Based on the Virtual Screening of Multiple Pharmacophores, Docking and Molecular Dynamics Simulation Approaches toward the Discovery of Novel HPPD Inhibitors


Abstract
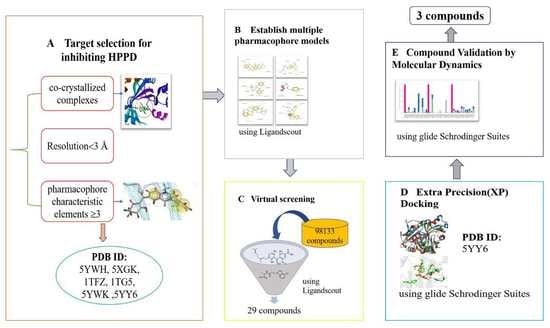
1. Introduction
2. Results
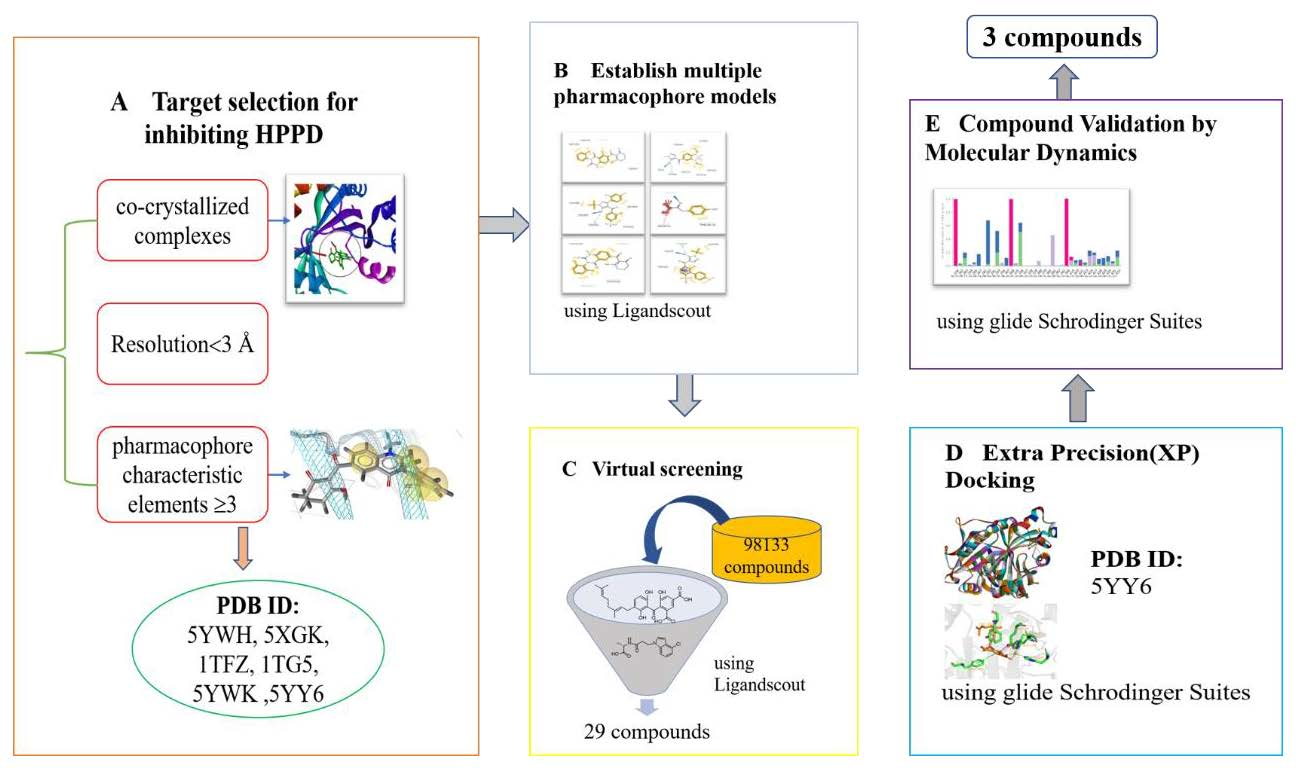
2.1. Pharmacophores for Virtual Screening
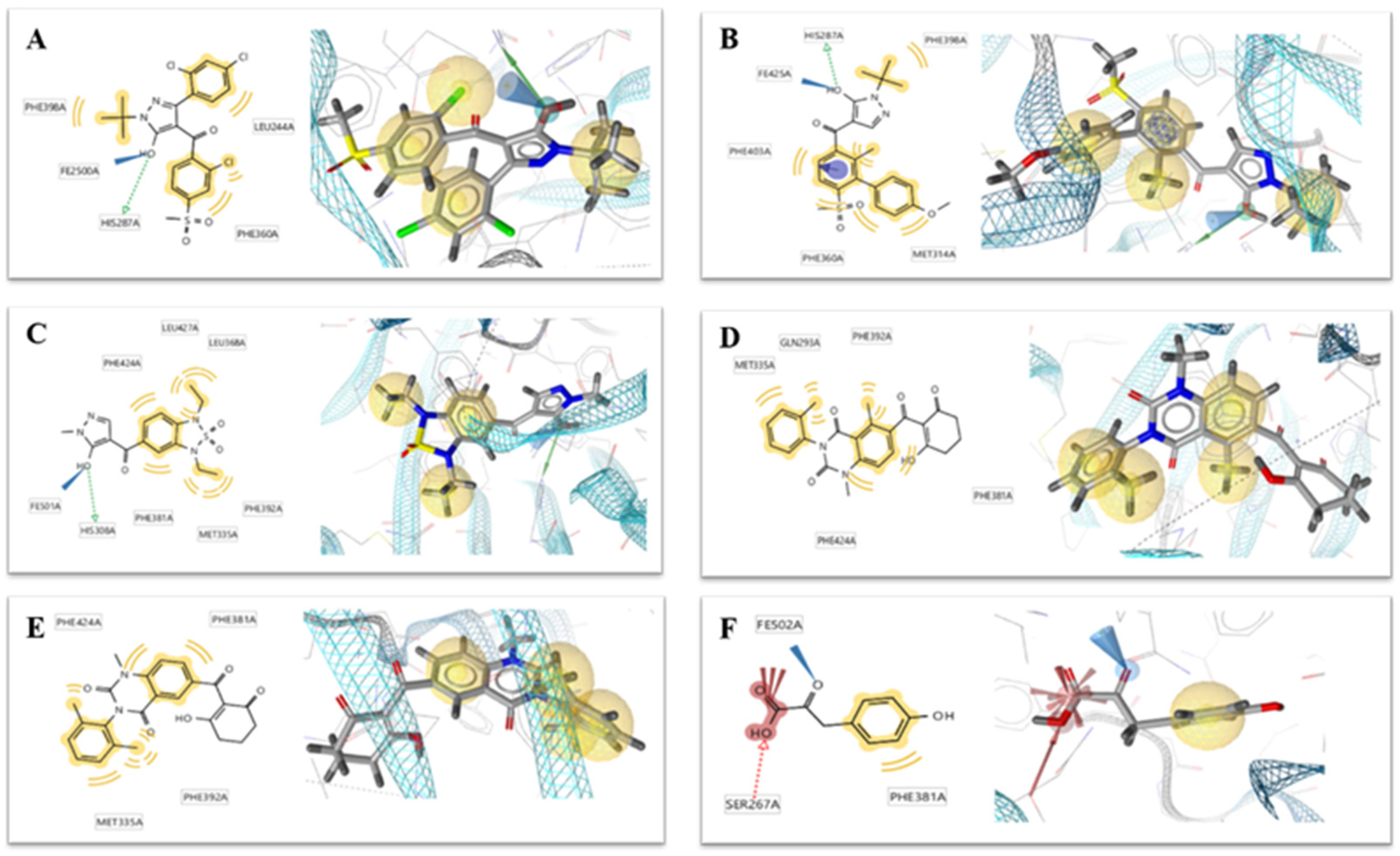
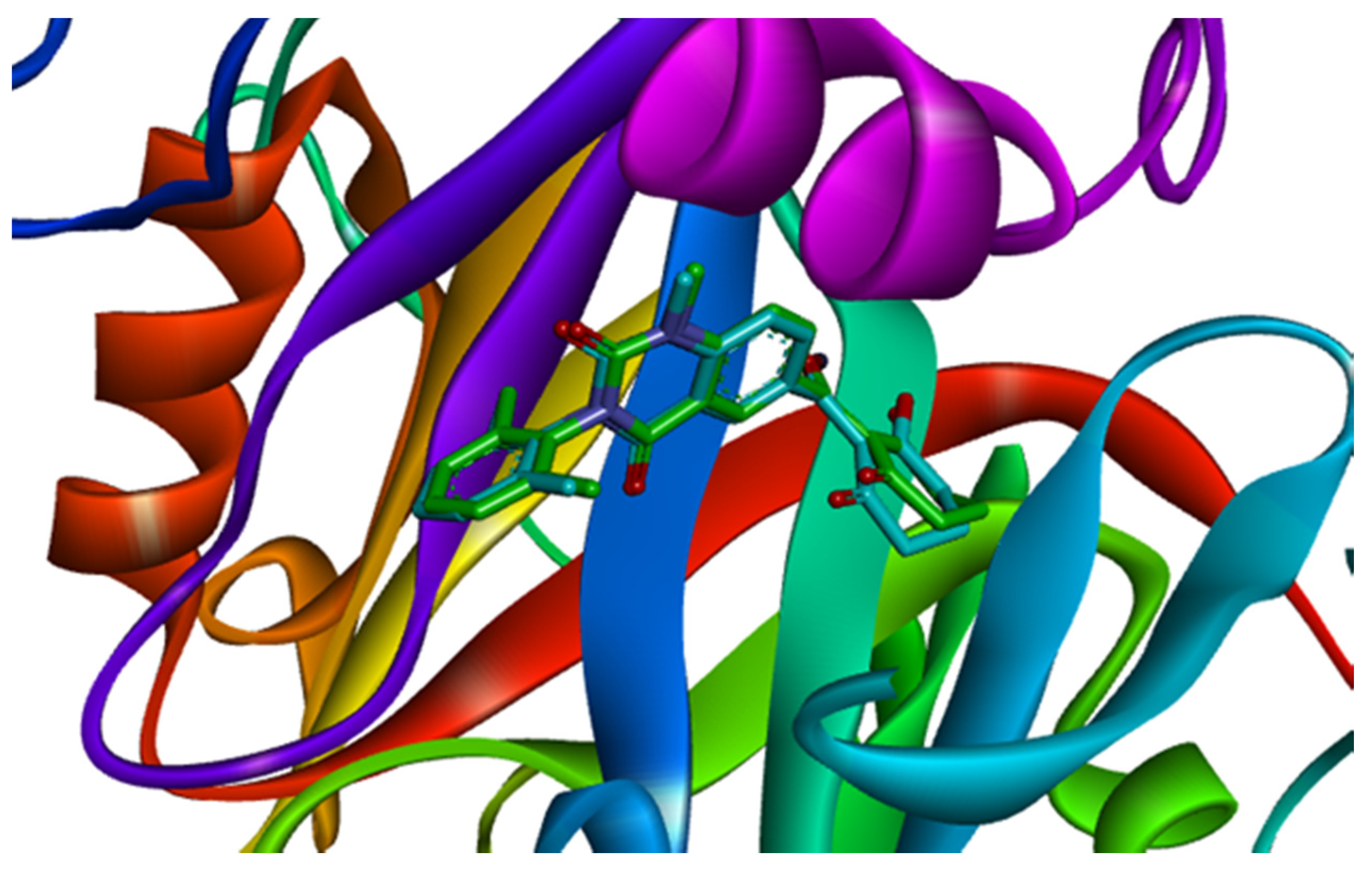
2.2. Docking Result
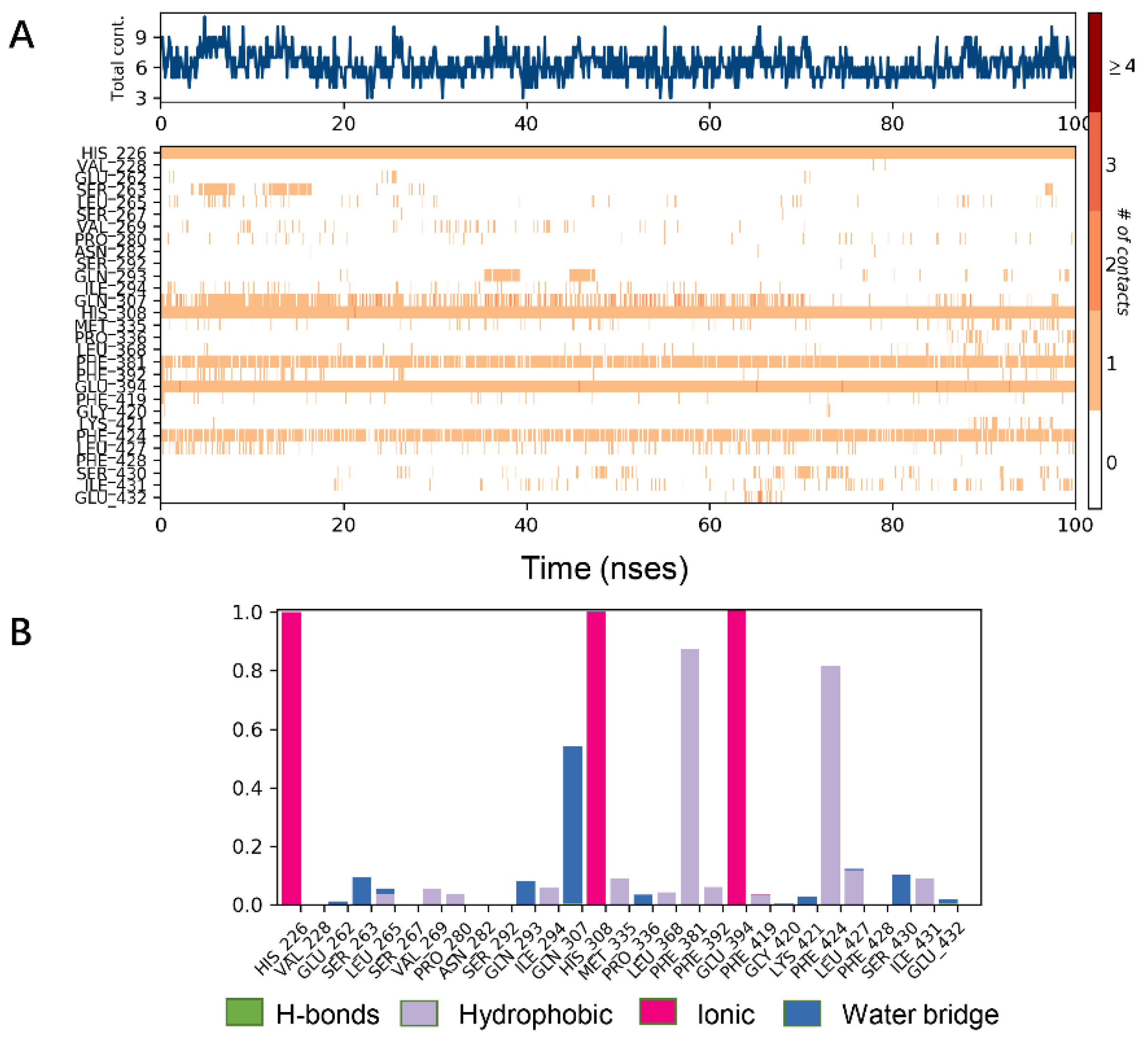
2.3. MD Simulations
2.4. MM/GBSA Calculation
3. Discussion
4. Materials and Methods
4.1. Pharmacophore Model Generation and Theoretical Validation
4.2. Multiple Receptor Conformers Based Molecular Docking
4.2.1. Ligand Preparation
4.2.2. Protein Preparation
4.2.3. Docking Simulation
4.3. MD Simulation
Author Contributions
Funding
Conflicts of Interest
References
- Alberto, D.; Serra, A.; Sulmon, C.; Gouesbet, G.; Couee, I. Herbicide-related signaling in plants reveals novel insights for herbicide use strategies, environmental risk assessment and global change assessment challenges. Sci. Total Environ. 2016, 569, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Dong, J.; Lin, H.Y.; Wang, M.Y.; Li, X.K.; Zheng, B.F.; Chen, Q.; Hao, G.F.; Yang, W.C.; Yang, G.F. Pyrazole-isoindoline-1,3-dione hybrid: A promising scaffold for 4-hydroxyphenylpyruvate dioxygenase inhibitors. J. Agric. Food Chem. 2019, 67, 10844–10852. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.E.; Kim, J.T.; Kim, E.; Ko, Y.K.; Knag, N.S. 12 HPPD inhibitors as herbicide. Bull. Korean Chem. Soc. 2013, 34, 2909–2914. