Glucagon-Like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
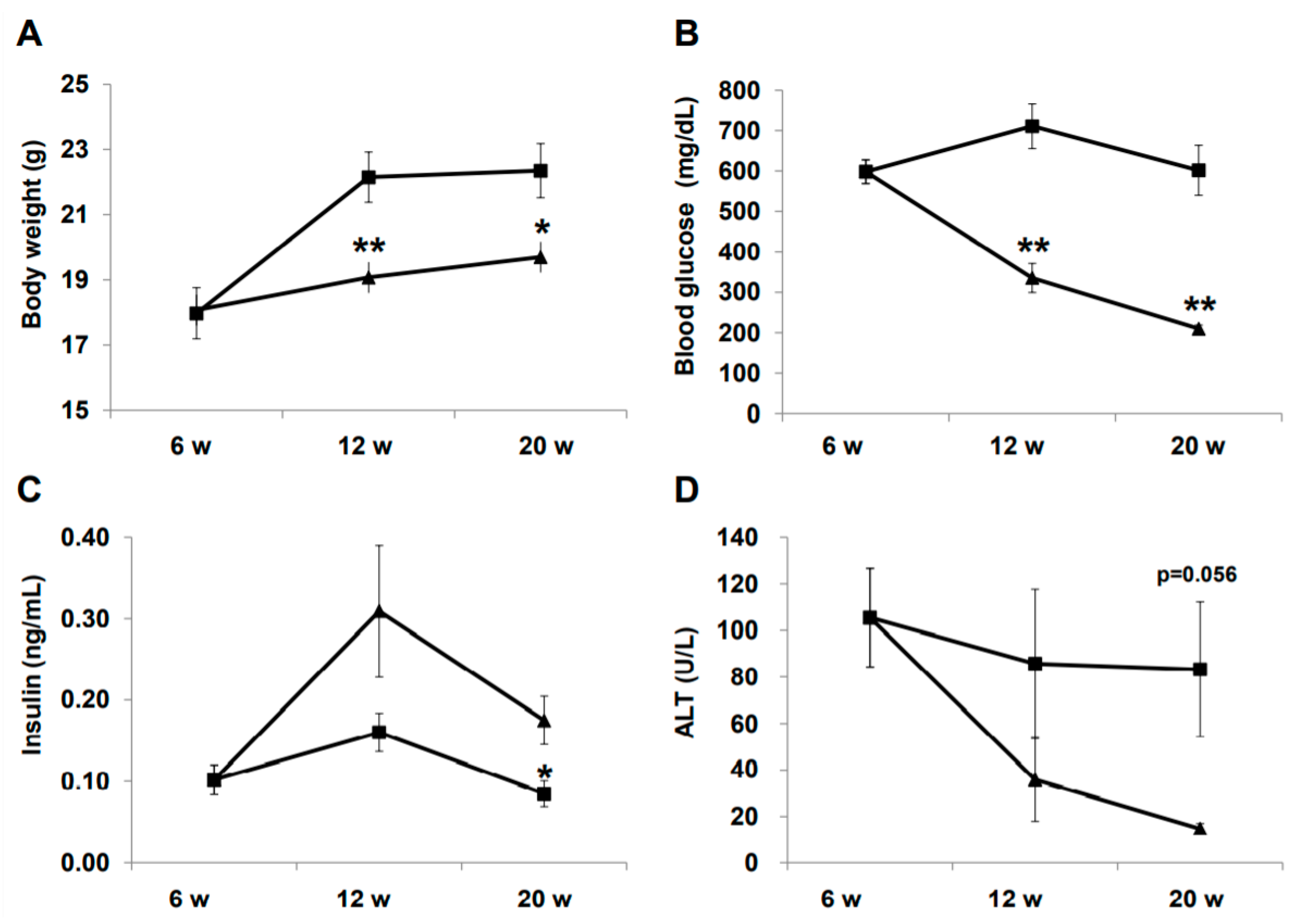
2.1. Effect of Liraglutide on Physiology and Biochemistry
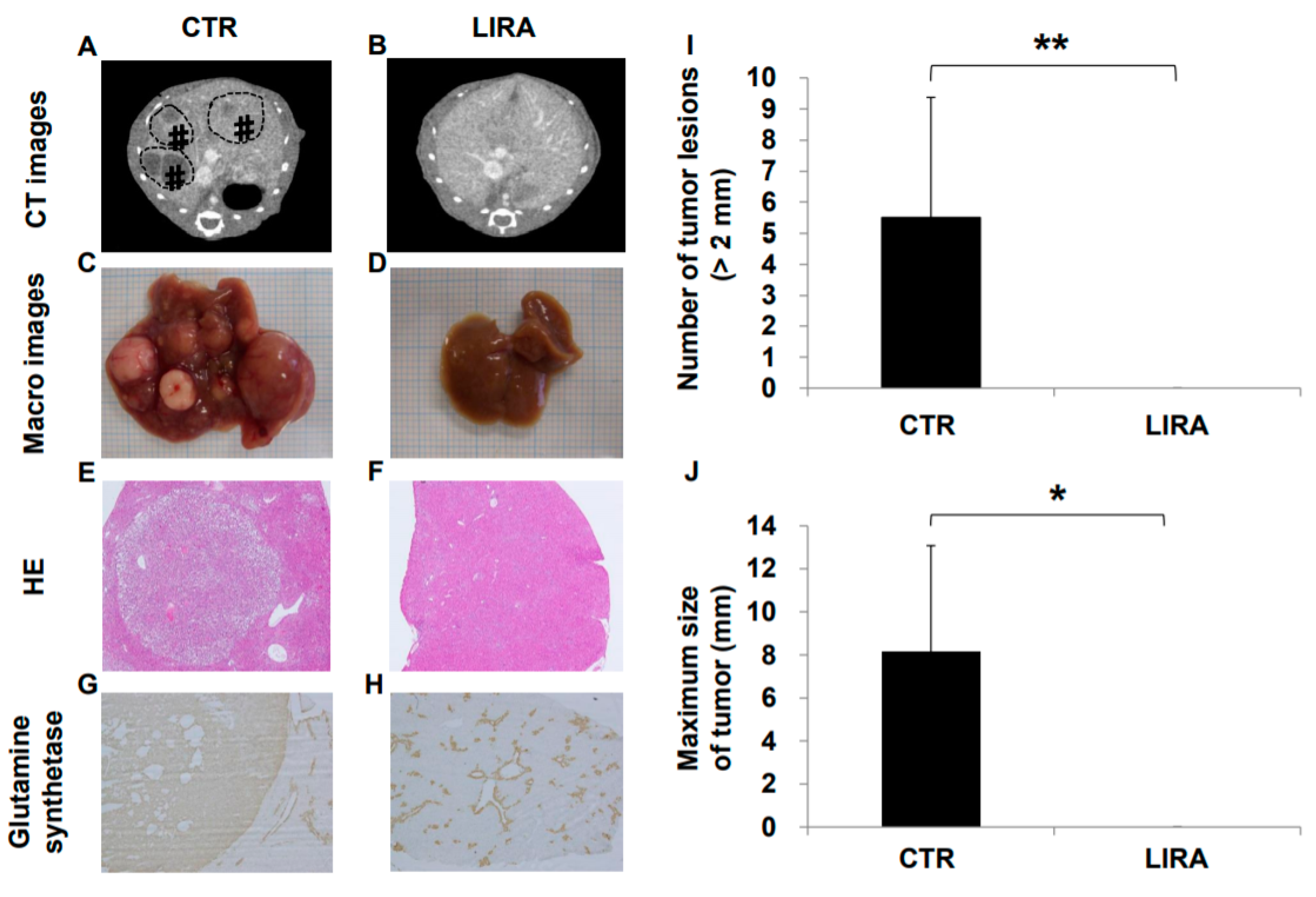
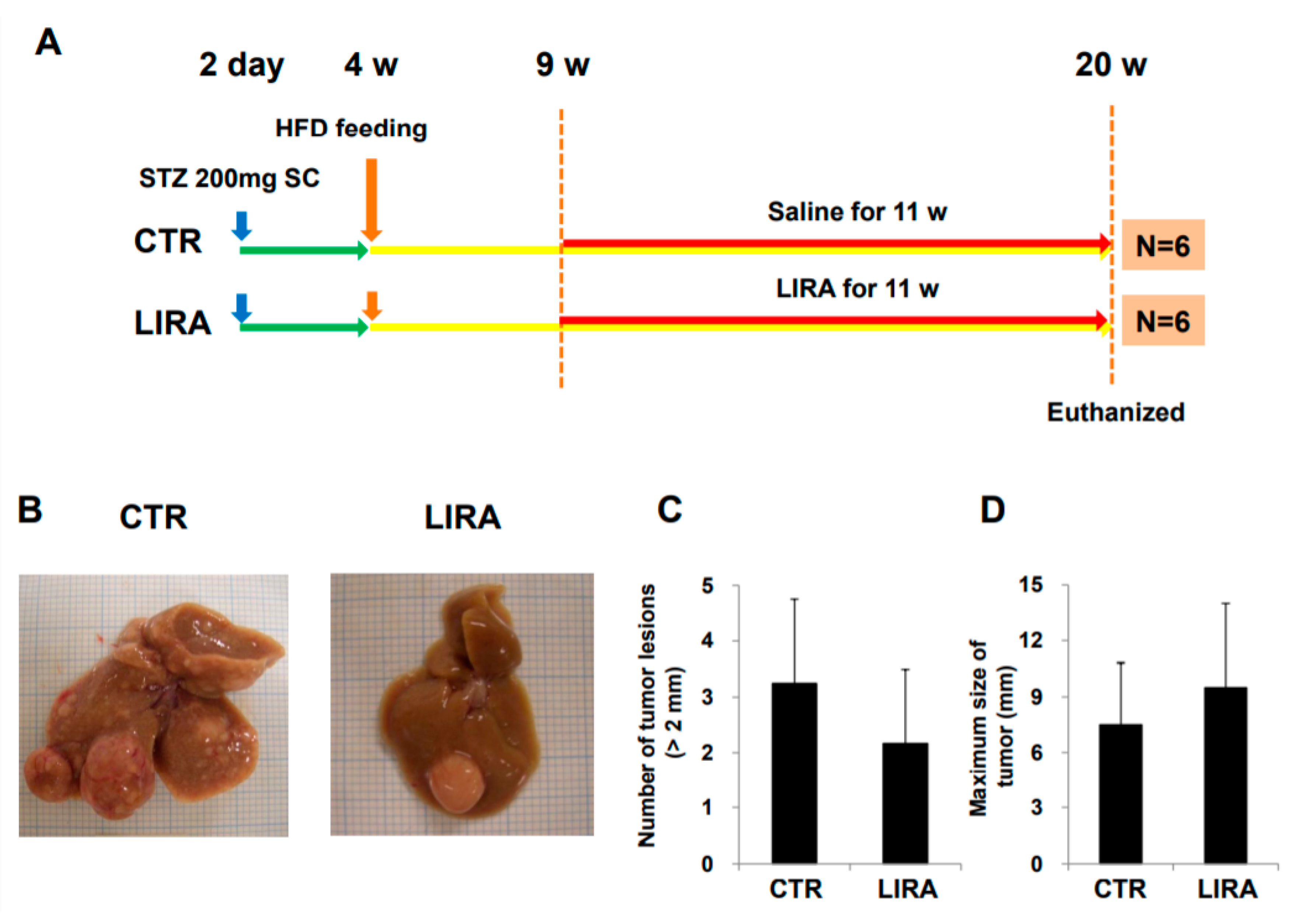
2.2. Development of HCC
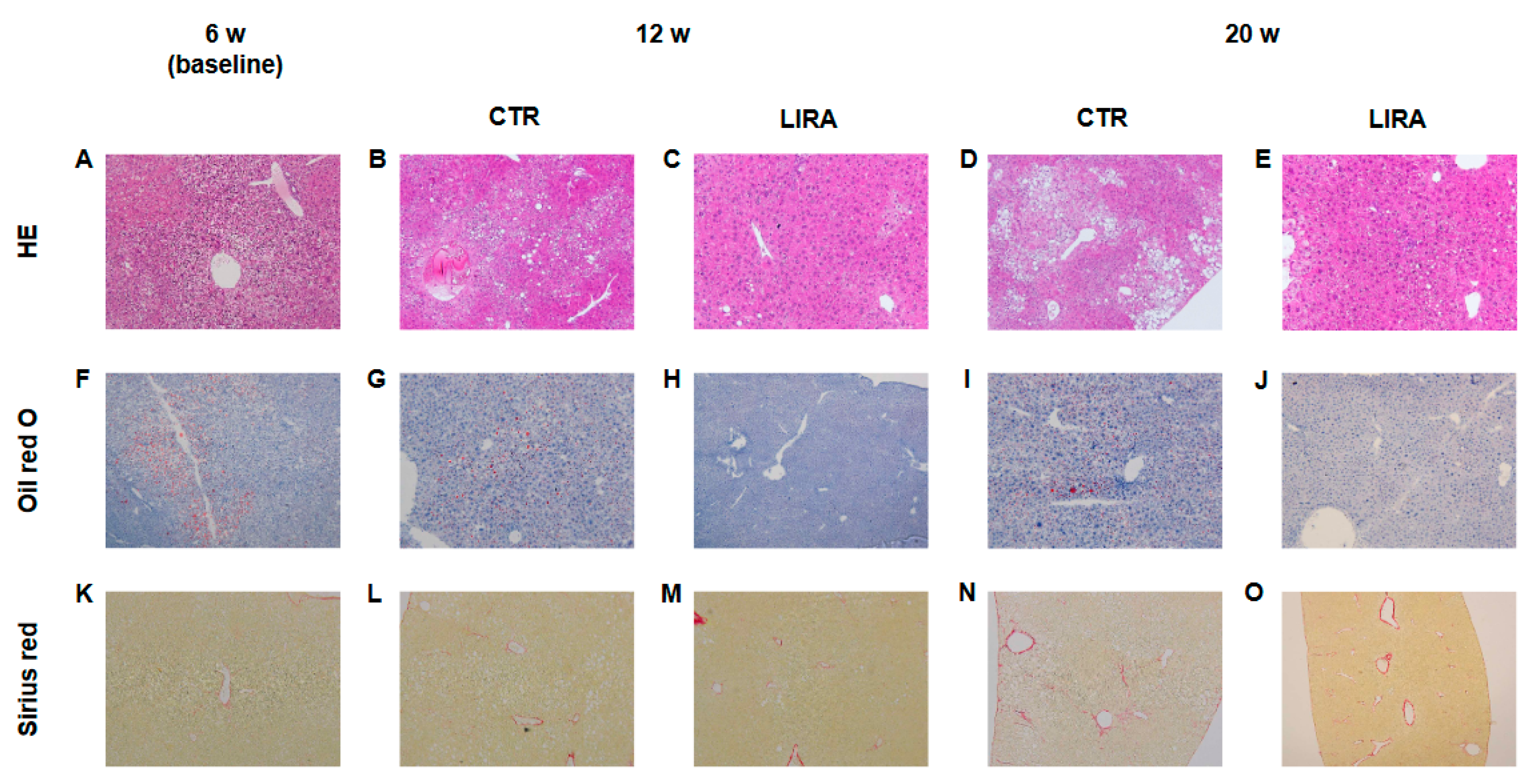
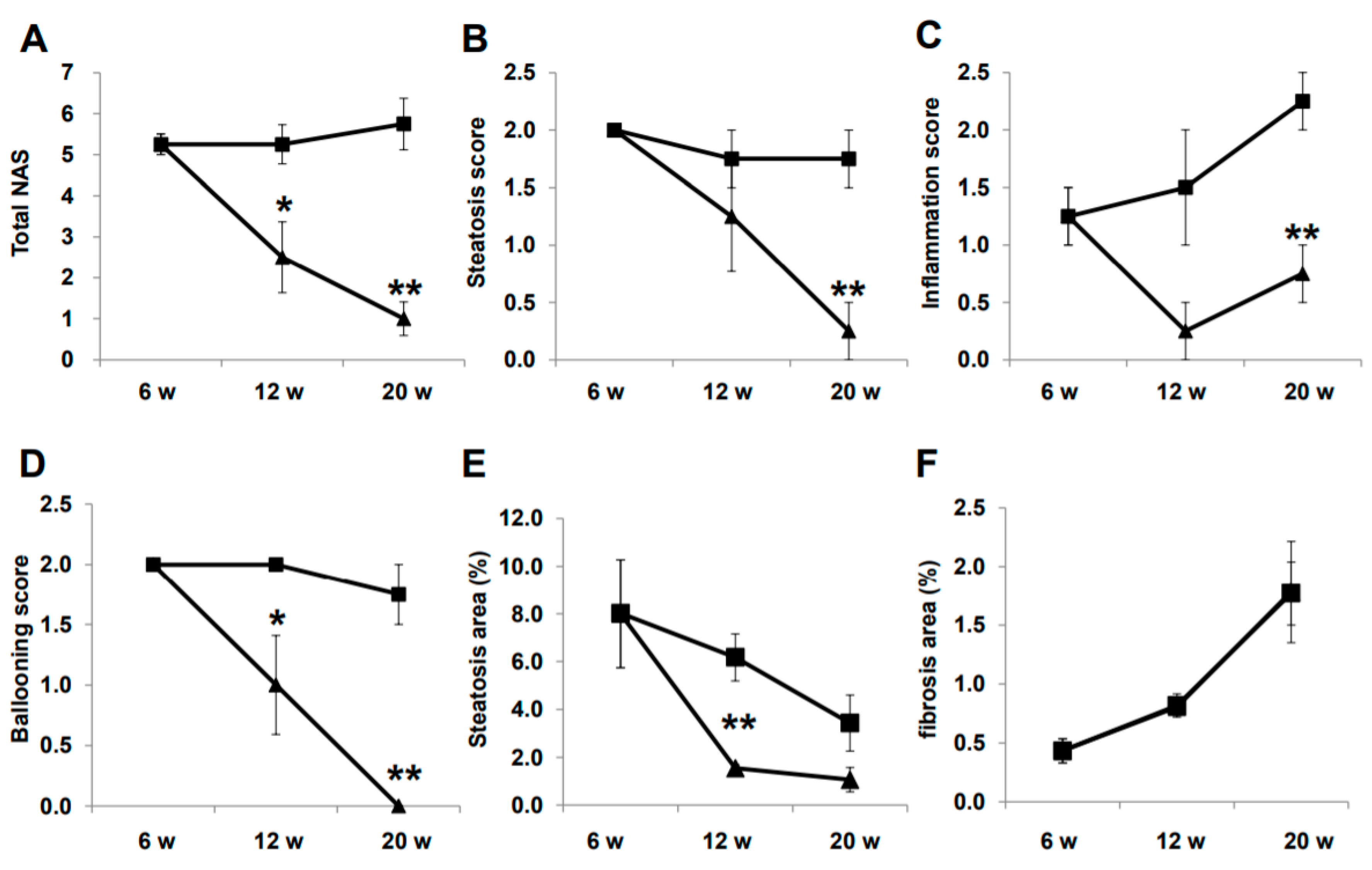
2.3. Histological Evaluation of NASH
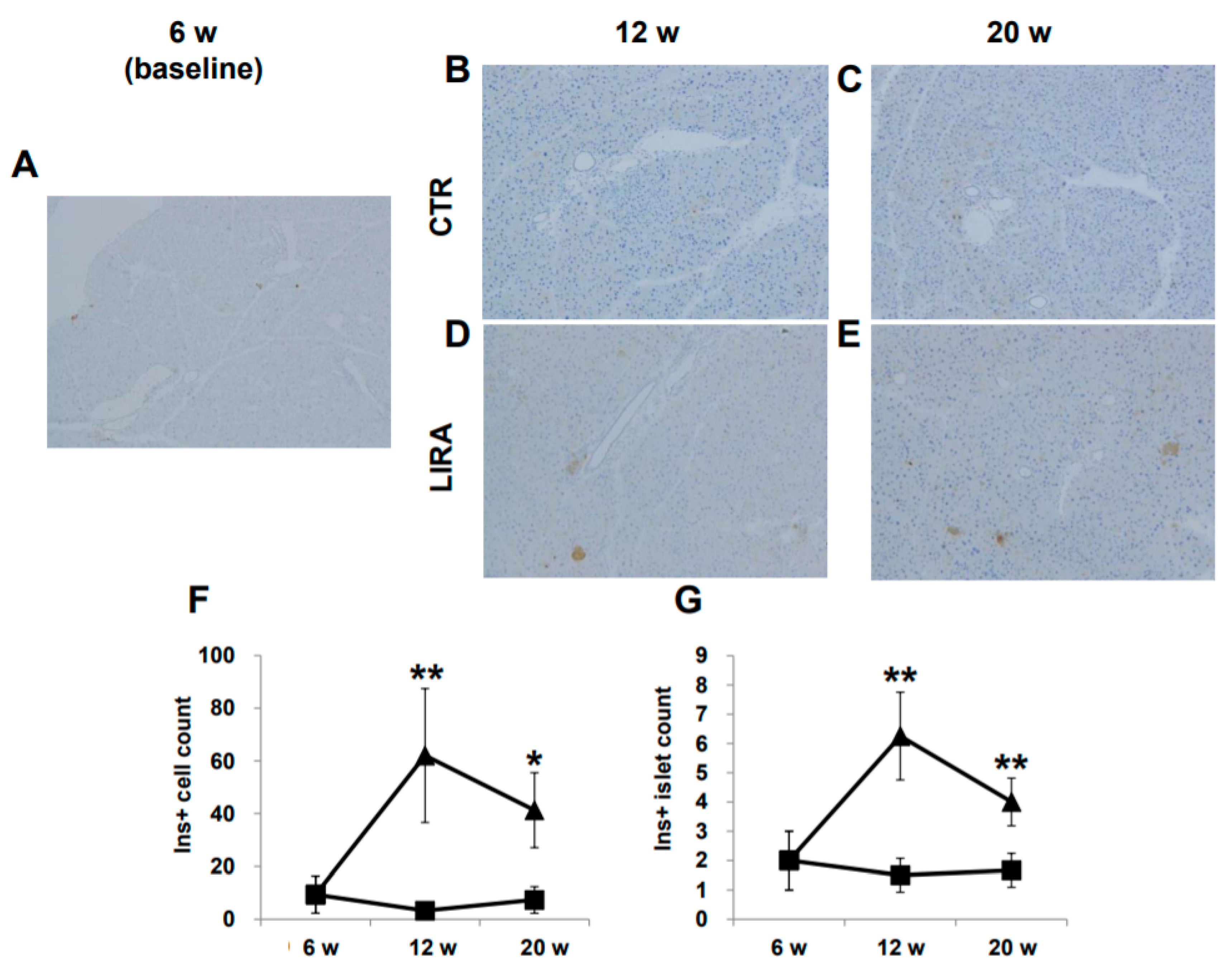
2.4. Histological Evaluation of Pancreatic β-Cells
2.5. Development of HCC in the Delayed Therapy
3. Discussion
4. Materials and Methods
4.1. Animal Model
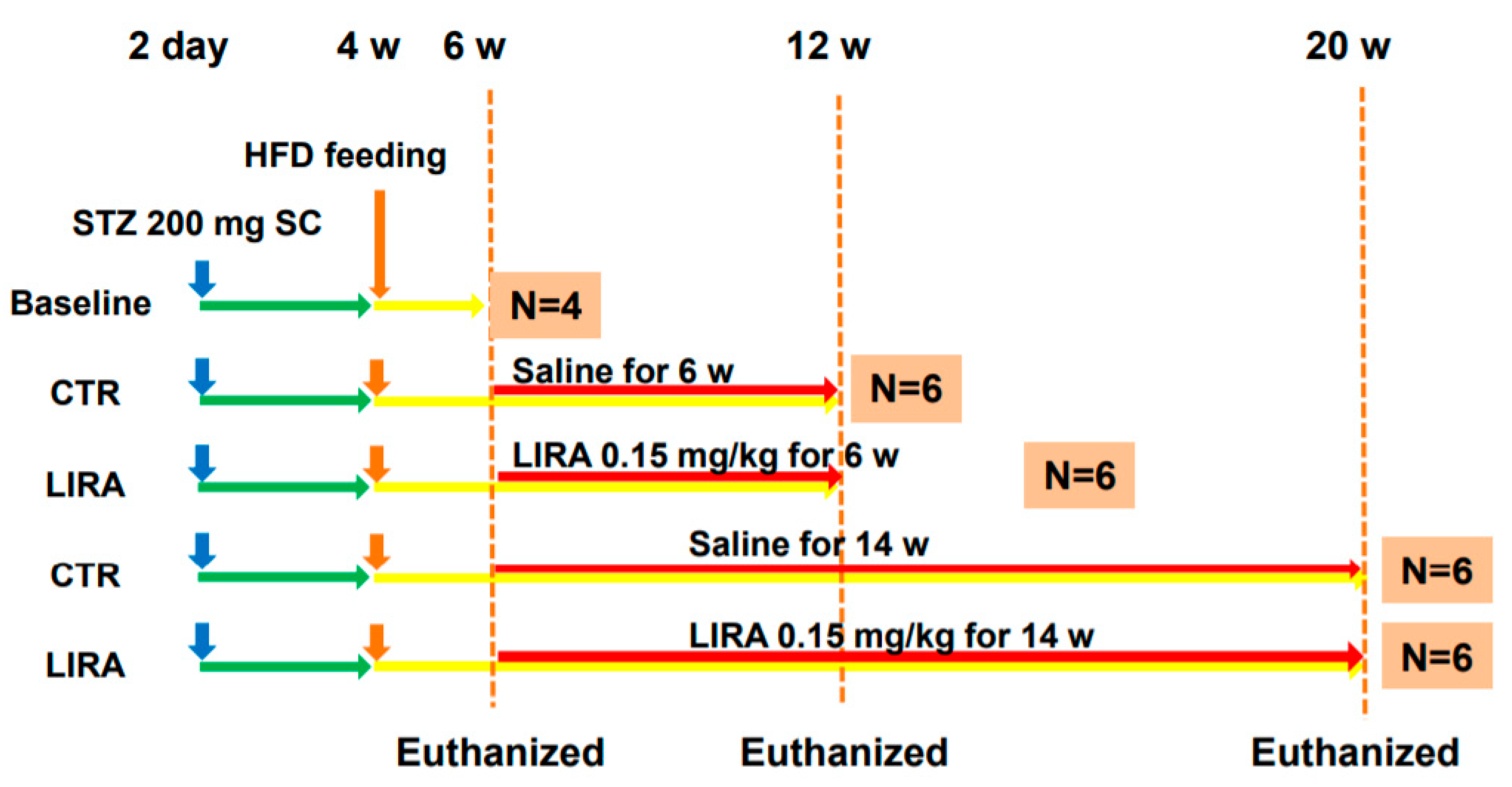
4.2. Experimental Design
4.3. Biochemistry Analysis
4.4. Histological Analysis
4.5. Imaging Analysis Using Computer Tomography
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander, S.B.; Mostafa, I.; Bugianesi, E.; Wai-Sun, W.V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Non-alcoholic Fatty Liver Disease and Non-alcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Caldwell, S.H. Nonalcoholic steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003, 37, 1202–1219. [Google Scholar] [CrossRef] [PubMed]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar] [CrossRef]
- Bugianesi, E.; Leone, N.; Vanni, E.; Marchsini, G.; Brunello, F.; Carucci, P.; Musso, A.; Paolis, P.; Capussotti, L.; Salizzoni, M.; et al. Expanding the natural history of nonalcholic steatohepatitis: From cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 2002, 123, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Marrero, J.A.; Fontana, R.J.; Su, G.L.; Conjeearam, H.S.; Emick, D.M.; Lok, A.S. NAFLD may be a common underlying liver disease in patients with hepatocellular carcinoma in the United States. Hepatology 2002, 36, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef]
- Phillips, L.K.; Prins, J.B. Update on incretin hormones. Ann. N. Y. Acad. Sci. 2011, 1243, 55–74. [Google Scholar] [CrossRef]
- Meier, J.J.; Gallwitz, B.; Salmen, S.; Goetze, O.; Holst, J.J.; Schmidt, W.E.; Nauck, M.A. Normalization of glucose concentrations and deceleration of gastric emptying after solid meals during intravenous glucagon-like peptide 1 in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2003, 88, 2719–2725. [Google Scholar] [CrossRef]
- Bode, B. An overview of the pharmacokinetics, efficacy and safety of liraglutide. Diabetes Res. Clin. Pract. 2012, 97, 27–42. [Google Scholar] [CrossRef]
- Peterson, G.E.; Pollom, R.D. Liraglutide in clinical practice: Dosing, safety and efficacy. Int. J. Clin. Pract. 2010, 64, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Mells, J.E.; Fu, P.P.; Sharma, S.; Olson, D.; Cheng, L.; Handy, J.A.; Saxena, N.K.; Sorescu, D.; Anania, F.A. Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in C57BL/6J mice fed a Western diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G225–G235. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Kitajima, Y.; Hyogo, H.; Takahashi, H.; Kojima, M.; Ono, M.; Araki, N.; Tanaka., K.; Yamaguchi, M.; Matsuda, Y.; et al. Pilot study of liraglutide effects in non-alcoholic steatohepatitis and non-alcoholic fatty liver disease with glucose intolerance in Japanese patients (LEAN-J). Hepatol. Res. 2015, 45, 269–278. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN Trial Team; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Greene, M.W.; Burrington, C.M.; Ruhouff, M.S.; Johnson, A.K.; Chongkrairatanakul, T.; Kangwanpornsiri, A. PKCδ is activated in a dietary model of steatohepatitis and regulates endoplasmic reticulum stress and cell death. J. Biol. Chem. 2010, 285, 42115–42129. [Google Scholar] [CrossRef]
- Jiang, J.X.; Chen, X.; Fukuda, H.; Serizawa, N.; Devaraj, S.; Torok, N.J. Advanced glycation endproducts induce fibrogenic activity in nonalcoholic steatohepatitis by modulating TNF-α-converting enzyme activity in mice. Hepatology 2013, 58, 1339–1348. [Google Scholar] [CrossRef]
- Hyogo, H.; Yamagishi, S.; Iwamoto, K.; Arihiro, K.; Takeuchi, M.; Sato, T.; Ochi, H.; Nonaka, M.; Nabeshima, Y.; Inoue, M.; et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2007, 22, 1112–1119. [Google Scholar] [CrossRef]
- Drucker, D.J. The Cardiovascular Biology of Glucagon-like Peptide-1. Cell Metab. 2016, 24, 15–30. [Google Scholar] [CrossRef]
- Kwon, D.Y.; Kim, Y.S.; Ahn, I.S.; Kim, D.S.; Kang, S.; Hong, S.M.; Park, S. Exendin-4 potentiates insulinotropic action partly via increasing beta-cell proliferation and neogenesis and decreasing apoptosis in association with the attenuation of endoplasmic reticulum stress in islets of diabetic rats. J. Pharmacol. Sci. 2009, 111, 361–371. