Extrahepatic Drug Transporters in Liver Failure: Focus on Kidney and Gastrointestinal Tract




Abstract
1. Introduction
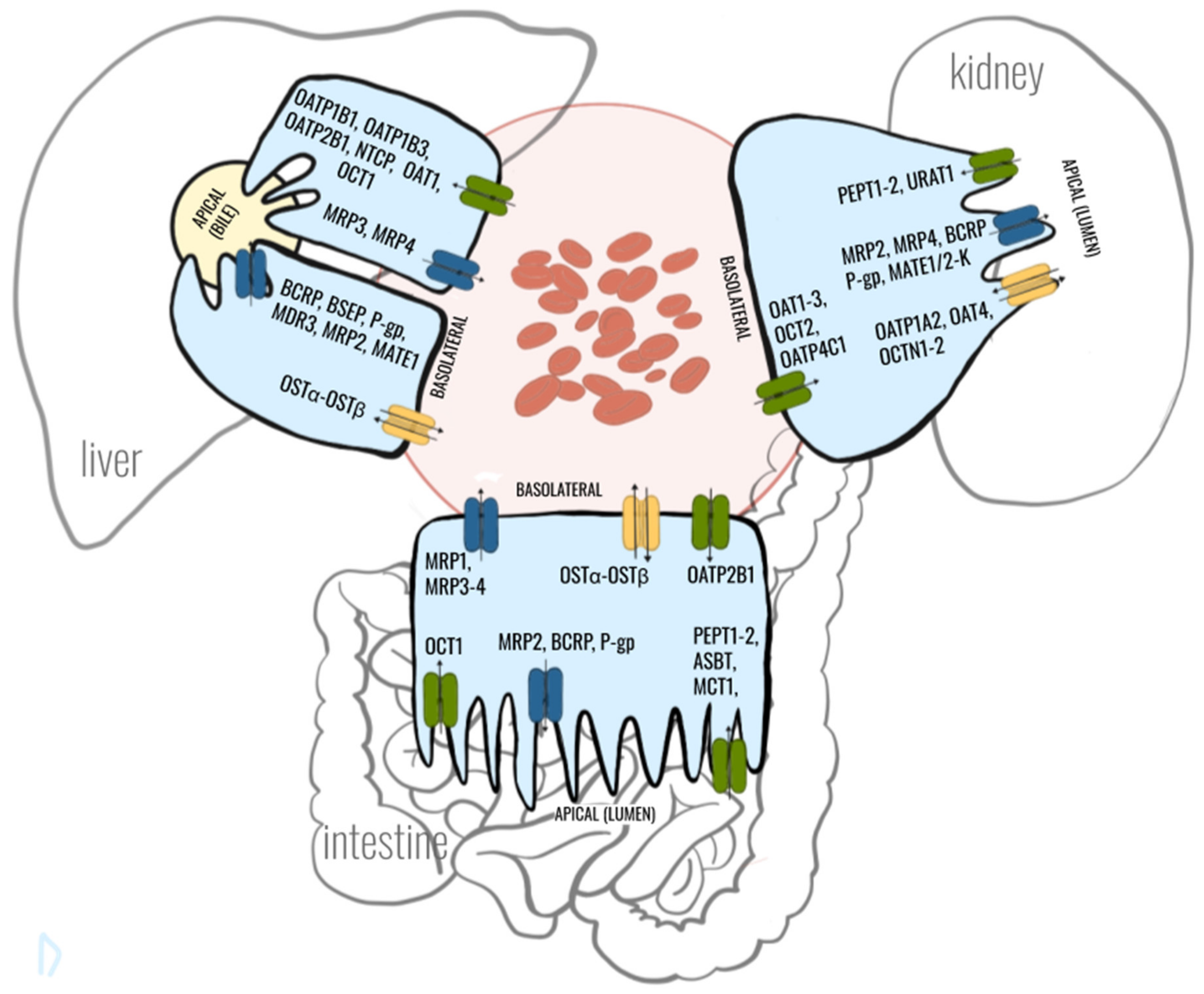
2. Crosstalk between Gut-Liver-Kidney-Axis in Context with Drug Transporters
3. Effects of Liver Failure on Hepatic Drug Transporters
4. Effects of Liver Failure on Drug Transporters in the Gastrointestinal Tract
4.1. In vitro Studies
4.2. Animal Models
4.3. Clinical Studies
5. Effects of Liver Failure on Drug Transporters in the Kidney
5.1. In vitro Studies
5.2. Animal Models
5.3. Clinical Studies
6. Pharmacokinetics and Drug Actions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| AhR | aryl-hydrocarbon receptor |
| ALF | acute liver failure |
| ASBT | apical sodium-dependent bile acid transporter |
| BCRP | breast cancer resistance protein |
| BDL | bile duct ligation |
| CCl4 | carbon tetrachloride |
| EHBR | Eisai hyperbilirubinemic rats |
| FPTC | fresh proximal tubular cells |
| FXR | farnesoid X receptor |
| HNF | hepatocyte nuclear factor |
| MATE | multidrug and toxin extrusion protein |
| MCT | monocarboxylic acid transporter |
| MDR | multidrug resistance |
| MRP | multidrug resistance protein |
| NASH | nonalcoholic steatohepatitis |
| OAT | organic anion transporter |
| OATP | organic anion transporting polypeptide |
| OCT | organic cation transporter |
| OCTN | organic cation/carnitine transporter |
| OST | organic solute transporter |
| PEPT | peptide transporter |
| PXR | pregnane X receptor |
| RPTEC | human renal proximal tubular epithelial cells; |
| SHP | small heterodimer partner |
| SLC | solute carrier |
References
- Morrissey, K.M.; Stocker, S.L.; Wittwer, M.B.; Xu, L.; Giacomini, K.M. Renal transporters in drug development. Annu. Rev. Pharmacol. Toxicol. 2012, 53, 503–529. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, K. Organic cation transporters in intestine, kidney, liver, and brain. Annu. Rev. Physiol. 