The Significance of Mitochondrial Dysfunction in Cancer

Abstract
1. Introduction
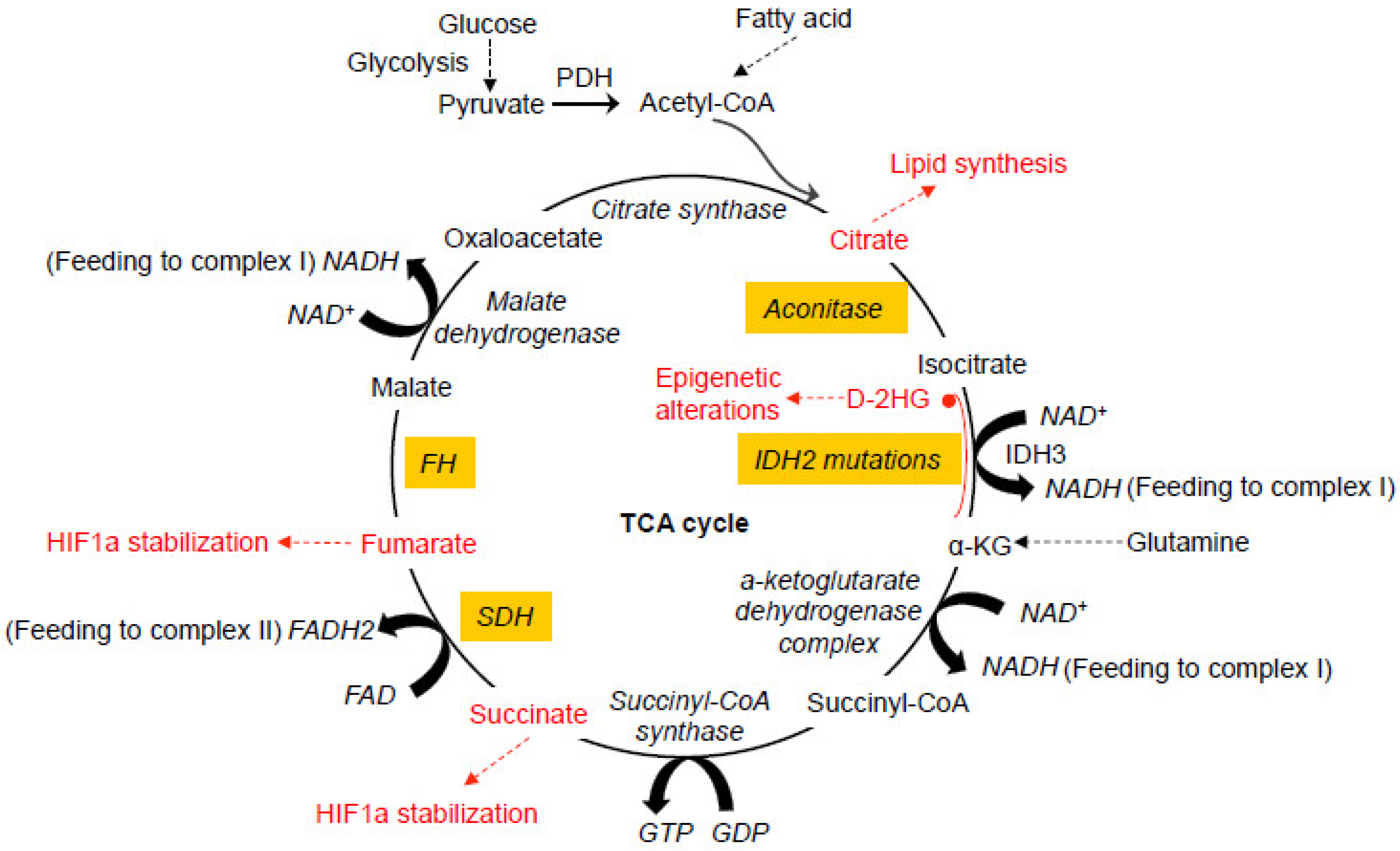
2. Mitochondrial TCA Cycle and Human Cancer
2.1. Gain-of-Function IDH Mutations
2.2. Loss-of-Function SDH Mutations
2.3. Loss-of-Function FH Mutations
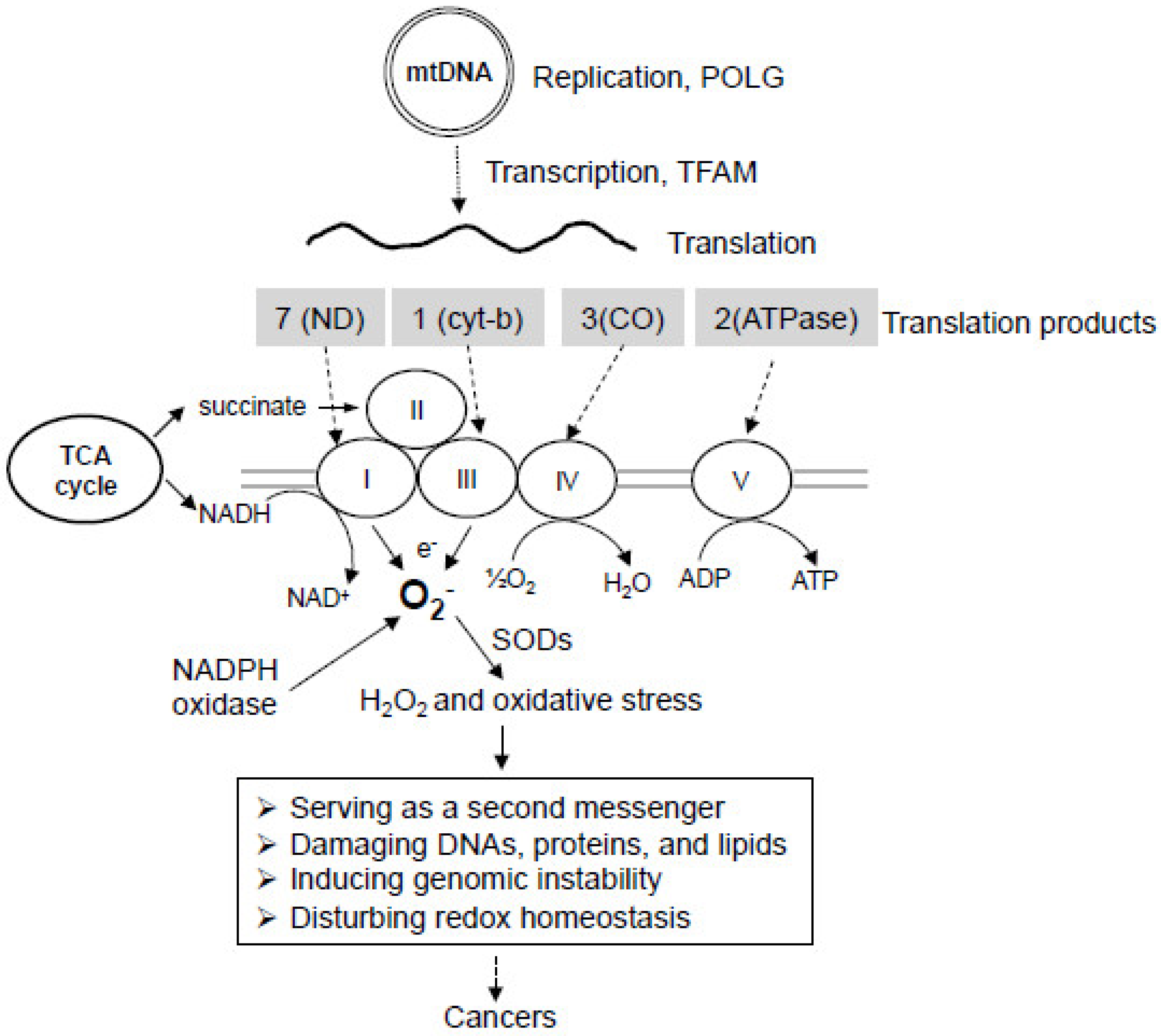
3. Mitochondrial DNA Mutations and Respiratory Chain Dysfunction in Cancer
3.1. Mitochondrial Genome
3.2. Mitochondrial DNA Mutations in Cancer
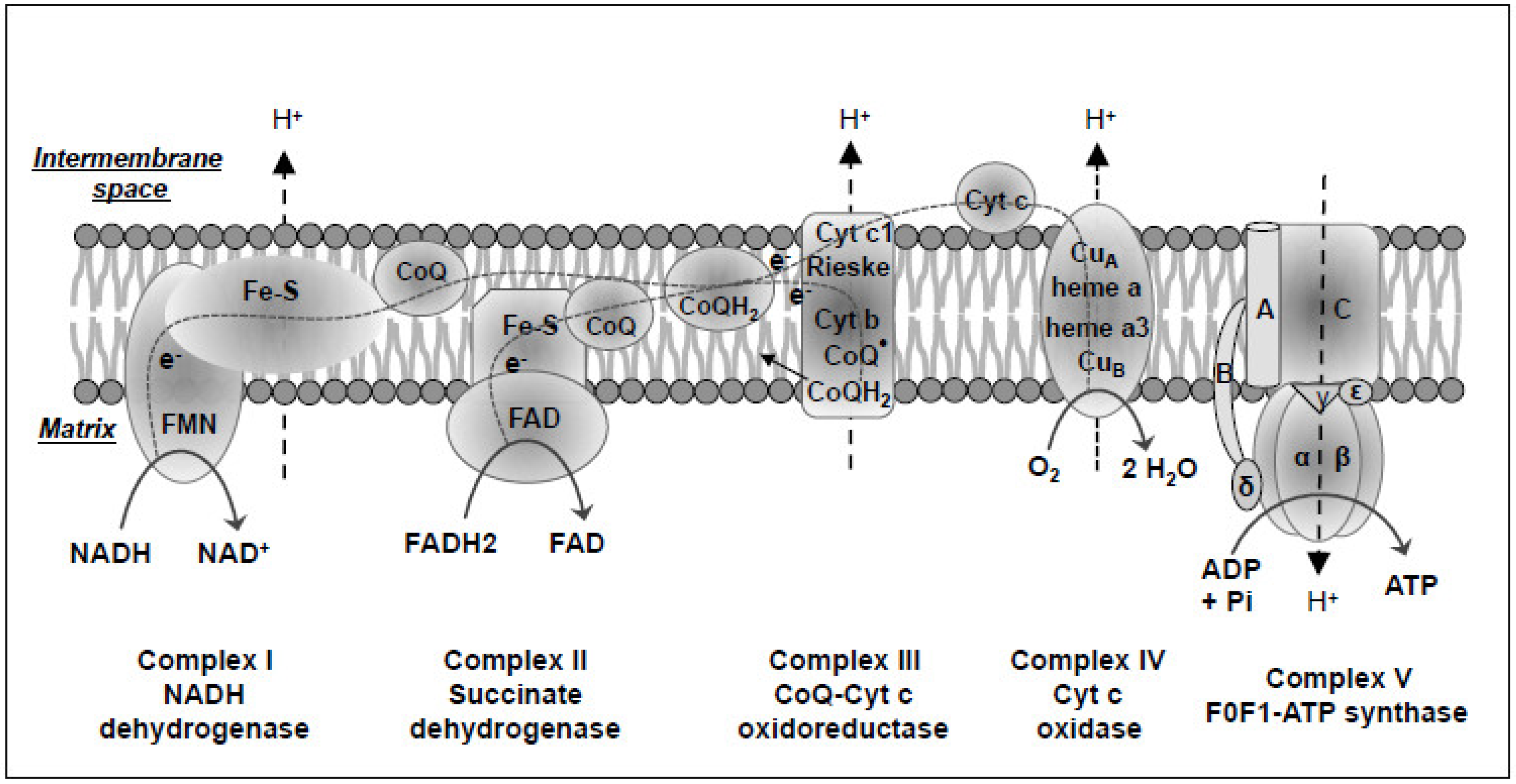
3.3. Mitochondrial Oxidative Phosphorylation Defects in Cancer
3.4. The Complexity in the Roles of mtDNA Mutations in Tumorigenesis
4. Mitochondrial Oxidative Stress and Cancer
5. Oncogene and Tumor Suppressor in Regulating Mitochondrial Function
5.1. RAS and Mitochondrial Function
5.2. HIF and Mitochondrial Function
5.3. c-Myc and Mitochondrial Function
5.4. p53 and Mitochondrial Function
5.5. NRF2 and Mitochondrial Function
6. Summary
Funding
Conflicts of Interest
References
- Munn, E.A. On the structure of mitochondria and the value of ammonium molybdate as a negative stain for osmotically sensitive structures. J. Ultrastruct. Res. 1968, 25, 362–380. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Newmeyer, D.D.; Ferguson-Miller, S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell 2003, 112, 481–490. [Google Scholar] [CrossRef]
- Bonawitz, N.D.; Clayton, D.A.; Shadel, G.S. Initiation and beyond: Multiple functions of the human mitochondrial transcription machinery. Mol. Cell 2006, 24, 813–825. [Google Scholar] [CrossRef]
- Thyagarajan, B.; Padua, R.A.; Campbell, C. Mammalian mitochondria possess homologous DNA recombination activity. J. Biol. Chem. 1996, 271, 27536–27543. [Google Scholar] [CrossRef]
- Schatz, G. The protein import system of mitochondria. J. Biol. Chem. 1996, 271, 31763–31766. [Google Scholar] [CrossRef]
- Krebs, H.A. The history of the tricarboxylic acid cycle. Perspect. Biol. Med. 1970, 14, 154–170. [Google Scholar] [CrossRef]
- Krebs, H.A. Rate control of the tricarboxylic acid cycle. Adv. Enzyme Regul. 1970, 8, 335–353. [Google Scholar] [CrossRef]
- Akram, M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 2014, 68, 475–478. [Google Scholar] [CrossRef]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brustle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Losman, J.A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef]
- Li, F.; He, X.; Ye, D.; Lin, Y.; Yu, H.; Yao, C.; Huang, L.; Zhang, J.; Wang, F.; Xu, S.; et al. NADP(+)-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance. Mol. Cell 2015, 60, 661–675. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Skoldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, W.; Xiang, B.; Li, N.; Huang, S.; Zhou, W.; Sun, Y.; Wang, X.; Ma, J.; Li, G.; et al. Reduced succinate dehydrogenase B expression is associated with growth and de-differentiation of colorectal cancer cells. Tumour Biol. 2013, 34, 2337–2347. [Google Scholar] [CrossRef]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nose, V.; Rustin, P.; Gaal, J.; Dahia, P.L.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef]