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, C.; Choung, J.S.; Jung, H.S.; Jun, H.S. Glucagon-Like Peptide 1 Increases β-Cell Regeneration by Promoting α- to β-Cell Transdifferentiation. Diabetes 2018, 67, 2601–2614. [Google Scholar] [CrossRef] [PubMed]
- Hui, H.; Wright, C.; Perfetti, R. Glucagon-like peptide 1 induces differentiation of islet duodenal homeobox-1-positive pancreatic ductal cells into insulin-secreting cells. Diabetes 2001, 50, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Jojima, T.; Wakamatsu, S.; Kase, M.; Iijima, T.; Maejima, Y.; Shimomura, K.; Kogai, T.; Tomaru, T.; Usui, I.; Aso, Y. The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 5237. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Nakano, D.; Koga, H.; Torimura, T. Effects of a DPP4 Inhibitor on Progression of NASH-related HCC and the p62/Keap1/Nrf2-Pentose Phosphate Pathway in a Mouse Model. Liver Cancer 2019, 8, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Samir, S.; Kirby, M.; Berger, N.G.; Shroyer, N.F.; Woods, S.C.; Kohli, R. Insulin Concentration Modulates Hepatic Lipid Accumulation in Mice in Part via Transcriptional Regulation of Fatty Acid Transport Proteins. PLoS ONE 2012, 7, e38952. [Google Scholar]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5·24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef]
