Abstract
The eukaryotic and archaeal translation factor IF5A requires a post-translational hypusine modification, which is catalyzed by deoxyhypusine synthase (DHS) at a single lysine residue of IF5A with NAD+ and spermidine as cofactors, followed by hydroxylation to form hypusine. While human DHS catalyzed reactions have been well characterized, the mechanism of the hypusination of archaeal IF5A by DHS is not clear. Here we report a DHS structure from Pyrococcus horikoshii OT3 (PhoDHS) at 2.2 Å resolution. The structure reveals two states in a single functional unit (tetramer): two NAD+-bound monomers with the NAD+ and spermidine binding sites observed in multi-conformations (closed and open), and two NAD+-free monomers. The dynamic loop region V288–P299, in the vicinity of the active site, adopts different positions in the closed and open conformations and is disordered when NAD+ is absent. Combined with NAD+ binding analysis, it is clear that PhoDHS can exist in three states: apo, PhoDHS-2 equiv NAD+, and PhoDHS-4 equiv NAD+, which are affected by the NAD+ concentration. Our results demonstrate the dynamic structure of PhoDHS at the NAD+ and spermidine binding site, with conformational changes that may be the response to the local NAD+ concentration, and thus fine-tune the regulation of the translation process via the hypusine modification of IF5A.
1. Introduction
Hypusine (N-(4-amino-2-hydroxybutyl) lysine), an unusual amino acid, is formed by the post-translational modification of a lysine residue with the addition of the aminobutyl moiety from the polyamine spermidine. The hypusine modification uniquely occurs in eukaryotic translation factor 5A (eIF5A) precursor proteins [1]. Although eIF5A was originally designed as a translation initial factor, it has been shown that eIF5A is more directly related to elongation rather than the initiation step. The hypusine-modification of eIF5A is involved in the mRNA transportation and translation elongation on the ribosome [2,3]. The binding of eIF5A to translating ribosomes in a hypusine-dependent manner indicates modifications of the specific binding to the translational machinery [4,5], suggesting the important role of eIF5A receiving hypusine modifications in translation processes, which was supported by evidence that the hypusine modification of eIF5A is vital for the growth and survival of eukaryotes, from yeast to mammals [6,7,8]. Previous studies also reported that the hypusine residue is essential for the homodimerization of eIF5A and affects its subcellular location [1,2,9,10].
The hypusine modification of eIF5A is catalyzed by deoxyhypusine synthase (DHS), followed by the hydroxylation of deoxyhypusine by deoxyhypusine hydroxylase (DOHH) [1,11]. DHS catalyzes a NAD+-dependent reaction that synthesizes deoxyhypusine (N-(4-aminobutyl) lysine) by transferring the butylamine moiety of spermidine to a specific lysine residue of eIF5A [12]. Then, deoxyhypusine is hydroxylated by DOHH to form hypusine [13,14,15]. Like eIF5A, DHS is an essential gene, indicating the importance of hypusine modification [16]. Since eIF5A is associated with many diseases, such as diabetes, cancers, malaria, and HIV-1 infections [16], trials aimed at blocking the hypusine modification of eIF5A have been attempted. Specific inhibitors of DHS or DOHH have been reported to affect the arrest of cellular growth, and also alter the proliferation and differentiation of tumor cells [17,18,19]. An extensive biochemical characterization of DHS has been carried out, establishing it as a 40–43 kDa enzyme (the exact size being species-dependent) requiring NAD+ as a cofactor. The mechanism of human deoxyhypusine synthesis by DHS has been proposed as follows [20]: firstly, a hydrogen of spermidine is transferred to NAD+ to generate dehydrospermidine and NADH; the dehydrospermidine then links covalently to a lysine residue (K329 in human DHS) of DHS, forming an enzyme-imine intermediate; finally, DHS catalyzes the formation of deoxyhypusine through the conjugation of the butylamine moiety of dehydrospermidine to the ε-amino group of the lysine residue (K37) of the eIF5A precursor protein.
The reported crystal structures of human DHS show a homotetramer consisting of two tightly associated dimers with four active sites—two near each tight dimer interface [21,22]. Residues from both monomers of a dimer contribute to each of its active sites. In human DHS, four active sites accommodate four molecules of NAD+ (PDB:1DHS) [21,22]. Interestingly, the catalytic sites are blocked by a projecting N-terminal two-turn helix (described as a “ball and chain”) in a cross-monomer manner in the first crystal form of human DHS, obtained upon crystallization at high ionic strengths and a low pH. However, the catalytic site is not obstructed in a structure of human DHS in a ternary complex with NAD+ and GC7 (PDB:1RQD), an analog of spermidine [22]. In this ternary complex structure, GC7 is also located at the interface of the tight dimers and is separated from NAD+ by a loop structure (T307–A319) [22]. The absence of a blockage by the ball and chain structure might suggest that this dynamic part of the enzyme could be involved in the regulation of activity within the functional tetramer. However, this human DHS complex structure without any bound GC7 (PDB:1ROZ), determined upon crystallization at low ionic strengths and pH 8.0 (nearer physiological conditions), lacked the electron density for the N-terminal ball and chain region [22]. In recent research, it has been reported that the N-terminal ball and chain region is important for the assembly of a functional DHS tetramer [23]. This N-terminal region is, therefore, clearly highly dynamic, with the flexibility being for DHS function. Whether it is regulating the active site accessibility in a substrate-dependent manner, binding eIF5A, or conferring sensitivity to cellular conditions, remains unclear. Different from human DHS, the DHS from Trypanosoma brucei exists as a heterotetramer formed by two paralogs, DHSc and DHSp (dimer of heterodimer), with DHSc homologous to human DHS. Each heterodimer contains two NAD+ binding sites, one located in the functional catalytic site while the other is located in a remnant site lacking key catalytic residues [24].
