 2020; 21(16):5788. https://doi.org/10.3390/ijms21165788
Chicago/Turabian StyleSulaiman, Andrew, Sarah McGarry, Jason Chambers, Emil Al-Kadi, Alexandra Phan, Li Li, Karan Mediratta, Jim Dimitroulakos, Christina Addison, Xuguang Li, and et al. 2020. "Targeting Hypoxia Sensitizes TNBC to Cisplatin and Promotes Inhibition of Both Bulk and Cancer Stem Cells" International Journal of Molecular Sciences 21, no. 16: 5788. https://doi.org/10.3390/ijms21165788
APA StyleSulaiman, A., McGarry, S., Chambers, J., Al-Kadi, E., Phan, A., Li, L., Mediratta, K., Dimitroulakos, J., Addison, C., Li, X., & Wang, L. (2020). Targeting Hypoxia Sensitizes TNBC to Cisplatin and Promotes Inhibition of Both Bulk and Cancer Stem Cells. International Journal of Molecular Sciences, 21(16), 5788. https://doi.org/10.3390/ijms21165788