- Tobari, M.; Hashimoto, E.; Taniai, M.; Ikarashi, Y.; Kodama, K.; Kogiso, T.; Tokushige, K.; Takayoshi, N.; Hashimoto, N. Characteristics of non-alcoholic steatohepatitis among lean patients in Japan: Not uncommon and not always benign. J. Gastroenterol. Hepatol. 2019, 34, 1404–1410. [Google Scholar] [CrossRef]
- Fan, J.G.; Kim, S.U.; Wong, V.W. New trends on obesity and NAFLD in Asia. J. Hepatol. 2017, 67, 862–873. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Cheang, K.L.; Luketic, V.A.; Boyett, S.; Idowu, M.O.; Patidar, K.; Puri, P.; Matherly, S.; Stravitz, R.T.; Sterling, R.K.; et al. Nonalcoholic Steatohepatitis (NASH) Is Associated with a Decline in Pancreatic Beta Cell (β-Cell) Function. Dig. Dis. Sci. 2015, 60, 2529–2537. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Biroli, G.; Carello, M.; Faga, E.; Pacini, G.; De Michieli, F.; Cassader, M.; Durazzo, M.; Rizzetto, M.; et al. Hypoadiponectinemia predicts the severity of hepatic fibrosis and pancreatic beta-cell dysfunction in nondiabetic nonobese patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2005, 100, 2438–2446. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Miyake, S.; Yano, M.; Ueki, Y.; Yamaguchi, Y.; Akazawa, S.; Tominaga, Y. Glucose tolerance, insulin secretion, and insulin sensitivity in nonobese and obese Japanese subjects. Diabetes Care 1997, 20, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Gupta, N.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef]