Consistent with archaea sharing a requirement for hypusine for the function of translation factor aIF5A, both the modification [24] and DHS gene homologues [1] have been identified. Furthermore, a study has shown the arrest of archaeal growth in G1 upon treatment with an inhibitor of DHS [25], indicating that DHS is also essential in archaea. However, the mechanism of hypusination in archaea is unclear yet [26]. Homologues of the non-essential second enzyme required for hypusine formation, DOHH, have not been identified. While some archaea may function with a deoxyhypusine modification (rather than hypusine), species are known that form hypusine in the absence of oxygen, which implies a distinct mechanism of hypusine modification in archaea. The structure of an archaeal DHS could provide a better understanding of the process of hypusine modification of aIF5A.
In this study, we determined the crystal structure of DHS from Pyrococcus horikoshii OT3 (PhoDHS), with NAD+-bound, at 2.2 Å resolution. PhoDHS is a tetramer, each monomer sharing a similar fold with its human homologue (PDB ID:1DHS). Interestingly, unlike human and T. brucei DHS, PhoDHS contains only two bound NAD+ molecules and shows a difference in the NAD+-bound and NAD+-free monomeric conformations. In particular, NAD+-bound monomers in the PhoDHS tetramer display different conformations in the region of V288–P299 around the NAD+ binding site. The observations are suggestive of a distinct manner in which NAD+ binds to DHS that could be of relevance to the mechanism and regulation of enzyme activity. Having the states of NAD+-bound and NAD+-free also sheds light on the NAD+-dependent spermidine binding to DHS and raises a possibility that dynamic NAD+ binding in PhoDHS may fine-tune the translation activity of aIF5A via hypusine modification.
2. Results
2.1. The Overall Structure of PhoDHS
We determined the crystal structure of PhoDHS at 2.2 Å resolution by using a molecular replacement (Table 1). In an asymmetric unit, the structure of PhsDHS formed a tetramer in accordance with its known biological assembly [21,22], with unbuilt N-terminal 14 and C-terminal 6 of the total 342 residues in each monomer. Additionally, we could not build the parts of two loops in each monomer (E122-D125 and G306–K311 in molA, W290–K311 in molB, E122–V123 and W290–A310 in molC, and W121–K130 and E301–K311 in molD, respectively), because of the lack of electron density. Similar to DHS from other organisms, this tetramer is a dimer of dimers with a point group of D2, generated by two local 2-fold axes that are perpendicular to each other (Figure 1). The monomers of PhoDHS tetramers are more related to each other with a root mean squared deviation (r.m.s.d) between 0.544 Å and 0.606 Å for 276–284 Cα atoms. Based on the buried areas of intermolecular interactions, the tightly bound dimers are defined as molAB and molCD (buried areas > 2700 Å2), while the buried area is less than 1800 Å2 between molA and molD (or between molB and molC). PhoDHS is very similar to human DHS (PDB ID:1DHS), with a r.m.s.d of 1.195 Å for 289 Cα, and is also similar to T. brucei DHS (PDB:6DFT) [24] with a r.m.s.d of 1.39 Å for 275 Cα (Figure 1b). Differences include the N-terminal region (M1–P26) and an insertion (L77–L94) in human DHS. Unlike the inactive human DHS structure, wherein the N-terminal two-turn α helix (A9–L18) blocks the entrance tunnel to the spermidine active site [21,22], the N-terminal region (M1–I14) of PhoDHS is not built due to the poor electron density for this region, suggesting that it is highly flexible and different from human DHS, despite the crystallization conditions being similar to those of inactive human DHS [21]. Given that the active sites are accessible, we assume that the structure of PhoDHS obtained is illustrative of an active form.

Table 1.
Summary of data collection and refinement statistics.
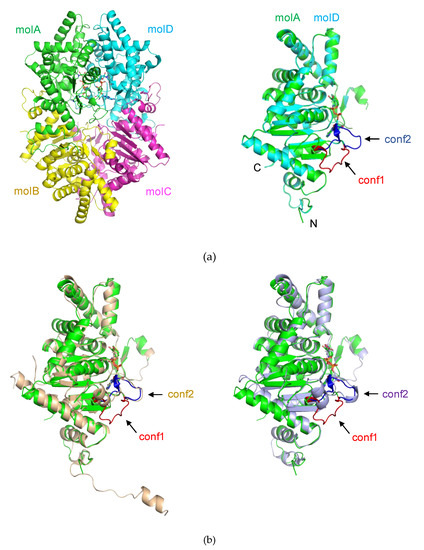
Figure 1.