1998, 60, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Drozdzik, M.; Busch, D.; Lapczuk, J.; Müller, J.; Ostrowski, M.; Kurzawski, M.; Oswald, S. Protein Abundance of Clinically Relevant Drug Transporters in the Human Liver and Intestine: A Comparative Analysis in Paired Tissue Specimens. Clin. Pharmacol. Ther. 2019, 105, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Wu, W.; Bush, K.T.; Hoenig, M.P.; Blantz, R.C.; Bhatnagar, V. Handling of Drugs, Metabolites, and Uremic Toxins by Kidney Proximal Tubule Drug Transporters. Clin. J. Am. Soc. Nephrol. 2015, 10, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Crowe, A.; Wang, X.; Zhang, P.; Ding, K.; Li, L.; Yue, W. Regulation of Organic Anion Transporting Polypeptides (OATP) 1B1- and OATP1B3-Mediated Transport: An Updated Review in the Context of OATP-Mediated Drug-Drug Interactions. Int. J. Mol. Sci. 2018, 19, 855. [Google Scholar] [CrossRef]
- Shen, H.; Lai, Y.; Rodrigues, D.A. Organic Anion Transporter 2 (OAT2): An Enigmatic Human Solute Carrier. Drug Metab. Dispos. 2017, 45, 228–236. [Google Scholar] [CrossRef]
- Varma, M.V.; Rotter, C.J.; Chupka, J.; Whalen, K.M.; Duignan, D.B.; Feng, B.; Litchfield, J.; Goosen, T.C.; El-Kattan, A.F. pH-Sensitive Interaction of HMG-CoA Reductase Inhibitors (Statins) with Organic Anion Transporting Polypeptide 2B1. Mol. Pharmaceutics 2011, 8, 1303–1313. [Google Scholar] [CrossRef]
- Oswald, S.; Gröer, C.; Drozdzik, M.; Siegmund, W. Mass spectrometry-based targeted proteomics as a tool to elucidate the expression and function of intestinal drug transporters. AAPS J. 2013, 15, 1128–1140. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Galetin, A.; Huang, S.M. The International Transporter Consortium: Summarizing Advances in the Role of Transporters in Drug Development. Clin. Pharmacol. Ther. 2018, 104, 766–771. [Google Scholar] [CrossRef]
- In Vitro Drug Interaction Studies - Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research. 2020. Available online: https://www.fda.gov/drugs/guidance-compliance-regulatory-information/guidances-drugs (accessed on 9 August 2020).