- Ben-Shlomo, S.; Zvibel, I.; Shnell, M.; Shlomai, A.; Chepurko, E.; Halpern, Z.; Barzilai, N.; Oren, R.; Fishman, S. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol. 2011, 54, 1214–1223. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK: A target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Mok, M.T.; Sun, H.; Chan, A.W.; Huang, Y.; Cheng, A.S.; Xu, G. The anti-diabetic drug exenatide, a glucagon-like peptide-1 receptor agonist, counteracts hepatocarcinogenesis through cAMP-PKA-EGFR-STAT3 axis. Oncogene 2017, 36, 4135–4149. [Google Scholar] [CrossRef] [PubMed]
- Koehler, J.A.; Kain, T.; Drucker, D.J. Glucagon-like peptide-1 receptor activation inhibits growth and augments apoptosis in murine CT26 colon cancer cells. Endocrinology 2011, 152, 3362–3372. [Google Scholar] [CrossRef] [PubMed]
- Ligumsky, H.; Wolf, I.; Israeli, S.; Haimsohn, M.; Ferber, S.; Karasik, A.; Kaufman, B.; Rubinek, T. The peptide-hormone glucagon-like peptide-1 activates cAMP and inhibits growth of breast cancer cells. Breast Cancer Res. Treat. 2012, 132, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Feng, P.P.; Zhao, Z.B.; Zhu, W.; Gong, J.P.; Du, H.M. Liraglutide protects against inflammatory stress in non-alcoholic fatty liver by modulating Kupffer cells M2 polarization via cAMP-PKA-STAT3 signaling pathway. Biochem. Biophys. Res. Commun. 2019, 510, 20–26. [Google Scholar] [CrossRef]
- Wan, S.; Sun, H. Glucagon-like peptide-1 modulates RAW264.7 macrophage polarization by interfering with the JNK/STAT3 signaling pathway. Exp. Ther. Med. 2019, 17, 3573–3579. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef]