- Xekouki, P.; Pacak, K.; Almeida, M.; Wassif, C.A.; Rustin, P.; Nesterova, M.; de la Luz Sierra, M.; Matro, J.; Ball, E.; Azevedo, M.; et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: A new association for SDH? J. Clin. Endocrinol. Metab. 2012, 97, E357–E366. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Zhang, S.; Zhou, J.; Wang, Y.; Di, W. Succinate dehydrogenase subunit B inhibits the AMPK-HIF-1alpha pathway in human ovarian cancer in vitro. J. Ovarian Res. 2014, 7, 115. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwarzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Pollard, P.J. Succinate: A new epigenetic hacker. Cancer Cell 2013, 23, 709–711. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kiuru, M.; Launonen, V.; Hietala, M.; Aittomaki, K.; Vierimaa, O.; Salovaara, R.; Arola, J.; Pukkala, E.; Sistonen, P.; Herva, R.; et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am. J. Pathol. 2001, 159, 825–829. [Google Scholar] [CrossRef]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef]
- Yong, C.; Stewart, G.D.; Frezza, C. Oncometabolites in renal cancer. Nat. Rev. Nephrol. 2020, 16, 156–172. [Google Scholar] [CrossRef]
- Castro-Vega, L.J.; Buffet, A.; De Cubas, A.A.; Cascon, A.; Menara, M.; Khalifa, E.; Amar, L.; Azriel, S.; Bourdeau, I.; Chabre, O.; et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 2014, 23, 2440–2446. [Google Scholar] [CrossRef]
- Clark, G.R.; Sciacovelli, M.; Gaude, E.; Walsh, D.M.; Kirby, G.; Simpson, M.A.; Trembath, R.C.; Berg, J.N.; Woodward, E.R.; Kinning, E.; et al. Germline FH mutations presenting with pheochromocytoma. J. Clin. Endocrinol. Metab. 2014, 99, E2046–E2050. [Google Scholar] [CrossRef]
- Zhang, J.; Nichols, K.E.; Downing, J.R. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef]
- Fieuw, A.; Kumps, C.; Schramm, A.; Pattyn, F.; Menten, B.; Antonacci, F.; Sudmant, P.; Schulte, J.H.; Van Roy, N.; Vergult, S.; et al. Identification of a novel recurrent 1q42.2-1qter deletion in high risk MYCN single copy 11q deleted neuroblastomas. Int. J. Cancer 2012, 130, 2599–2606. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.S.; Chihara, Y.; Yoon, H.Y.; Kim, Y.J.; Kim, T.H.; Woo, S.H.; Yun, S.J.; Kim, I.Y.; Hirao, Y.; Kim, W.J. Downregulation of fumarate hydratase is related to tumorigenesis in sporadic renal cell cancer. Urol. Int. 2013, 90, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Locasale, J.W.; Bielas, J.H.; O’Sullivan, J.; Sheahan, K.; Cantley, L.C.; Vander Heiden, M.G.; Vitkup, D. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat. Biotechnol. 2013, 31, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Hirsila, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genom. 2003, 16, 29–37. [Google Scholar] [CrossRef]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef]
- Shim, E.H.; Livi, C.B.; Rakheja, D.; Tan, J.; Benson, D.; Parekh, V.; Kho, E.Y.; Ghosh, A.P.; Kirkman, R.; Velu, S.; et al. L-2-Hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014, 4, 1290–1298. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Na, U.; Winge, D.R.; Rutter, J. Protein-mediated assembly of succinate dehydrogenase and its cofactors. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 168–180. [Google Scholar] [CrossRef]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Invest. 2013, 123, 3652–3658. [Google Scholar] [CrossRef]