- Burt, H.J.; Riedmaier, A.E.; Harwood, M.D.; Crewe, H.K.; Gill, K.L.; Neuhoff, S. Abundance of hepatic transporters in Caucasians: A meta-analysis. Drug Metab. Dispos. 2016, 44, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Drozdzik, M.; Szelag-Pieniek, S.; Post, M.; Zeair, S.; Wrzesinski, M.; Kurzawski, M.; Prieto, J.; Oswald, S. Protein Abundance of Hepatic Drug Transporters in Patients With Different Forms of Liver Damage. Clin. Pharmacol. Ther. 2020, 107, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- International Transporter Consortium; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Drozdzik, M.; Oswald, S. Expression and regulation of drug transporters and metabolizing enzymes in the human gastrointestinal tract. Curr. Med. Chem. 2016, 23, 4468–4489. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Keiser, M.; Drozdzik, M.; Oswald, S. Expression, regulation and function of intestinal drug transporters: An update. Biol. Chem. 2017, 398, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Drozdzik, M.; Gröer, C.; Penski, J.; Lapczuk, J.; Ostrowski, M.; Lai, Y.; Prasad, B.; Unadkat, J.D.; Siegmund, W.; Oswald, S. Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol. Pharm. 2014, 11, 3547–3555. [Google Scholar] [CrossRef]
- Prasad, B.; Johnson, K.; Billington, S.; Lee, C.; Chung, G.W.; Brown, C.D.; Kelly, E.J.; Himmelfarb, J.; Unadkat, J.D. Abundance of Drug Transporters in the Human Kidney Cortex as Quantified by Quantitative Targeted Proteomics. Drug Metab. Dispos. 2016, 44, 1920–1924. [Google Scholar] [CrossRef]
- Oswald, S.; Müller, J.; Neugebauer, U.; Schröter, R.; Herrmann, E.; Pavenstädt, H.; Ciarimboli, G. Protein Abundance of Clinically Relevant Drug Transporters in The Human Kidneys. Int. J. Mol. Sci. 2019, 24, 20. [Google Scholar] [CrossRef]
- Russel, F.G.; Masereeuw, R.; van Aubel, R.A. Molecular aspects of renal anionic drug transport. Annu. Rev. Physiol. 2002, 64, 563–594. [Google Scholar] [CrossRef]
- Rosenthal, S.B.; Bush, K.T.; Nigam, S.K. A Network of SLC and ABC transporter and DME genes involved in remote sensing and signaling in the Gut-Liver-Kidney axis. Sci. Rep. 2019, 9, 11879. [Google Scholar] [CrossRef]
- Van der Schoor, L.W.; Verkade, H.J.; Kuipers, F.; Jonker, J.W. New insights in the biology of ABC transporters ABCC2 and ABCC3: Impact on drug disposition. Expert. Opin. Drug Metab. Toxicol. 2015, 11, 273–293. [Google Scholar] [CrossRef]
- Bush, K.T.; Wu, W.; Lun, C.; Nigam, S.K. The drug transporter OAT3 (SLC22A8) and endogenous metabolite communication via the gut-liver-kidney axis. J. Biol. Chem. 2017, 292, 15789–15803. [Google Scholar] [CrossRef] [PubMed]
- Jetter, A.; Kullak-Ublick, G.A. Drugs and hepatic transporters: A review. Pharmacol Res. 2019, 154, 104234. [Google Scholar] [CrossRef] [PubMed]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef] [PubMed]
- Geier, A.; Martin, I.V.; Dietrich, C.G.; Balasubramaniyan, N.; Strauch, S.; Suchy, F.J.; Gartung, C.; Trautwein, C.; Ananthanarayanan, M. Hepatocyte nuclear factor-4alpha is a central transactivator of the mouse Ntcp gene. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, 226–233. [Google Scholar] [CrossRef]