Crystal structure of deoxyhypusine synthase (DHS). The protein and NAD+ is shown by the ribbon and stick, respectively. (a) Overall structure of Pyrococcus horikoshii OT3 DHS (PhoDHS): (left) A tetramer of PhoDHS in an asymmetric unit with monomeric units colored green (molA), yellow (molB), magenta (molC) and cyan (molD). (right) Superimposition of molD (cyan) onto molA (green). A loop (V288–P299) around the NAD+ binding site is observed in two different conformations (blue and red) in molA (conformation 1, conf1) and molD (conformation 2, conf2), respectively. While the conf1 in molA is away from NAD+, the conf2 in molD moves towards NAD+. (b) Structural superposition of PhoDHS (green) with human (wheat, PDBID:1DHS) and Trypanosoma brucei (light purple, PDBID:6DFT) DHS. The conf1 and conf2 loop in the comparison molecules are labeled (in their respective colors), and the loop in conf1 (red) of molA is added for comparison.
Although the overall structures of DHS are similar, there is a significant structural difference in a loop region (V288–P299) that forms part of the active site of PhoDHS. This active site loop is flexible and presents itself in multi-conformations, conf1 and conf2 in molA and molD, respectively, which are likely to represent two snapshots of the active site (Figure S1). In the molB and molC of PhoDHS, the loop region is either disordered, or partially visible. Although only one of two conformations, conformation 1 or conformation 2 (conf1, conf2), was assigned to each molA and molD, a weak density of the alternative conformations could be observed, further suggesting the flexibility of the loop region in PhoDHS. In contrast, this active site loop is stable and present in just one conformation (conf2) in the previously published human and T. brucei DHS structures [24].
Another striking structural difference seen in PhoDHS is at its C-terminus. The C-terminus of PhoDHS (Gly337–Gln342, in all four chains) is invisible in the structure, which could be observed in human and Trypanosoma brucei DHS [24]. A sequence alignment shows that the primary structure of this region is not conserved in PhoDHS, human DHS, and T. brucei DHS (Figure 2). The short helix predicted for PhoDHS is longer in human DHS. The helix is visible in the human DHS structure, though the region retains some intrinsic flexibility, indicating conformational dynamics in the C-terminal region across species. Removal of five residues from the C-terminus in human DHS or 39 residues from the C-terminus of S. cerevisiae DHS impairs activity [26,27]. It is probable that the C-terminus of PhoDHS is also required for full function, and that the intrinsic flexibility of this region, rather than the conserved sequences, is likely important for the activity.

Figure 2.
Sequence alignment of PhoDHS with DHS from human and T. brucei. The ESPript 3.0 server was used to output the alignment [28]. The blue box above the sequence indicates that the sequences of at least two out of three organisms share a similarity, while the sequences identical in all the three organisms are marked with red fills. The secondary structure shown by the respective crystal structures for PhoDHS and human DHS are labeled at the top and bottom, respectively. The loop 288–299 with alternative conformations is indicated with grey stars above.
2.2. NAD+ Binding Manner of PhoDHS
Two bulky electron density blocks shaped as NAD+ were distinguished at the interface between molA and molD of the PhoDHS tetramer after several cycles of refinement and were later assigned as NAD+ (Figure 3a and Figure S2). Those NAD+ molecules were naturally captured during the expression of the enzyme in Escherichia coli, since no NAD+ was added during purification and crystallization processes.
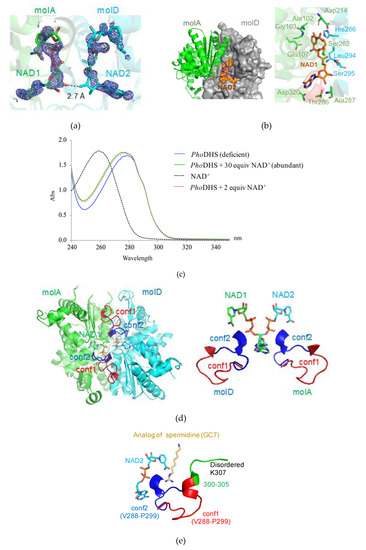
Figure 3.
Active site of PhoDHS-bound NAD+. (a) Electron density map for NAD+. NAD+ is superposed with the 2Fo-Fc electron density map contoured at the 1σ level (blue). (b) Binding geometry of NAD+ by PhoDHS. (left) NAD+ is bounded to the interface of the dimer in a sandwich manner. (right) NAD+ extensively interacts with residues from both monomers via a hydrogen bond and electronic interaction. (c) Quantification of NAD+ bound to PhoDHS in solution by UV absorbance spectroscopy. An obvious difference in the absorption curves for PhoDHS alone (no NAD+ added) and supplemented with NAD+ as PhoDHS + 30 equiv NAD+ (molar ratio PhoDHS:NAD of 1:30), shows the binding of NAD+ to vacant sites in purified PhoDHS. The absorption curve of PhoDHS + two equiv NAD+ (molar ratio 1:2) correlates well to the curve PhoDHS + 30 equiv NAD+, indicating that PhoDHS could only accommodate two additional NAD+ even if excessive NAD+ molecules are available. (d) (left) Closed (blue) and open (red) forms of the binding loop V288–P299 with NAD+ bound. (right) Alternative conformations of the loop involved in NAD+ and spermidine binding. (e) Superposition of GC7 onto PhoDHS. GC7 is superposed onto PhoDHS by structural alignment with human DHS in complex with GC7. The loop of two conformations (conf1 and conf2) in molA and molD was generated by the superposition of the two monomers.