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, S.Q.; Liu, Y.X.; Wang, J.Y.; Gao, S.; Zhao, L.X.; Ye, F. Design, synthesis, SAR and molecular docking of novel green niacin-triketone HPPD inhibitor. Ind. Crop Prod. 2019, 137, 566–575. [Google Scholar] [CrossRef]
- Neidig, M.L.; Kavana, M.; Moran, G.R.; Solomon, E.I. CD and MCD studies of the non-heme ferrous active site in (4-hydroxyphenyl) pyruvate dioxygenase: Correlation between oxygen activation in the extradiol and α-KG-dependent dioxygenases. J. Am. Chem. Soc. 2004, 126, 4486–4487. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Y.X.; Kang, T.; Sun, Y.N.; Li, J.Z.; Ye, F. Identification of novel inhibitors of p-hydroxyphenylpyruvate dioxygenase using receptor-based virtual screening. J. Taiwan Inst. Chem. E. 2019, 103, 33–34. [Google Scholar] [CrossRef]
- He, B.; Li, L.; Li, J.X.; Han, T.F.; He, J.L.; Zhu, Y.Q. Novel HPPD inhibitors: Triketone 2H-benzo[b][1,4]oxazin-3(4H)-one analogs. Pest Manag. Sci. 2018, 74, 579–589. [Google Scholar]
- Zhao, L.X.; Jiang, M.J.; Hu, J.J.; Zou, Y.L.; Cheng, Y.; Ren, T.; Gao, S.; Fu, Y.; Ye, F. Design, synthesis, and herbicidal activity of novel diphenyl ether derivatives containing fast degrading tetrahydrophthalimide. J. Agric. Food Chem. 2020, 68, 3729–3741. [Google Scholar] [CrossRef]