- Whelan, D.T.; Hill, R.E.; McClorry, S. Fumaric aciduria: A new organic aciduria, associated with mental retardation and speech impairment. Clin. Chim Acta 1983, 132, 301–308. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Mandavilli, B.S.; Santos, J.H.; Van Houten, B. Mitochondrial DNA repair and aging. Mutat. Res. 2002, 509, 127–151. [Google Scholar] [CrossRef]
- Ingman, M.; Kaessmann, H.; Paabo, S.; Gyllensten, U. Mitochondrial genome variation and the origin of modern humans. Nature 2000, 408, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Schon, E.A. Mitochondria. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Clayton, D.A. Molecular genetic aspects of human mitochondrial disorders. Ann. Rev. Genet. 1995, 29, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef]
- Graziewicz, M.A.; Longley, M.J.; Copeland, W.C. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem Rev. 2006, 106, 383–405. [Google Scholar] [CrossRef] [PubMed]
- Kaguni, L.S. DNA polymerase gamma, the mitochondrial replicase. Ann. Rev. Biochem. 2004, 73, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Vanderstraeten, S.; Van den Brule, S.; Hu, J.; Foury, F. The role of 3′-5′ exonucleolytic proofreading and mismatch repair in yeast mitochondrial DNA error avoidance. J. Biol. Chem. 1998, 273, 23690–23697. [Google Scholar] [CrossRef] [PubMed]
- Farr, C.L.; Wang, Y.; Kaguni, L.S. Functional interactions of mitochondrial DNA polymerase and single-stranded DNA-binding protein. Template-primer DNA binding and initiation and elongation of DNA strand synthesis. J. Biol. Chem. 1999, 274, 14779–14785. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.E.; Longley, M.J.; Copeland, W.C. The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J. Biol. Chem. 1999, 274, 38197–38203. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A. Transcription and replication of mitochondrial DNA. Human Reprod. 2000, 15, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A. Vertebrate mitochondrial DNA-a circle of surprises. Exp. Cell Res. 2000, 255, 4–9. [Google Scholar] [CrossRef]
- Falkenberg, M.; Gaspari, M.; Rantanen, A.; Trifunovic, A.; Larsson, N.G.; Gustafsson, C.M. Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat. Genet. 2002, 31, 289–294. [Google Scholar] [CrossRef]
- Gaspari, M.; Falkenberg, M.; Larsson, N.G.; Gustafsson, C.M. The mitochondrial RNA polymerase contributes critically to promoter specificity in mammalian cells. EMBO J. 2004, 23, 4606–4614. [Google Scholar] [CrossRef]
- Gaspari, M.; Larsson, N.G.; Gustafsson, C.M. The transcription machinery in mammalian mitochondria. Biochim. Biophys. Acta 2004, 1659, 148–152. [Google Scholar] [CrossRef]
- Fisher, R.P.; Lisowsky, T.; Parisi, M.A.; Clayton, D.A. DNA wrapping and bending by a mitochondrial high mobility group-like transcriptional activator protein. J. Biol. Chem. 1992, 267, 3358–3367. [Google Scholar] [PubMed]
- Fisher, R.P.; Topper, J.N.; Clayton, D.A. Promoter selection in human mitochondria involves binding of a transcription factor to orientation-independent upstream regulatory elements. Cell 1987, 50, 247–258. [Google Scholar] [CrossRef]
- Fisher, R.P.; Parisi, M.A.; Clayton, D.A. Flexible recognition of rapidly evolving promoter sequences by mitochondrial transcription factor 1. Genes Dev. 1989, 3, 2202–2217. [Google Scholar] [CrossRef]