Bound NAD+ is proximal to a loop (V288–P299 (hereafter named “the binding loop”)) that displays very different conformations in each monomer of the PhoDHS tetramer. Notably, in molA and molD, observed to bind NAD+, the binding loop adopts two different conformations (Figure 1a, right): in molA, the binding loop positions away from NAD+ (conf1), while in molD, the binding loop adopts another conformation to access NAD+ (conf2). In contrast, only the second conformation (conf2) of the binding loop was observed in the structures of human and T. brucei DHS (Figure 1b). In the molB and molC of the PhoDHS tetramer, where NAD+ is absent, the binding loop is disordered or only partially visible. We propose that NAD+ binding to PhoDHS induces the stabilization of the flexible loop structure, but that the alternative conformations indicate the action of the loop as a “lid.” In an open conformation (conf1), the active site is exposed to receive NAD+, and the lid is closed over NAD+ in conf2. This controlled switched direction of the binding loop in the presence of NAD+ may be important for the regulation of NAD+ and/or spermidine binding, the “lid” potentially opening and closing in response to the surrounding NAD+ content. In the absence of NAD+, the binding loop is presumably much more flexible in structure and thus invisible in molB and molC. Our PhoDHS structure is the first structure showing distinct differences in the NAD+-bound state of the DHS active site, differences additional to those in the NAD+-free state. This may be an additional pointer to a complex role for conformational dynamics in the function of DHS in the cell, potentially linked to the regulation of the translation process via a hypusine modification of IF5A.
The binding geometry of NAD+ in PhoDHS is similar to that in human DHS [21]. The two NAD+ molecules (NAD1 and NAD2) are embraced individually by molA and molD of PhoDHS in a sandwich binding manner and insert their ends into the pockets of molA, and molD with 2-fold symmetry (Figure 3b). The closest distance between NAD1 and NAD2 is 2.7 Å, where a hydrogen bond can form between the ribose hydroxyl group of each adenosine moiety (Figure 3a). Such interaction between NAD1 and NAD2 stabilizes their binding to DHS, which may explain why, when only two NAD+s are bound, these bind to molAD but not molAB. A cluster of residues from molA and molD compose a binding site for NAD+: NAD1 forms hydrogen bonds with Thr101, Ala102, Gly103, Glu107, Asp214, Ser262, Thr286, Ala287 and Asp320 from molA, and stacks with the imidazole ring of His266 from molD. Interestingly, the main chain groups of Leu294 and Ser295 of the binding loop from molD in conf2 are also involved in a hydrogen bonding network to NAD1. To further characterize the binding mode, we generate the two alternative conformations (conf1, conf2) in both molA and molD by the superimposition of the molA and molD, which shows that the binding loop in conf2 of molA is more tightly associated with the NAD2 (which shares a similarly bound geometry to NAD1) of molD than with NAD1. Except for Ser262, all residues that contact NAD+ are conserved (Figure 2). The crossover of interactions would suggest that molA and molD enhance NAD+ binding to each other, and there is likely to be a synergistic effect between the binding of NAD1 and NAD2, mediated by the flexible loop.
In the previously reported DHS structures [21,22,23], four NAD+ were accommodated individually in four active sites of the tetramer. In our PhoDHS structure, however, the occupation of only two active sites by NAD+ is observed for the first time. To further investigate the NAD+ binding state of the PhoDHS tetramer, we carried out a UV absorption scan using PhoDHS in solution (Figure 3c). There is an obvious difference in the absorption curves of PhoDHS alone and PhoDHS supplemented with NAD+. The match between the absorption scan for PhoDHS + 30 equiv NAD+ (molar ratio of PhoDHS:NAD+ = 1:30) and PhoDHS + two equiv NAD+ (molar ratio 1:2) indicates that PhoDHS can only accommodate two additional NAD+ even if excess NAD+ molecules are available, suggesting that there are two vacant sites in the purified PhoDHS, which become occupied in both conditions. The spectroscopy confirms that there are likely only two out of four binding sites that are occupied by NAD+ in purified PhoDHS in solution, validating the observations from crystallography. The fact that human DHS tetramers bind four NAD+ molecules at high concentrations of NAD+ [21,22] does not exclude the binding of only two NAD+ molecules when the NAD+ concentration is low. Conversely, our spectroscopy data show that PhoDHS can likely bind four NAD+ molecules when sufficient NAD+ is present. The lower binding ratio of NAD+ observed in PhoDHS could infer that DHS may also adopt distinct conformations for NAD+ binding in other species, probably regulated by the local concentration of cellular NAD+, which may offer a control mechanism for IF5A modification and, therefore, activity in cell processes.