- Canbay, A.; Friedman, S.; Gores, G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology 2004, 39, 273–278. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Wang, C.; Li, Q.; Wang, W.; Guo, L.; Guo, C.; Sun, Y.; Zhang, J. GLP-1 contributes to increases in PGC-1α expression by downregulating miR-23a to reduce apoptosis. Biochem. Biophys. Res. Commun. 2015, 466, 33–39. [Google Scholar] [CrossRef]
- Yao, Y.; Li, Q.; Wang, W.; Zhang, J.; Gao, P.; Xu, Y. Glucagon-Like Peptide-1 Modulates Cholesterol Homeostasis by Suppressing the miR-19b-Induced Downregulation of ABCA1. Cell Physiol. Biochem. 2018, 50, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.K.; Liu, Y.C.; Shi, L.L.; Lu, K.D. Glucagon-like peptide-1 receptor agonists inhibit hepatic stellate cell activation by blocking the p38 MAPK signaling pathway. Genet. Mol. Res. 2015, 14, 19087–19093. [Google Scholar] [CrossRef] [PubMed]
- De Mesquita, F.C.; Guixé-Muntet, S.; Fernández-Iglesias, A.; Maeso-Díaz, R.; Vila, S.; Hide, D.; Ortega-Ribera, M.; Rosa, J.L.; García-Pagán, J.C.; Bosch, J.; et al. Liraglutide improves liver microvascular dysfunction in cirrhosis: Evidence from translational studies. Sci. Rep. 2017, 7, 3255. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Natta, M.V.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Dal bello, B.; Rosa, L.; Campanini, N.; Tinelli, C.; Viera, F.T.; D’Ambrosio, G.; Rossi, S.; Silini, M. Glutamine synthetase immunostaining correlates with pathologic features of hepatocellular