- Navasa, M.; Follo, A.; Filella, X.; Jiménez, W.; Francitorra, A.; Planas, R.; Rimola, A.; Arroyo, V.; Rodés, J. Tumor Necrosis Factor and interleukin-6 in Spontaneous Bacterial Peritonitis in Cirrhosis: Relationship With the Development of Renal Impairment and Mortality. Hepatology. 1998, 27, 1227–1232. [Google Scholar] [CrossRef]
- Fukui, H. Gut-liver axis in liver cirrhosis: How to manage leaky gut and endotoxemia. World J. Hepatol. 2015, 7, 425–442. [Google Scholar] [CrossRef]
- Teng, S.; Piquette-Miller, M. The involvement of the pregnane X receptor in hepatic gene regulation during inflammation in mice. J. Pharmacol. Exp. Ther. 2005, 312, 841–848. [Google Scholar] [CrossRef]
- Acharya, C.; Esra Sahingur, S.; Bajaj, J. Microbiota, cirrhosis, and the emerging oral-gut-liver axis. JCI Insight. 2017, 5, e94416. [Google Scholar] [CrossRef]
- Kuno, T.; Hirayama-Kurogi, M.; Ito, S.; Ohtsuki, S. Effect of Intestinal Flora on Protein Expression of Drug-Metabolizing Enzymes and Transporters in the Liver and Kidney of Germ-Free and Antibiotics-Treated Mice. Mol. Pharmaceutics. 2016, 13, 2691–2701. [Google Scholar] [CrossRef]
- Licorresponding, T.; Apte, U. Bile acid metabolism and signaling in cholestasis, inflammation and cancer. Adv. Pharmacol. 2015, 74, 263–302. [Google Scholar] [CrossRef]
- Jansen, J.; Jansen, K.; Neven, E.; Poesen, R.; Othman, A.; van Mil, A.; Sluijter, J.; Sastre Torano, J.; Zaal, E.; Berkers, C.; et al. Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome-derived organic anion secretion. PNAS 2019, 116, 16105–16110. [Google Scholar] [CrossRef] [PubMed]
- Atilano-Roque, A.; Roda, G.; Fogueri, U.; Kiser, J.J.; Joy, M.S. Effect of Disease Pathologies on Transporter Expression and Function. J. Clin. Pharmacol. 2016, 56, S205–S221. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, B. Drug uptake systems in liver and kidney: Historic perspective. Clin. Pharmacol. Ther. 2010, 87, 39. [Google Scholar] [CrossRef]
- Prasad, B.; Bhatt, D.; Johnson, K.; Chapa, R.; Chu, X.; Salphati, L.; Xiao, G.; Lee, C.; Hop, C.; Mathias, A.; et al. Abundance of Phase 1 and 2 Drug-Metabolizing Enzymes in Alcoholic and Hepatitis C Cirrhotic Livers: A Quantitative Targeted Proteomics Study. Drug Metab. Dispos. 2018, 46, 943–952. [Google Scholar] [CrossRef]
- Li, R.; Barton, H.A.; Maurer, T.S. A Mechanistic pharmacokinetic model for liver transporter substrates under liver cirrhosis conditions. CPT Pharmacometrics Syst. Pharmacol. 2015, 4, 338–349. [Google Scholar] [CrossRef]
- Templeton, I.; Eichenbaum, G.; Sane, R.; Zhou, J. Case study 5. Deconvoluting hyperbilirubinemia: Differentiating between hepatotoxicity and reversible inhibition of UGT1A1, MRP2, or OATP1B1 in drug development. Methods Mol. Biol. 2014, 1113, 471–483. [Google Scholar] [CrossRef]
- Rochling, F.A. Evaluation of abnormal liver tests. Clin. Cornerstone. 2001, 3, 1–12. [Google Scholar] [CrossRef]
- Liang, Y.; Li, S.; Chen, L. The physiological role of drug transporters. Protein. Cell. 2015, 6, 334–350. [Google Scholar] [CrossRef]