2.3. Dynamic Conformation of the Region for Both NAD+ and Spermidine Binding
As described above, the comparison of PhoDHS monomers reveals the conformational dynamics near the active site. The region V288–K311, located in a valley of the DHS tetramer center, shows either free mobility (and therefore disorder) in the absence of bound NAD+ or two distinct conformations in its presence. In all four monomers, the electron density was lacking for G306–K311, which includes the catalytic Lys307, the imine bond acceptor with spermidine, and thus the aminobutyl donor to aIF5A. In a crystal structure of a human DHS ternary complex, this region is visualized and interacts with NAD+ and a spermidine analog, GC7 [22]. In a recent published paper, it was suggested that no conformational changes occur in the DHS structure upon binding with spermidine [23]. Our identification of the binding loop region V288–P299, acting as a lid over the active site to give open (conf1) and closed (conf2) forms, is indicative of a significant conformational change, at least with respect to NAD binding (Figure 3d). Upon the superposition of the PhoDHS and human DHS (complexed with GC7) structures, we found that GC7 has very close access (about 4.4 Å) to the V288–P299 loop in conf2, but is substantially far away (about 8.2 Å) in conf1 (Figure 3e). Thus, both NAD+ and spermidine can interact with the binding loop in conf2 but not in conf1, suggesting that the flexible binding loop plays a role in accepting substrates, even when NAD+ has bound. We can, therefore, consider three conformational states of this loop in relation to its function (Figure 4): (I) apo form: the disordered structure observed in molB and molC of PhoDHS, prior to NAD+ binding; (II) open form (conf1), the lid is away from the bound NAD+ (observed in molA) in PhoDHS, and is awaiting substrate binding; (III) reaction/closed form (conf2), the lid closes to contact the bound NAD+ (observed in molD) and also spermidine (as indicated in human DHS). Since PhoDHS interacts with NAD+, spermidine, and IF5A via this region, the conformational dynamics are perhaps an unsurprising potential contributor to the achievement of the different conformations for binding different partners. It is also possible, as stated earlier, that the conformational dynamics are a mechanism by which DHS can respond to the concentration of NAD+ and fine-tune the regulation of translation in organisms via the hypusine modification of IF5A.
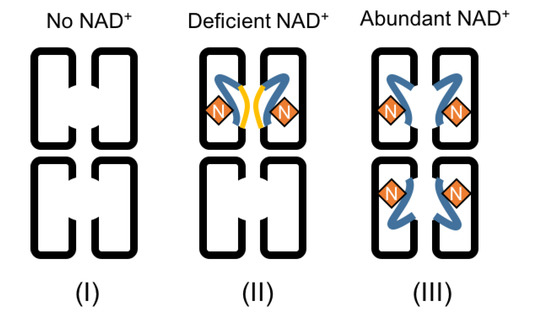
Figure 4.
A model of the conformational dynamics in DHS, regulated by NAD+. Three binding conformations are shown. On the left, the spermidine binding loop is disordered when there is no NAD+ binding to DHS. In the middle, the loop is partially stabilized and structured in either conf1 or conf2 when NAD+ binds to DHS. The binding of one NAD+ to DHS could promote a second NAD+ to bind to another monomer of the dimer, and binding of both NAD+s would be stabilized via the formation of the hydrogen bond between them. The spermidine binding loop is still disordered in another dimer of the tetramer which remains NAD+-free. On the right, the spermidine binding loop of all four monomers of PhoDHS will be ordered in conf2 in the presence of abundant NAD+, based on the current structure and human DHS structure. Yellow lines: loop in conf1 away from NAD+; blue lines: loop in conf2 towards NAD+; N: NAD+ molecules.
3. Discussion
Taking the results as a whole, we propose that the binding of NAD+ to PhoDHS is regulated by the NAD+ concentration, which might itself be linked to the requirement for IF5A modification and the subsequent participation in translation. One PhoDHS tetramer binds two NAD+s at a low concentration of NAD+, but may be able to bind two additional NAD+s when supplemented with higher NAD+ concentrations. This new model for NAD+ binding by DHS is shown in Figure 4 with PhoDHS existing in three states: no NAD+, one PhoDHS tetramer binding two NAD+s on one side (molAD or molBC), and one PhoDHS tetramer binding four NAD+s. Our structural data definitively demonstrate the different conformations of loop V288–P299 in each monomer of PhoDHS, with the significant structural stabilization induced by NAD+ binding, which, with the help of a comparison to the human DHS:GC7 structure, is likely stabilized into the final closed conformation (conf2) upon binding spermidine. Indeed, the different conformations of the active site “lid,” or binding loop, observed in our structure are likely to reflect the absence of stabilizing interactions that spermidine binding would provide. Our structure provides a potential explanation for the NAD+-dependent binding of spermidine to DHS [29]. The conformation diversity observed in this region in our structure is compatible with an independence of NAD+ binding to each monomer, suggesting that monomers of DHS may behave equally whether in a tetramer, dimer or heterodimers. Strikingly, our structure presents a simultaneous combination of three different conformations at the spermidine binding site that are reflective of a three-state conformational regulation of DHS by NAD+: the apo form, the open form, and the closed/reaction form. The structure thus highlights the role of conformational dynamics in the regulation of DHS and hypusine modification of aIF5A.
To further address the relationship between the conformational changes of region V288–P299 observed here and the binding of spermidine and IF5A to generate hypusinated IF5A, the complex structure of IF5A-DHS is an important future goal.