carcinoma and better survival after radiofrequency thermal ablation. Clin. Cancer Res. 2010, 16, 2157–2166. [Google Scholar] [CrossRef]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Levy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of b-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kojima, M.; Takahashi, H.; Kuwashiro, T.; Tanaka, K.; Mori, H.; Ozaki, I.; Kitajima, Y.; Matsuda, Y.; Ashida, K.; Eguchi, Y.; et al. Glucagon-Like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5722. https://doi.org/10.3390/ijms21165722
Kojima M, Takahashi H, Kuwashiro T, Tanaka K, Mori H, Ozaki I, Kitajima Y, Matsuda Y, Ashida K, Eguchi Y, et al. Glucagon-Like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis. International Journal of Molecular Sciences. 2020; 21(16):5722. https://doi.org/10.3390/ijms21165722
Chicago/Turabian StyleKojima, Motoyasu, Hirokazu Takahashi, Takuya Kuwashiro, Kenichi Tanaka, Hitoe Mori, Iwata Ozaki, Yoichiro Kitajima, Yayoi Matsuda, Kenji Ashida, Yuichiro Eguchi, and et al. 2020. "Glucagon-Like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis" International Journal of Molecular Sciences 21, no. 16: 5722. https://doi.org/10.3390/ijms21165722
APA StyleKojima, M., Takahashi, H., Kuwashiro, T., Tanaka, K., Mori, H., Ozaki, I., Kitajima, Y., Matsuda, Y., Ashida, K., Eguchi, Y., & Anzai, K. (2020). Glucagon-Like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis. International Journal of Molecular Sciences, 21(16), 5722. https://doi.org/10.3390/ijms21165722