- Fickert, P.; Wagner, M. Biliary bile acids in hepatobiliary injury - What is the link? J. Hepatol. 2017, 67, 619–631. [Google Scholar] [CrossRef]
- Billington, S.; Ray, S.A.; Salphati, L.; Xiao, G.; Chu, X.; Humphreys, G.W.; Liao, M.; Lee, C.A.; Mathias, C.; Hop, A.C.E.C. Transporter expression in noncancerous and cancerous liver tissue from donors with hepatocellular carcinoma and chronic Hepatitis C infection quantified by LC-MS/MS proteomics. Drug Metab. Dispos. 2018, 46, 189–196. [Google Scholar] [CrossRef]
- Prasad, B.; Lai, Y.; Lin, Y.; Unadkat, J.D. Interindividual variability in the hepatic expression of the human breast cancer resistance protein (BCRP/ABCG2): Effect of age, sex, and genotype. J. Pharm. Sci. 2013, 102, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Prasad, B.; Salphati, L.; Chu, X.; Gupta, A.; Hop, C.; Evers, R.; Unadkat, J.D. Interspecies variability in expression of hepatobiliary transporters across human, dog, monkey, and rat as determined by quantitative proteomics. Drug Metab. Dispos. 2015, 43, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Schaefer, O.; Kawakami, H.; Inoue, T.; Liehner, S.; Saito, A.; Ishiguro, N.; Kishimoto, W.; Ludwig-Schwellinger, E.; Ebner, T.; et al. Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: Comparison with mRNA levels and activities. Drug Metab. Dispos. 2012, 40, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Vildhede, A.; Kimoto, E.; Pelis, R.M.; Rodrigues, D.A.; Varma, M.V.S. Quantitative Proteomics and Mechanistic Modeling of Transporter-Mediated Disposition in Nonalcoholic Fatty Liver Disease. Clin. Pharmacol. Ther. 2020, 107, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Collins, C.; Kelly, E.J.; Chu, X.; Ray, A.S.; Salphati, L.; Xiao, G.; Lee, C.; Lai, Y.; Liao, M.; et al. Transporter expression in liver tissue from subjects with alcoholic or hepatitis C cirrhosis quantified by targeted quantitative proteomics. Drug Metab. Dispos. 2016, 44, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Drozdzik, M.; Czekawy, I.; Oswald, S.; Drozdzik, A. Intestinal drug transporters in pathological states: An overview. Pharmacol. Rep 2020, in press. [Google Scholar] [CrossRef]
- Evers, R.; Piquette-Miller, M.; Polli, J.W.; Russel, F.G.; Sprowl, J.A.; Tohyama, K.; Ware, J.A.; de Wildt, S.N.; Xie, W.; Brouwer, K.L. Disease-Associated Changes in Drug Transporters May Impact the Pharmacokinetics and/or Toxicity of Drugs: A White Paper from the International Transporter Consortium. Clin. Pharmacol. Ther. 2018, 104, 900–915. [Google Scholar] [CrossRef]
- Minemura, M.; Tajiri, K.; Shimizu, Y. Systemic abnormalities in liver disease. World, J. Gastroenterol. 2009, 15, 2960–2974. [Google Scholar] [CrossRef]
- Huang, Z.H.; Murakami, T.; Okochi, A.; Yumoyo, R.; Nagai, J.; Takano, M. Expression and function of P-glycoprotein in rats with carbon tetrachloride-induced acute hepatic failure. J. Pharm. Pharmacol. 2001, 53, 873–881. [Google Scholar] [CrossRef]