4. Materials and Methods
4.1. Expression and Purification of PhoDHS
The gene encoding DHS (1–342 aa, 39 kDa) from P. horikoshii OT3 (UniProtKB-O50105) was cloned into a modified pET26b expression vector with a hexahistidine (His6) tag and a TEV cleavage sequence (Glu-Asn-Leu-Tyr-Phe-Gln-Gly) at the N-terminus. For overexpression, the recombinant plasmid containing PhoDHS was transformed into E. coli strain B834 (DE3) pRARE2 by electroporation. The transformants were inoculated into Luria–Bertani (LB) containing 25 μg/mL kanamycin and 34 μg/mL chloramphenicol and cultivated at 30 °C until the optical density at 600 nm (OD600) reached about 0.6. After adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, the cells were grown for an additional 16 h.
The cells were harvested by centrifugation at 4500× g at 25 °C for 20 min, and then washed with cell-wash-buffer (50 mM Tris-HCl, pH 8.0, 50 mM NaCl). The washed cells were disrupted by sonication with cell-lysis buffer (50 mM Tris-HCl, pH 8.5, 500 mM NaCl, 5% glycerol, 0.25 mM DTT, 25 mM imidazole) in the presence of 100 μg/mL deoxyribonuclease I and 50 μg/mL lysozyme, and the cell debris was then removed by centrifugation at 40,000× g for 30 min. The cell lysate was heat-treated at 70 °C for 30 min and centrifuged at 40,000× g for 30 min. The supernatant was filtered through a 0.22 μm filter and then loaded onto a Ni-affinity chromatography column (1 mL Histrap™ HP; GE Healthcare, Chicago, IL, USA) equilibrated with buffer A (50 mM Tris-HCl, pH 8.5, 500 mM NaCl, 5% glycerol, 0.25 mM DTT, 25 mM imidazole). The samples were eluted with a gradient of 25–400 mM imidazole in buffer A. The eluted fractions of the proteins were treated with TEV protease and dialyzed against buffer B (50 mM Tris-HCl, pH 8.5, 25 mM NaCl, 5% glycerol, 0.25 mM DTT, 25 mM imidazole) to remove the cleaved His tag. The dialyzed proteins were purified again using the Ni-affinity column, and the flow-through fraction was collected. The proteins were then purified using a 1 mL RESOURCETM Q column (GE Healthcare) with buffer C (20 mM Tris-HCl, pH 8.5, 25 mM NaCl, 5% glycerol, 0.25 mM DTT). The bound protein was eluted with a linear gradient of NaCl (25–2000 mM). The fractions containing DHS were pooled and then load on to a Superdex 75 16/60 column (GE Healthcare, Chicago, IL, USA) in buffer D (20 mM HEPES, pH 8.0, 200 mM NaCl, 5% glycerol, and 1 mM DTT), and DHS was eluted as a single peak with an estimated molecular weight around 130 kDa, corresponding to the tetramer. Fractions containing purified tetramer were combined and concentrated using VIVAPORE 10/20 (2–20 mL). All steps were monitored by 15% SDS-PAGE.
4.2. Crystallization and Data Collection
Preliminary crystallization trials were performed using a crystallization screening kit from QIAGEN (JCSG I-IV, JCSG + and PEG I&II, Hilden, Germany). A crystallization drop containing 1 μL of protein solution and 1 μL reservoir solution was set up and equilibrated against 100 μL of reservoir solution at 20 °C using the sitting-drop method. Initial crystals were grown in a condition containing 100 mM phosphate–citrate pH 4.2, 40% ethanol, 5% PEG 1000. After the optimization of crystallization conditions with respect to pH and precipitant concentration, well diffracted crystals were obtained in a condition containing 100 mM phosphate–citrate pH 4.8, 32% Ethanol, 5% PEG 1000.
For diffraction data collection, the crystals of PhoDHS were transferred into a cryo-protectant solution containing 15% glycerol in the reservoir solution, then mounted in a loop and flash-cooled in a stream of nitrogen gas at −173 °C. The X-ray diffraction experiment was performed with a wavelength of 1.0 Å at beamline BL41XU in SPring-8 (Hyogo, Japan). The diffraction data were indexed, integrated, scaled, and merged using the HKL2000 package [30]. The crystals belong to space group P212121, with the unit-cell parameters of a = 87.3 Å, b = 89.9 Å, c = 164.8 Å. Using a calculated molecular weight of 38.9 Da, the Matthews coefficient VM value was estimated to be 2.07 Å3Da−1 with 4 PhoDHS molecules in the asymmetric unit, corresponding to a solvent content of 40.56%. A summary of data collection and processing statistics is given in Table 1.