- Fisher, R.P.; Clayton, D.A. Purification and characterization of human mitochondrial transcription factor 1. Mol. Cell. Biol. 1988, 8, 3496–3509. [Google Scholar] [CrossRef] [PubMed]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; van der Bliek, A.; Spelbrink, J.N. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 2003, 14, 1583–1596. [Google Scholar] [CrossRef]
- Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef]
- Lu, W.; Hu, Y.; Chen, G.; Chen, Z.; Zhang, H.; Wang, F.; Feng, L.; Pelicano, H.; Wang, H.; Keating, M.J.; et al. Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PLoS Biol. 2012, 10, e1001326. [Google Scholar] [CrossRef]
- Lu, W.; Chen, Z.; Zhang, H.; Wang, Y.; Luo, Y.; Huang, P. ZNF143 transcription factor mediates cell survival through upregulation of the GPX1 activity in the mitochondrial respiratory dysfunction. Cell Death Dis. 2012, 3, e422. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Singh, K.K. Mitochondrial dysfunction in cancer. Mitochondrion 2004, 4, 755–762. [Google Scholar] [CrossRef]
- Verma, M.; Kagan, J.; Sidransky, D.; Srivastava, S. Proteomic analysis of cancer-cell mitochondria. Nat. Rev. 2003, 3, 789–795. [Google Scholar] [CrossRef]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Huang, K.H.; Yeh, T.S.; Chi, C.W. Somatic alterations in mitochondrial DNA and mitochondrial dysfunction in gastric cancer progression. World J. Gastroenterol. 2014, 20, 3950–3959. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.H.; Wu, C.C.; Lin, J.C.; Chi, C.W.; Wei, Y.H.; Lee, H.C. Somatic mutations of mitochondrial genome in hepatocellular carcinoma. Mitochondrion 2010, 10, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.M.; Yin, P.H.; Yang, C.W.; Tsai, Y.F.; Hsu, C.Y.; Chi, C.W.; Lee, H.C. Somatic mutations of the mitochondrial genome in human breast cancers. Genes Chromosom. Cancer 2011, 50, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.Y.; Wu, C.W.; Yin, P.H.; Chang, C.J.; Li, A.F.; Chi, C.W.; Wei, Y.H.; Lee, H.C. Somatic mutations in mitochondrial genome and their potential roles in the progression of human gastric cancer. Biochim. Biophys. Acta 2010, 1800, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun 2017, 8, 656. [Google Scholar] [CrossRef] [PubMed]
- Larman, T.C.; DePalma, S.R.; Hadjipanayis, A.G.; Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; Cancer Genome Atlas Research Network. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 14087–14091. [Google Scholar] [CrossRef]
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293. [Google Scholar] [CrossRef]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef]
- Habano, W.; Nakamura, S.; Sugai, T. Microsatellite instability in the mitochondrial DNA of colorectal carcinomas: Evidence for mismatch repair systems in mitochondrial genome. Oncogene 1998, 17, 1931–1937. [Google Scholar] [CrossRef]
- Grzybowska-Szatkowska, L.; Slaska, B.; Rzymowska, J.; Brzozowska, A.; Florianczyk, B. Novel mitochondrial mutations in the ATP6 and ATP8 genes in patients with breast cancer. Mol. Med. Rep. 2014, 10, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Nishiguchi, S.; Shiomi, S.; Tamori, A.; Koh, N.; Takeda, T.; Kubo, S.; Hirohashi, K.; Kinoshita, H.; Sato, E.; et al. Somatic mutation of mitochondrial DNA in cancerous and noncancerous liver tissue in individuals with hepatocellular carcinoma. Cancer Res. 2001, 61, 1843–1845. [Google Scholar] [PubMed]