- Wang, F.; Miao, M.X.; Sun, B.B.; Wang, Z.J.; Tang, X.G.; Chen, Y.; Zhao, K.-J.; Liu, X.-D.; Liu, L. Acute liver failure enhances oral plasma exposure of zidovudine in rats by downregulation of hepatic UGT2B7 and intestinal P-gp. Acta Pharmacol. Sin. 2017, 38, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, R.; Murakami, T.; Takano, M. Differential effect of acute hepatic failure on in vivo and in vitro P-glycoprotein functions in the intestine. Pharm. Res. 2003, 20, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Mennone, A.; Soroka, C.J.; Harry, K.M.; Boyer, J.L. Role of breast cancer resistance protein in the adaptive response to cholestasis. Drug Metab. Dispos. 2010, 38, 1673–1678. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.; Geier, A.; Salein, N.; Lammert, F.; Roeb, E.; Oude Elferink, R.; Matern, S.; Gartung, C. Consequences of Bile Duct Obstruction on Intestinal Expression and Function of Multidrug Resistance-Associated Protein 2. Gastroenterology. 2004, 126, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, C.; Hruz, P.; Gutmann, H.; Terracciano, L.; Beuers, U.; Lehmann, F.; Beglinger, C.; Drewe, J. Decreased Expression of Breast Cancer Resistance Protein in the Duodenum in Patients With Obstructive Cholestasis. Digestion. 2006, 74, 101–108. [Google Scholar] [CrossRef]
- Yokooji, T.; Murakami, T.; Yumoto, R.; Nagai, J.; Takano, M. Function of multidrug resistance-associated protein 2 in acute hepatic failure rats. Eur. J. Pharmacol. 2006, 546, 152–160. [Google Scholar] [CrossRef]
- Kamisako, T.; Ogawa, H. Alteration of the expression of adenosine triphosphate-binding cassette transporters associated with bile acid and cholesterol transport in the rat liver and intestine during cholestasis. J. Gastroenterol. Hepatol. 2005, 20, 1429–1434. [Google Scholar] [CrossRef]
- Mindikoglu, A.L.; Pappas, S.C. New Developments in Hepatorenal Syndrome. Clin. Gastroenterol. Hepatol. 2018, 16, 162–177. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T. Uraemic syndrome of chronic kidney disease: Altered remote sensing and signalling. Nat. Rev. Nephrol. 2019, 15, 301–316. [Google Scholar] [CrossRef]
- Canet, M.J.; Hardwick, R.N.; Lake, A.; Dzierlenga, A.; Clarke, J.; Goedken, M.; Cherrington, N.J. Renal Xenobiotic Transporter Expression Is Altered in Multiple Experimental Models of Nonalcoholic Steatohepatitis. Drug Metab. Dispos. 2015, 43, 266–272. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kobayashi, Y.; Gabazza, E.C.; Higuchi, K.; Kamisako, T.; Kuroda, M.; Takeuchi, K.; Iwasa, M.; Kaito, M.; Adachi, Y. Increased renal expression of bilirubin glucuronide transporters in a rat model of obstructive jaundice. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G656–G662. [Google Scholar] [CrossRef]
- Khemawoot, P.; Maruyama, C.; Tsukada, H.; Noda, H.; Ishizaki, J.; Yokogawa, K.; Miyamoto, K. Influence of Chronic Hepatic Failure on Disposition Kinetics of Valproate Excretion Through a Phase II Reaction in Rats Treated With Carbon Tetrachloride. Biopharm. Drug Dispos. 2007, 28, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Azzaroli, F.; Wang, L.; Soroka, C.J.; Gigliozzi, A.; Setchell, K.D.; Kramer, W.; Boyer, J.L. Adaptive regulation of bile salt transporters in kidney and liver in obstructive cholestasis in the rat. Gastroenterology. 2001, 121, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Kobayashi, Y.; Tanaka, Y.; Itani, T.; Mifuji, R.; Araki, J.; Kaito, M.; Adachi, Y. Increased hepatic and renal expressions of multidrug resistance-associated protein 3 in Eisai hyperbilirubinuria rats. J. Gastroenterol. Hepatol. 2004, 19, 146–153. [Google Scholar] [CrossRef]