4.3. Structural Determination and Refinement
The structure of PhoDHS was solved by molecular replacement with MOLREP [31], using the protein structure of human DHS (PDB ID:1DHS) as a search model [21], which shows 39.5% amino acid sequence identity with PhoDHS. Initial rigid body refinement was performed with the REFMAC [32] of the CCP4 program suite [33] and the structure was then rebuilt using the program LAFIRE with CNS automatically [34,35]. Structural refinements were further performed using the program phenix_refine of the phenix program suite [36], followed by manual modifications with Coot [37]. After several refinement cycles, the NAD+ molecules were manually built based on 2Fo-Fc and Fo-Fc electron density maps. During the refinement of PhoDHS in complex with NAD+, the NAD+ molecules were rebuilt based on the omit-map several times and the occupancy of NAD+ molecules was adjusted manually following check of Fo-Fc and 2Fo-Fc maps. We also rebuilt the binding loop (V288–P299) several times based on the omit-map and the manually fine-tuned residues occupancy. The Rwork and Rfree factors were monitored during the refinement process, and the latter was calculated from 5% of the reflections. The final structure of PhoDHS was refined to a Rwork/Rfree = 17.52/22.95%, respectively. The summary of refinement statistics is shown in Table 1. The atomic coordinates of PhoDHS have been deposited in the Protein Data Bank under the accession number 7CMC. The buried areas of inter monomer were calculated using PDBePISA [38].
4.4. Analysis of NAD+ Binding by UV Absorbance Spectroscopy
The purified PhoDHS was mixed with NAD+ (Roche, Nutley, New Jersey, USA) at the molar ratio of 1:30. The complex of PhoDHS and NAD+ was dialyzed in buffer C (20 mM HEPES, pH 8.0, 200 mM NaCl, 5% glycerol, and 1 mM DTT) by using EasySep (TOMY, Tokyo, Japan) to remove free, excess NAD+. The final concentration of the PhoDHS tetramer was about 5.87 μM. The UV absorption spectra of the dialyzed samples were measured by using a DU800 Spectrophotometer (Beckman, Brea, California, USA) at the scanning speed of 1200 nm/min. The dialyzed buffer was taken as the blank. NAD+ at 106 μM in buffer D (20 mM HEPES, pH 8.0, 200 mM NaCl, 5% glycerol, and 1 mM DTT) was used as control. The absorption curve of 2*NAD+ (11.74 μM), a 1:2 molar ratio of PhoDHS:NAD+ was similarly measured using the control curve of NAD+ (106 μM) as the reference.
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/21/15/5509/s1.
Author Contributions
Conceptualization, M.C., C.O. and M.Y.; Investigation, C.O., Z.G., J.Y.; validation, Z.G., Y.Y. and M.C.; writing—original draft preparation, M.C., Z.G., Y.Y.; writing—review and editing, M.C., M.Y.; supervision, M.Y.; project administration, M.Y.; funding acquisition, M.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from Japan Agency for Medical Research and Development (AMED) under Grant Number JP18am0101071.
Acknowledgments
We would like to thank H. Wakabayashi for her work on sample preparation. We thank the beamline staff of Spring-8 for their help with data collection.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Greganova, E.; Altmann, M.; Buetikofer, P. Unique modifications of translation elongation factors. FEBS J. 2011, 278, 2613–2624. [Google Scholar] [CrossRef] [PubMed]
- Saini, P.; Eyler, D.E.; Green, R.; Dever, T.E. Hypusine-containing protein eIF5A promotes translation elongation. Nature 2009, 459, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Landau, G.; Bercovich, Z.; Park, M.H.; Kahana, C. The Role of Polyamines in Supporting Growth of Mammalian Cells Is Mediated through Their Requirement for Translation Initiation and Elongation. J. Biol. Chem. 2010, 285, 12474–12481. [Google Scholar] [CrossRef] [PubMed]
- Jao, D.L.E.; Chen, K.Y. Tandem affinity purification revealed the hypusine-dependent binding of eukaryotic initiation factor 5A to the translating 80S ribosomal complex. J. Cell. Biochem. 2006, 97, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Zanelli, C.F.; Maragno, A.L.; Gregio, A.P.; Komili, S.; Pandolfi, J.R.; Mestriner, C.A.; Lustri, W.R.; Valentini, S.R. eIF5A binds to translational machinery components and affects translation in yeast. Biochem. Biophys. Res. Commun. 2006, 348, 1358–1366. [Google Scholar] [CrossRef]
- Park, M.H.; Joe, Y.A.; Kang, K.R. Deoxyhypusine synthase activity is essential for cell viability in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 1677–1683. [Google Scholar] [CrossRef]
- Nishimura, K.; Lee, S.B.; Park, J.H.; Park, M.H. Essential role of eIF5A-1 and deoxyhypusine synthase in mouse embryonic development. Amino Acids 2012, 42, 703–710. [Google Scholar] [CrossRef]
- Patel, P.H.; Costa-Mattioli, M.; Schulze, K.L.; Bellen, H.J. The Drosophila deoxyhypusine hydroxylase homologue nero and its target elF5A are required for cell growth and the regulation of autophagy. J. Cell Biol. 2009, 185, 1181–1194. [Google Scholar] [CrossRef]