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Moyle, J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature 1967, 213, 137–139. [Google Scholar] [CrossRef]
- Skulachev, V.P. Chemiosmotic concept of the membrane bioenergetics: What is already clear and what is still waiting for elucidation? J. Bioenerg. Biomembr. 1994, 26, 589–598. [Google Scholar] [CrossRef]
- Zhu, J.; Vinothkumar, K.R.; Hirst, J. Structure of mammalian respiratory complex I. Nature 2016, 536, 354–358. [Google Scholar] [CrossRef]
- Hirst, J. Mitochondrial complex I. Annu Rev. Biochem 2013, 82, 551–575. [Google Scholar] [CrossRef]
- Lazarou, M.; Thorburn, D.R.; Ryan, M.T.; McKenzie, M. Assembly of mitochondrial complex I and defects in disease. Biochim. Biophys. Acta 2009, 1793, 78–88. [Google Scholar] [CrossRef]
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Leone, G.; Abla, H.; Gasparre, G.; Porcelli, A.M.; Iommarini, L. The Oncojanus Paradigm of Respiratory Complex I. Genes 2018, 9, 243. [Google Scholar] [CrossRef]
- Nouws, J.; Nijtmans, L.G.; Smeitink, J.A.; Vogel, R.O. Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: Cause, pathology and treatment options. Brain 2012, 135, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Marco-Brualla, J.; Al-Wasaby, S.; Soler, R.; Romanos, E.; Conde, B.; Justo-Mendez, R.; Enriquez, J.A.; Fernandez-Silva, P.; Martinez-Lostao, L.; Villalba, M.; et al. Mutations in the ND2 Subunit of Mitochondrial Complex I Are Sufficient to Confer Increased Tumorigenic and Metastatic Potential to Cancer Cells. Cancers 2019, 11, 1027. [Google Scholar] [CrossRef] [PubMed]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondrial complex III: An essential component of universal oxygen sensing machinery? Respir. Physiol. Neurobiol. 2010, 174, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Esser, L.; Tang, W.-K.; Zhou, F.; Zhou, Y.; Yu, L.; Yu, C.-A. Structural analysis of cytochrome bc1 complexes: Implications to the mechanism of function. Biochim. Biophys. Acta 2013, 1827, 1278–1294. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Hoque, M.O.; Upadhyay, S.; Sidransky, D. Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Res. 2008, 68, 700–706. [Google Scholar] [CrossRef]
- Kassauei, K.; Habbe, N.; Mullendore, M.E.; Karikari, C.A.; Maitra, A.; Feldmann, G. Mitochondrial DNA mutations in pancreatic cancer. Int. J. Gastrointest. Cancer 2006, 37, 57–64. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Kadenbach, B.; Huttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Dong, D.W.; Whelan, K.A.; Ruthel, G.; Uchikado, Y.; Natsugoe, S.; Nakagawa, H.; Avadhani, N.G. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 2016, 35, 1585–1595. [Google Scholar] [CrossRef]
- Wallace, D.C. Genetics: Mitochondrial DNA in evolution and disease. Nature 2016, 535, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Griguer, C.E.; Cantor, A.B.; Fathallah-Shaykh, H.M.; Gillespie, G.Y.; Gordon, A.S.; Markert, J.M.; Radovanovic, I.; Clement-Schatlo, V.; Shannon, C.N.; Oliva, C.R. Prognostic relevance of cytochrome C oxidase in primary glioblastoma multiforme. PLoS ONE 2013, 8, e61035. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.S.; Sun, Q.; Sun, C.Q.; Richards, J.C.; O’Hearn, S.; Osunkoya, A.O.; Wallace, D.C.; Petros, J.A. An inherited heteroplasmic mutation in mitochondrial gene COI in a patient with prostate cancer alters reactive oxygen, reactive nitrogen and proliferation. Biomed. Res. Int. 2013, 2013, 239257. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef]