- Denk, G.; Soroka, C.J.; Takeyama, Y.; Chen, W.-S.; Schuetz, J.D.; Boyer, J.L. Multidrug Resistance-Associated Protein 4 Is Up-Regulated in Liver but Down-Regulated in Kidney in Obstructive Cholestasis in the Rat. J. Hepatol. 2004, 40, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Schlattjan, J.H.; Winter, C.; Greven, J. Regulation of Renal Tubular Bile Acid Transport in the Early Phase of an Obstructive Cholestasis in the Rat. Nephron. Physiol. 2003, 95, 49–56. [Google Scholar] [CrossRef]
- Chen, C.; Slitt, A.L.; Dieter, M.Z.; Tanaka, Y.; Scheffer, G.L.; Klaassen, C.D. Up-regulation of Mrp4 expression in kidney of Mrp2-deficient TR- rats. Biochem. Pharmacol. 2005, 70, 1088–1095. [Google Scholar] [CrossRef]
- Brandoni, A.; Villar, S.R.; Picena, J.C.; Anzai, N.; Endou, H.; Torres, A.M. Expression of rat renal cortical OAT1 and OAT3 in response to acute biliary obstruction. Hepatology. 2006, 43, 1092–1100. [Google Scholar] [CrossRef]
- Brandoni, A.; Anzai, N.; Kanai, Y.; Endou, H.; Torres, A.M. Renal elimination of p-aminohippurate (PAH) in response to three days of biliary obstruction in the rat. The role of OAT1 and OAT3. Biochim. Biophys. Acta 2006, 1762, 673–682. [Google Scholar] [CrossRef]
- Chen, J.; Terada, T.; Ogasawara, K.; Katsura, T.; Inui, K. Adaptive responses of renal organic anion transporter 3 (OAT3) during cholestasis. Am. J. Physiol. Renal Physiol. 2008, 295, F247–F252. [Google Scholar] [CrossRef]
- Brookman, L.J.; Rolan, P.E.; Benjamin, I.S.; Palmer, K.R.; Wyld, P.J.; Lloyd, P.; Flesch, G.; Waldmeier, F.; Sioufi, A.; Mullins, F. Pharmacokinetics of valsartan in patients with liver disease. Clin. Pharmacol. Ther. 1997, 62, 272–278. [Google Scholar] [CrossRef]
- Stangier, J.; Su, C.A.; Schöndorfer, G.; Roth, W. Pharmacokinetics and safety of intravenous and oral telmisartan 20 mg and 120 mg in subjects with hepatic impairment compared with healthy volunteers. J. Clin. Pharmacol. 2000, 40, 1355–1364. [Google Scholar]
- Simonson, S.G.; Martin, P.D.; Mitchell, P.; Schneck, D.W.; Lasseter, K.C.; Warwick, M.J. Pharmacokinetics and pharmacodynamics of rosuvastatin in subjects with hepatic impairment. Eur. J. Clin. Pharmacol. 2003, 58, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Brandoni, A.; Torres, A.M. Characterization of the mechanisms involved in the increased renal elimination of bromosulfophthalein during cholestasis: Involvement of Oatp1. J. Histochem Cytochem. 2009, 57, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Kurata, T.; Muraki, Y.; Mizutani, H.; Iwamoto, T.; Okuda, M. Elevated systemic elimination of cimetidine in rats with acute biliary obstruction: The role of renal organic cation transporter OCT2. Drug Metab. Pharmacokinet. 2010, 25, 328–334. [Google Scholar] [CrossRef]