- Ndikuryayo, F.; Moosavi, B.; Yang, W.C.; Yang, G.F. 4-Hydroxyphenylpyruvate dioxygenase inhibitors: From chemical biology to agrochemicals. J. Agric. Food Chem. 2017, 65, 8523–8537. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, D.; Zhang, S.Q.; Liu, Y.X.; Guo, Y.Y.; Wang, M.X.; Gao, S.; Zhao, L.X.; Ye, F. Discovery of N-aroyl diketone/triketone derivatives as novel 4-hydroxyphenylpyruvate dioxygenase inhibiting-based herbicides. J. Agric. Food Chem. 2019, 67, 11839–11847. [Google Scholar] [CrossRef]
- Ahrens, H.; Lange, G.; Muller, T.; Rosinger, C.; Willms, L.; van Almsick, A. 4-Hydroxyphenylpyruvate dioxygenase inhibitors in combination with safeners: Solutions for modern and sustainable agriculture. Angew. Chem. Int. Ed. 2013, 52, 9388–9398. [Google Scholar] [CrossRef] [PubMed]
- Siehl, D.L.; Tao, Y.M.; Albert, H.; Dong, Y.X.; Heckert, M.; Madrigal, A.; Lincoln-Cabatu, B.; Lu, J.; Fenwick, T.; Bermudez, E. Broad 4-hydroxyphenylpyruvate dioxygenase inhibitor herbicide tolerance in soybean with an optimized enzyme and expression cassette. Plant Physiol. 2014, 166, 1162–1176. [Google Scholar] [CrossRef] [PubMed]
- Tukur, S.; Shallangwa, G.; Adamu, I.A. Theoretical QSAR modelling and molecular docking studies of some 4-hydroxyphenylpyruvate dioxygenase (HPPD) enzyme inhibitors potentially used as herbicides. Heliyon 2019, 5, e02859. [Google Scholar] [CrossRef] [PubMed]