- Guo, X.G.; Liu, C.T.; Dai, H.; Guo, Q.N. Mutations in the mitochondrial ATPase6 gene are frequent in human osteosarcoma. Exp. Mol. Pathol 2012, 94, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef]
- Cavalli, L.R.; Varella-Garcia, M.; Liang, B.C. Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 1997, 8, 1189–1198. [Google Scholar]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017, 6. [Google Scholar] [CrossRef]
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubackova, S.; Endaya, B.; Judasova, K. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab. 2019, 29, 399–416. [Google Scholar] [CrossRef]
- Ye, C.; Shu, X.O.; Wen, W.; Pierce, L.; Courtney, R.; Gao, Y.T.; Zheng, W.; Cai, Q. Quantitative analysis of mitochondrial DNA 4977-bp deletion in sporadic breast cancer and benign breast diseases. Breast Cancer Res. Treat. 2008, 108, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Ericson, N.G.; Kulawiec, M.; Vermulst, M.; Sheahan, K.; O’Sullivan, J.; Salk, J.J.; Bielas, J.H. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet. 2012, 8, e1002689. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Henning, M.S.; Starkov, A.A.; Manfredi, G. The mitochondrial respiratory chain is a modulator of apoptosis. J. Cell Biol. 2007, 179, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Romeo, G.; Rugolo, M.; Porcelli, A.M. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim. Biophys. Acta 2011, 1807, 633–642. [Google Scholar] [CrossRef]
- Joshi, S.; Tolkunov, D.; Aviv, H.; Hakimi, A.; Yao, M.; Hsieh, J.J.; Ganesan, S.; Chan, C.S.; White, E. The Genomic Landscape of Renal Oncocytoma Identifies a Metabolic Barrier to Tumorigenesis. Cell Rep. 2015, 13, 1895–1908. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Scandalios, J.G. The rise of ROS. Trends Biochem. Sci. 2002, 27, 483–486. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Le Bras, M.; Clement, M.V.; Pervaiz, S.; Brenner, C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 2005, 20, 205–219. [Google Scholar] [CrossRef]
- Szibor, M.; Richter, C.; Ghafourifar, P. Redox control of mitochondrial functions. Antioxid. Redox Signal. 2001, 3, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-Nitrosylation of mitochondrial caspases. J. Cell Biol. 2001, 154, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Goettsch, W.; Lattmann, T.; Amann, K.; Szibor, M.; Morawietz, H.; Munter, K.; Muller, S.P.; Shaw, S.; Barton, M. Increased expression of endothelin-1 and inducible nitric oxide synthase isoform II in aging arteries in vivo: Implications for atherosclerosis. Biochem. Biophys. Res. Commun. 2001, 280, 908–913. [Google Scholar] [CrossRef]
- Khan, S.R. Hyperoxaluria-induced oxidative stress and antioxidants for renal protection. Urol. Res. 2005, 33, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.G. Review: The role of glutathione in the regulation of apoptosis. Eur. J. Clin. Invest. 1999, 29, 238–245. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Drose, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidase. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003, 101, 4098–4104. [Google Scholar] [CrossRef] [PubMed]