- González-Martin, G.; Arancibia, A.; Fajuri, M.; Chesta, J.; Novoa, X. Pharmacokinetics of Cimetidine in Patients With Liver Disease. Int. J. Clin. Pharmacol. Ther. Toxicol. 1985, 23, 355–358. [Google Scholar]
- Pinzani, M.; Daskalopoulos, G.; Laffi, G.; Gentilini, P.; Zipser, R. Altered furosemide pharmacokinetics in chronic alcoholic liver disease with ascites contributes to diuretic resistance. Gastroenterology. 1987, 92, 294–298. [Google Scholar] [CrossRef]
- Giacomini, K.M. The Use of Drug Transporters as Therapeutic Targets. Clin. Adv. Hematol. Oncol. 2016, 14, 869–871. [Google Scholar]
- Tan, P.K.; Ostertag, T.M.; Miner, J.N. Mechanism of high affinity inhibition of the human urate transporter URAT1. Sci. Rep. 2016, 6, 34995. [Google Scholar] [CrossRef]
- Kramer, W. Transporters, Trojan horses and therapeutics: Suitability of bile acid and peptide transporters for drug delivery. Biol. Chem. 2011, 392, 77–94. [Google Scholar] [CrossRef]


{kind=link}
| Transporter | Cell Models (ref) | Animal Studies (ref) | Clinical Studies (ref) |
|---|---|---|---|
| P-gp | ↓ Caco-2 [51] ↓ Caco-2 [52] | ↓ CCl4-ALF rats [53] ↑ CCl4-ALF rats [53] ↔ BDL rats [54] | ↔ cholestasis [55] |
| BCRP | ↔ Caco-2 [52] | ↑ BDL rats [54] | ↓ cholestasis [56] ↔ cholestasis [55] |
| MRP2 | ↓ ↔ CCl4-ALF rats [57] ↓ BDL rats [58] ↓ BDL rats [55] | ↓ cholestasis [55] | |
| MRP3 | ↔ BDL rats [58] | ↔ cholestasis [55] | |
| ASBT | ↔ BDL rats [57] | ||
| OSTα | ↓ BDL rats [57] | ||
| OSTβ | ↓ BDL rats [57] |
| Transporter | Cell Models (ref) | Animal Studies (ref) |
|---|---|---|
| P-gp | ↑ NASH mice, rats [61] | |
| BCRP | ↔ NASH mice, rats [61] | |
| MRP2 | ↑ RPTEC [62] | ↑ CCl4—ALF rats [63] ↑ BDL rats [64] ↑ BDL rats [62] ↑ NASH mice, rats [61] |
| MRP3 | ↑ EHBR [65] | |
| MRP4 | ↑ NASH mice, rats [61] ↓ BDL rats [66] | |
| ASBT | ↔ FPTC [67] | |
| OCT2 | ↓ NASH mice, rats [61] | |
| OCT3 | ↔ NASH mice, rats [61] | |
| OAT1 | ↔ NASH mice, rats [49] ↔ EHBR [68] ↑ BDL rats [69] ↓ BDL rats [70] | |
| OAT3 | ↑ NASH mice, rats [61] ↑ EHBR [68] ↑ BDL rats [69] | |
| OATP1 | ↑ ↔ NASH mice, rats [61] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Droździk, M.; Oswald, S.; Droździk, A. Extrahepatic Drug Transporters in Liver Failure: Focus on Kidney and Gastrointestinal Tract. Int. J. Mol. Sci. 2020, 21, 5737. https://doi.org/10.3390/ijms21165737
Droździk M, Oswald S, Droździk A. Extrahepatic Drug Transporters in Liver Failure: Focus on Kidney and Gastrointestinal Tract. International Journal of Molecular Sciences. 2020; 21(16):5737. https://doi.org/10.3390/ijms21165737
Chicago/Turabian StyleDroździk, Marek, Stefan Oswald, and Agnieszka Droździk. 2020. "Extrahepatic Drug Transporters in Liver Failure: Focus on Kidney and Gastrointestinal Tract" International Journal of Molecular Sciences 21, no. 16: 5737. https://doi.org/10.3390/ijms21165737
APA StyleDroździk, M., Oswald, S., & Droździk, A. (2020). Extrahepatic Drug Transporters in Liver Failure: Focus on Kidney and Gastrointestinal Tract. International Journal of Molecular Sciences, 21(16), 5737. https://doi.org/10.3390/ijms21165737