- Kavana, M.; Moran, G.R. Interaction of (4-hydroxyphenyl)yruvate dioxygenase with the specific inhibitor 2-[2-nitro-4-(trifluoromethyl) benzoyl]-1,3-cyclohexanedione. Biochemistry 2003, 42, 10238–10245. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Liu, Y.X.; Wang, C.; Zhao, L.X.; Fu, Y.; Ye, F. Design, synthesis and biological activity of novel diazabicyclo derivatives as safeners. J. Agric. Food Chem. 2020, 68, 3403–3414. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Gao, S.; Hoang, M.T.; Wang, Z.W.; Ma, X.; Zhai, Y.; Li, N.; Zhao, L.X.; Fu, Y.; Ye, F. Protective efficacy of phenoxyacetyl oxazolidine derivatives as safeners against nicosulfuron toxicity in maize. Pest Manag. Sci. 2020, 7, 11. [Google Scholar] [CrossRef]
- Godar, A.S.; Varanasi, V.K.; Nakka, S.; Prasad, P.W.; Thompson, C.R.; Mithila, J. Physiological and molecular mechanisms of differential sensitivity of palmer amaranth (amaranthus palmeri) to mesotrione at varying growth temperatures. PLoS ONE 2015, 10, e0126731. [Google Scholar] [CrossRef]
- Ye, F.; Zhai, Y.; Guo, K.L.; Liu, Y.X.; Li, N.; Gao, S.; Zhao, L.X.; Fu, Y. Safeners improve maize tolerance under herbicide toxicity stress by increasing the activity of enzyme in vivo. J. Agric. Food Chem. 2019, 67, 11568–11576. [Google Scholar] [CrossRef]
- Gao, S.; Jiang, J.Y.; Li, X.M.; Liu, Y.Y.; Zhao, L.X.; Fu, Y.; Ye, F. Enhanced physicochemical properties and herbicidal activity of an environment-friendly clathrate formed by β-cyclodextrin and herbicide cyanazine. J. Mol. Liq. 2020, 305, 112858. [Google Scholar] [CrossRef]