- Baynes, J.W. The Maillard hypothesis on aging: Time to focus on DNA. Ann. N. Y. Acad. Sci. 2002, 959, 360–367. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Zastawny, T.H.; Dabrowska, M.; Jaskolski, T.; Klimarczyk, M.; Kulinski, L.; Koszela, A.; Szczesniewicz, M.; Sliwinska, M.; Witkowski, P.; Olinski, R. Comparison of oxidative base damage in mitochondrial and nuclear DNA. Free Radic. Biol. Med. 1998, 24, 722–725. [Google Scholar] [CrossRef]
- Olgun, A.; Akman, S.; Serdar, M.A.; Kutluay, T. Oxidative phosphorylation enzyme complexes in caloric restriction. Exp. Gerontol. 2002, 37, 639–645. [Google Scholar] [CrossRef]
- Lill, R.; Muhlenhoff, U. Iron-sulfur-protein biogenesis in eukaryotes. Trends Biochem. Sci. 2005, 30, 133–141. [Google Scholar] [CrossRef]
- Gardner, P.R.; Nguyen, D.D.; White, C.W. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc. Natl. Acad. Sci. USA 1994, 91, 12248–12252. [Google Scholar] [CrossRef]
- Hernanz, A.; de la Fuente, M. Characterization of aconitate hydratase from mitochondria and cytoplasm of ascites tumor cells. Biochem. Cell Biol. 1988, 66, 792–795. [Google Scholar] [CrossRef]
- Parlo, R.A.; Coleman, P.S. Continuous pyruvate carbon flux to newly synthesized cholesterol and the suppressed evolution of pyruvate-generated CO2 in tumors: Further evidence for a persistent truncated Krebs cycle in hepatomas. Biochim. Biophys. Acta 1986, 886, 169–176. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Spaargaren, M.; Bischoff, J.R.; McCormick, F. Signal transduction by Ras-like GTPases: A potential target for anticancer drugs. Gene Expr. 1995, 4, 345–356. [Google Scholar] [PubMed]
- Yakubovskaya, M.S.; Spiegelman, V.; Luo, F.C.; Malaev, S.; Salnev, A.; Zborovskaya, I.; Gasparyan, A.; Polotsky, B.; Machaladze, Z.; Trachtenberg, A.C. High frequency of K-ras mutations in normal appearing lung tissues and sputum of patients with lung cancer. Int. J. Cancer J. Int. Cancer 1995, 63, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Ohashi, M.; Shiratori, Y.; Okudaira, T.; Komatsu, Y.; Kawabe, T.; Yoshida, H.; Machinami, R.; Kishi, K.; Omata, M. Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology 1996, 110, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernandez-Porras, I.; Canamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 2017, 168, 817–829. [Google Scholar] [CrossRef]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020. [Google Scholar] [CrossRef]
- Logsdon, C.D.; Lu, W. The Significance of Ras Activity in Pancreatic Cancer Initiation. Int. J. Biol. Sci. 2016, 12, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Sharp, F.R.; Bernaudin, M. HIF1 and oxygen sensing in the brain. Nat. Rev. Neurosci. 2004, 5, 437–448. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef]
- Thomas, D.D.; Miranda, K.M.; Colton, C.A.; Citrin, D.; Espey, M.G.; Wink, D.A. Heme proteins and nitric oxide (NO): The neglected, eloquent chemistry in NO redox signaling and regulation. Antioxid. Redox Signal. 2003, 5, 307–317. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef]
- Little, C.D.; Nau, M.M.; Carney, D.N.; Gazdar, A.F.; Minna, J.D. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 1983, 306, 194–196. [Google Scholar] [CrossRef]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef]