- Gupta, C.L.; Khan, M.B.; Ampasala, D.R.; Akhtar, S.; Dwivedi, U.N.; Bajpa, P. Pharmacophore-based virtual screening approach for identification of potent natural modulatory compounds of human Toll-like receptor 7. J. Biomol. Struct. Dyn. 2019, 37, 4721–4736. [Google Scholar] [CrossRef]
- Wang, E.C.; Sun, H.Y.; Wang, J.M.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T.J. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sun, Y.N.; Yi, K.H.; Li, M.Q.; Cao, H.F.; Li, J.Z.; Ye, F. Combination of virtual screening protocol by in silico toward the discovery of novel 4-hydroxyphenylpyruvate dioxygenase inhibitors. Front. Chem. 2018, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X.; Gao, S.; Ye, T.; Li, J.Z.; Ye, F.; Fu, Y. Combined 3D-quantitative structure–activity relationships and topomer technology-based molecular design of human 4-hydroxyphenylpyruvate dioxygenase inhibitors. Future Med. Chem. 2020, 12, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Purpero, V.M.; Moran, G.R. Catalytic, noncatalytic, and inhibitory phenomena: Kinetic analysis of (4-hydroxyphenyl) pyruvate dioxygenase from Arabidopsis thaliana. Biochemistry 2006, 45, 6044–6055. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Y.X.; Yi, K.H.; Li, M.Q.; Li, J.Z.; Ye, F. Quantitative structure activity relationship studies and molecular dynamics simulations of 2-(aryloxyacetyl) cyclohexane-1,3-diones derivatives as 4-hydroxyphenylpyruvate dioxygenase inhibitors. Front. Chem. 2019, 7, 556. [Google Scholar] [CrossRef]
- He, B.; Wang, D.W.; Yang, W.C.; Chen, Q.; Yang, G.F. Advances in research on 4-hydroxyphenylpyruvate dioxygenase (HPPD) structure and pyrazole-containing herbicides. Chin. J. Org. Chem. 2017, 37, 2895–2904. [Google Scholar] [CrossRef]
- Yang, C.; Pflugrath, J.W.; Camper, D.L.; Foster, M.L.; Pemich, D.J.; Wlsh, T.A. Inhibitor selectivity revealed by comparison of crystal structures of plant and mammalian 4-hydroxyphenylpyruvate dioxygenases. Biochemistry 2004, 43, 10414–10423. [Google Scholar] [CrossRef]
- Fritze, I.M.; Linden, L.; Freigang, J.; Auerbach, G.; Huber, R.; Steinbacher, S. The crystal structures of Zea mays and Arabidopsis 4-hydroxyphenylpyruvate dioxygenase. Plant. Physiol. 2004, 134, 1388–1400. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chen, X.; Chen, J.N.; Wang, D.W.; Wu, F.X.; Lin, S.Y.; Zhan, C.G.; Wu, J.W.; Yang, W.C.; Yang, G.F. Crystal structure of 4-hydroxyphenylpyruvate dioxygenase in complex with substrate reveals a new starting point for herbicide discovery. Research 2019, 2019, 2602414. [Google Scholar] [CrossRef]
- Lin, H.Y.; Yang, J.F.; Wang, D.W.; Hao, G.F.; Dong, J.Q.; Wang, Y.X.; Yang, W.C.; Wu, J.W.; Zhan, C.G.; Yang, G.F. Molecular insights into the mechanism of 4-hydroxyphenylpyruvate dioxygenase inhibition: Enzyme kinetics, X-ray crystallography and computational simulations. FEBS J. 2019, 286, 975–990. [Google Scholar] [CrossRef]
- Yang, W.C.; Yang, W.C.; Lin, H.Y. Crystal structure of Arabidopsis thaliana HPPD complexed with Y13508. To be published.