- Dang, C.V.; Li, F.; Lee, L.A. Could MYC induction of mitochondrial biogenesis be linked to ROS production and genomic instability? Cell Cycle 2005, 4, 1465–1466. [Google Scholar] [CrossRef]
- De Souza, G.A.; Godoy, L.M.; Teixeira, V.R.; Otake, A.H.; Sabino, A.; Rosa, J.C.; Dinarte, A.R.; Pinheiro, D.G.; Silva, W.A., Jr. Proteomic and SAGE profiling of murine melanoma progression indicates the reduction of proteins responsible for ROS degradation. Proteomics 2006, 6, 1460–1470. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Itahana, Y.; Itahana, K. Emerging Roles of p53 Family Members in Glucose Metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. p53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17, 2074. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Shaw, R.J. Glucose metabolism and cancer. Curr. Opin. Cell. Biol. 2006, 18, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; Rojo de la Vega, M.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 2015, 43, 680–686. [Google Scholar] [CrossRef]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef]
- Hamada, S.; Taguchi, K.; Masamune, A.; Yamamoto, M.; Shimosegawa, T. Nrf2 promotes mutant K-ras/p53-driven pancreatic carcinogenesis. Carcinogenesis 2017, 38, 661–670. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Ludtmann, M.H.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Genes | Main Features and Mechanism | Cancer Types | References |
|---|---|---|---|
| IDH1/2 mutations | Accumulation of oncometabolites D-2HG, epigenetic alterations, and impaired mitochondrial respiration | Gliomas, glioblastomas, and AML | [11,12,13,14,15,16,17,18,19,20] |
| SDH mutations | Accumulation of succinate, inhibition of PHDs, and HIF1a stabilization | Pheochromocytoma, paraganglioma, and other cancers | [21,22,23,24,25,26,27,28,29,30,31,32,33,34] |
| FH mutations | Abnormal accumulation of fumarate, inhibition of PHDs, and HIF1a stabilization. Glutamine is used for heme synthesis. Glutamine is converted to aKG for citrate formation. Glutamine is also used to fuel TCA cycle and ATP production | Papillary renal cell cancer, leiomyosarcoma, pheochromocytoma, paragangliomas, and other cancers | [35,36,37,38,39,40,41,42,43,44,45,46] |
| SOD1-/- | Accumulation of O2-, abnormal mitochondria, oxidation of DNA, protein, and lipid, and decreased aconitase activity | Hepatocellular carcinoma | [47] |
| SOD2+/- | Mitochondrial oxidative damage, nuclear DNA/ mtDNA oxidation, and decreased mitochondrial membrane potential | Lymphoma, pituitary adenoma | [48] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Y.; Ma, J.; Lu, W. The Significance of Mitochondrial Dysfunction in Cancer. Int. J. Mol. Sci. 2020, 21, 5598. https://doi.org/10.3390/ijms21165598
Luo Y, Ma J, Lu W. The Significance of Mitochondrial Dysfunction in Cancer. International Journal of Molecular Sciences. 2020; 21(16):5598. https://doi.org/10.3390/ijms21165598
Chicago/Turabian StyleLuo, Yongde, Jianjia Ma, and Weiqin Lu. 2020. "The Significance of Mitochondrial Dysfunction in Cancer" International Journal of Molecular Sciences 21, no. 16: 5598. https://doi.org/10.3390/ijms21165598
APA StyleLuo, Y., Ma, J., & Lu, W. (2020). The Significance of Mitochondrial Dysfunction in Cancer. International Journal of Molecular Sciences, 21(16), 5598. https://doi.org/10.3390/ijms21165598