- Yang, W.C.; Lin, H.Y. Crystal structure of Arabidopsis thaliana HPPD Truncated mutant complexed with NTBC. To be published.
- Yang, W.C.; Chen, J.N. Crystal structure of Arabidopsis thaliana HPPD complexed with Benquitrione-Methyl. To be published.
- Lin, H.Y.; Yang, G.F. Structure of Y17107 complexed HPPD. To be published.
- Wang, M.X.; Wang, Y.; Kong, D.; Jiang, H.L.; Wang, J.; Cheng, M.S. In silico exploration of aryl sulfonamide analogs as voltage-gated sodium channel 1.7 inhibitors by using 3D-QSAR, molecular docking study, and molecular dynamics simulations. Comput. Biol. Chem. 2018, 77, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, B.; Garscha, U.; Temml, V.; Liers, J.; Werz, O.; Schuster, D.; Stuppner, H. Discovery of potent soluble epoxide hydrolase (sEH) inhibitors by pharmacophore-based virtual screening. J. Chem. Inf. Model. 2016, 56, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Nashev, L.G.; Odermatt, A.; Rollinger, J.M.; Schuster, D. Pharmacophore model refinement for 11β-hydroxysteroid dehydrogenase inhibitors: Search for modulators of intracellular glucocorticoid concentrations. Mol. Inf. 2014, 33, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.K.; Mehta, S.; Ember, S.W.J.; Zhu, J.Y.; Schonbrunn, E.; Ayad, N.G.; Schurer, S.C. Identification of a Novel Class of BRD4 Inhibitors by Computational Screening and Binding Simulations. ACS. Omega 2017, 2, 4760–4771. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, I.A.; Jiffri, E.H.; Ashraf, G.M.; Kamal, M.A. Structural insights into the camel milk lactoperoxidase: Homology modeling and molecular dynamics simulation studies. J. Mol. Graph. Model. 2018, 86, 43–51. [Google Scholar] [CrossRef]
- Aktulga, H.M.; Fogarty, J.C.; Pandit, S.A.; Grama, A.Y. Parallel reactive molecular dynamics: Numerical methods and algorithmic techniques. Parallel. Comput. 2006, 38, 245–259. [Google Scholar] [CrossRef]
- Slivashanmugam, M.; Sulochana, K.N.; Umashankar, V. Virtual screening of natural inhibitors targeting ornithine decarboxylase with pharmacophore scaffolding of DFMO and validation by molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2018, 37, 766–780. [Google Scholar] [CrossRef]
- Tao, H.Y.; Zhang, Y.; Huang, S.Y. Improving protein-peptide docking results via pose clustering and rescoring with a combined knowledge-based and MM-GBSA scoring function. J. Chem. Inf. Model. 2020, 60, 2377–2387. [Google Scholar] [CrossRef]
- Bello, M.; Mendez-Luna, D.; Sarmiento, V.; Basurto, J.C.; Najera, N.; Villarreal, F.; Ceballos, G. Structural and energetic basis for novel epicatechin derivatives acting as GPER agonists through the MMGBSA method. J. Steroid. Biochem. 2019, 189, 176–186. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | PDB ID | Resolution (Å) | Ligand | Metal Ion | Released |
|---|---|---|---|---|---|
| [27] | 1TFZ | 1.80 | DAS869 | Fe | 2004 |
| [27] | 1TG5 | 1.90 | DAS645 | Fe | 2004 |
| [27] | 1SQD | 1.80 | - | Fe | 2004 |
| [28] | 1SP9 | 3.00 | - | Fe | 2004 |
| [29] | 5XGK | 2.60 | HPPA | Fe | 2018 |
| [30] | 6ISD | 2.40 | sulcotrione | Co | 2018 |
| [30] | 5YWG | 2.60 | mesotrione | Co | 2019 |
| [30] | 6J63 | 2.62 | NTBC | Fe | 2019 |
| [31] | 5YWH | 2.72 | Y13508 | Fe | 2019 |
| [32] | 5YWI | 2.58 | NTBC | Co | 2019 |
| [33] | 5YWK | 2.80 | benquitrione-methyl | Co | 2019 |
| [30] | 5YY6 | 2.40 | benquitrione | Co | 2019 |
| [34] | 6JX9 | 1.80 | Y17107 | Co | 2020 |
| Compound | ΔGbind | ΔGbind Coulomb | ΔGbind Covalent | ΔGbind Hbond | ΔGbind Lipo | ΔGbind vdW |
|---|---|---|---|---|---|---|
| ZINC000049180836 | −55.129 | −48.956 | 6.930 | −0.530 | −26.162 | −41.324 |
| ZINC000040310216 | −48.339 | 0.322 | 8.513 | −0.482 | −25.319 | −48.749 |
| ZINC000003830381 | −30.081 | −26.801 | 9.483 | −1.109 | −27.654 | −41.861 |
| ZINC000002032320 | −43.629 | −15.992 | 8.294 | −0.565 | −23.267 | −49.487 |
| ZINC000035458722 | −50.129 | −13.244 | 3.182 | −0.592 | −26.8102 | −40.3212 |
| ZINC000012663485 | −29.341 | −45.45 | 7.568 | −0.449 | −24.507 | −34.352 |
| Benquitrione | −19.652 | −27.7484 | 3.5389 | −0.968 | −15.086 | −32.827 |
| Inhibitors | Structure | Docking Score | Glide Gscore | Glide Energy (kcal mol−1) |
|---|---|---|---|---|
| ZINC000049180836 |  | −12.206 | −12.294 | −52.002 |
| ZINC000040310216 |  | −11.449 | −11.449 | −47.096 |
| ZINC000003830381 |  | −10.762 | −10.765 | −31.596 |
| ZINC000002032320 |  | −9.504 | −10.859 | −30.080 |
| ZINC000035458722 |  | −8.865 | −10.903 | −51.623 |
| ZINC000012663485 |  | −8.549 | −8.549 | −32.895 |
| Benquitrione |  | −5.921 | −5.921 | −44.475 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Ye, T.; Liu, Y.-X.; Wang, J.; Ye, F. Based on the Virtual Screening of Multiple Pharmacophores, Docking and Molecular Dynamics Simulation Approaches toward the Discovery of Novel HPPD Inhibitors. Int. J. Mol. Sci. 2020, 21, 5546. https://doi.org/10.3390/ijms21155546
Fu Y, Ye T, Liu Y-X, Wang J, Ye F. Based on the Virtual Screening of Multiple Pharmacophores, Docking and Molecular Dynamics Simulation Approaches toward the Discovery of Novel HPPD Inhibitors. International Journal of Molecular Sciences. 2020; 21(15):5546. https://doi.org/10.3390/ijms21155546
Chicago/Turabian StyleFu, Ying, Tong Ye, Yong-Xuan Liu, Jian Wang, and Fei Ye. 2020. "Based on the Virtual Screening of Multiple Pharmacophores, Docking and Molecular Dynamics Simulation Approaches toward the Discovery of Novel HPPD Inhibitors" International Journal of Molecular Sciences 21, no. 15: 5546. https://doi.org/10.3390/ijms21155546
APA StyleFu, Y., Ye, T., Liu, Y.-X., Wang, J., & Ye, F. (2020). Based on the Virtual Screening of Multiple Pharmacophores, Docking and Molecular Dynamics Simulation Approaches toward the Discovery of Novel HPPD Inhibitors. International Journal of Molecular Sciences, 21(15), 5546. https://doi.org/10.3390/ijms21155546