- Lee, S.B.; Park, J.H.; Kaevel, J.; Sramkova, M.; Weigert, R.; Park, M.H. The effect of hypusine modification on the intracellular localization of eIF5A. Biochem. Biophys. Res. Commun. 2009, 383, 497–502. [Google Scholar] [CrossRef]
- Gentz, P.M.; Blatch, G.L.; Dorrington, R.A. Dimerization of the yeast eukaryotic translation initiation factor 5A requires hypusine and is RNA dependent. FEBS J. 2009, 276, 695–706. [Google Scholar] [CrossRef]
- Wolff, E.C.; Kang, K.R.; Kim, Y.S.; Park, M.H. Posttranslational synthesis of hypusine: Evolutionary progression and specificity of the hypusine modification. Amino Acids 2007, 33, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Lee, Y.B.; Joe, Y.A. Hypusine is essential for eukaryotic cell proliferation. Biol. Signals 1997, 6, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Cooper, H.L.; Folk, J.E. The biosynthesis of protein-bound hypusine (N epsilon—(4-amino-2-hydroxybutyl)lysine). Lysine as the amino acid precursor and the intermediate role of deoxyhypusine (N epsilon—(4-aminobutyl) lysine). J. Biol. Chem. 1982, 257, 7217–7222. [Google Scholar] [PubMed]
- Park, M.H.; Wolff, E.C. Cell-free synthesis of deoxyhypusine-separation of protein substrate and enzyme and identification of 1,3-diaminopropane as aproduct of spermidine cleavage. J. Biol. Chem. 1988, 263, 15264–15269. [Google Scholar]
- Wolff, E.C.; Lee, Y.B.; Chung, S.I.; Folk, J.E.; Park, M.H. Deoxyhypusine synthase from rat testis: Purification and characterization. J. Biol. Chem. 1995, 270, 8660–8666. [Google Scholar] [CrossRef]
- Kaiser, A. Translational control of eIF5A in various diseases. Amino Acids 2012, 42, 679–684. [Google Scholar] [CrossRef]
- Von Koschitzky, I.; Gerhardt, H.; Lämmerhofer, M.; Kohout, M.; Gehringer, M.; Laufer, S.; Pink, M.; Schmitz-Spanke, S.; Strube, C.; Kaiser, A. New insights into novel inhibitors against deoxyhypusine hydroxylase from plasmodium falciparum: Compounds with an iron chelating potential. Amino Acids 2015, 47, 1155–1166. [Google Scholar] [CrossRef]
- Preukschas, M.; Hagel, C.; Schulte, A.; Weber, K.; Lamszus, K.; Sievert, H.; Pällmann, N.; Bokemeyer, C.; Hauber, J.; Braig, M.; et al. Expression of eukaryotic initiation factor 5A and hypusine forming enzymes in glioblastoma patient samples: Implications for new targeted therapies. PLoS ONE 2012, 7, e43468. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kurasawa, O.; Yokota, A.; Klein, M.G.; Ono, K.; Saito, B.; Matsumoto, S.; Okaniwa, M.; Ambrus-Aikelin, G.; Morishita, D.; et al. Discovery of Novel Allosteric Inhibitors of Deoxyhypusine Synthase. J. Med. Chem. 2020, 63, 3215–3226. [Google Scholar]
- Wolff, E.C.; Folk, J.E.; Park, M.H. Enzyme-substrate intermediate formation at lysine 329 of human deoxyhypusine synthase. J. Biol. Chem. 1997, 272, 15865–15871. [Google Scholar] [CrossRef]
- Liao, D.I.; Wolff, E.C.; Park, M.H.; Davies, D.R. Crystal structure of the NAD complex of human deoxyhypusine synthase: An enzyme with a ball-and-chain mechanism for blocking the active site. Structure 1998, 6, 23–32. [Google Scholar] [CrossRef]
- Umland, T.C.; Wolff, E.C.; Park, M.H.; Davies, D.R. A new crystal structure of deoxyhypusine synthase reveals the configuration of the active enzyme and of an enzyme center dot NAD center dot inhibitor ternary complex. J. Biol. Chem. 2004, 279, 28697–28705. [Google Scholar] [CrossRef] [PubMed]
- Wątor, E.; Wilk, P.; Grudnik, P. Half Way to Hypusine—Structural Basis for Substrate Recognition by Human Deoxyhypusine Synthase. Biomolecules 2020, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Afanador, G.A.; Tomchick, D.R.; Phillips, A.M. Trypanosomatid Deoxyhypusine Synthase Activity Is Dependent on Shared Active-Site Complementation between Pseudoenzyme Paralogs. Structure 2018, 26, 1499–1512.e5. [Google Scholar] [CrossRef] [PubMed]
- Jansson, B.P.M.; Malandrin, L.; Johansson, H.E. Cell cycle arrest in archaea by the hypusination inhibitor N-1-guanyl-1,7-diaminoheptane. J. Bacteriol. 2000, 182, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.R.; Chung, S.I. Characterization of yeast deoxyhypusine synthase: PKC-dependent phosphorylation in vitro and functional domain identification. Exp. Mol. Med. 1999, 31, 210–216. [Google Scholar] [CrossRef]
- Joe, Y.A.; Wolff, E.C.; Park, M.H. Cloning and expression of human deoxyhypusine synthase cDNA—Structure-function studies with the recombinant enzyme and mutant proteins. J. Biol. Chem. 1995, 270, 22386–22392. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Lee, C.H.; Park, M.H. Human deoxyhypusine synthase: Interrelationship between binding of NAD and substrates. Biochem. J. 2000, 352, 851–857. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. In Macromolecular Crystallography, PT A. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Vagin, A.; Teplyakov, A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Struct. Biol. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yao, M.; Tanaka, I. New algorithm for protein model building: Extending a partial model in a map segment. J. Appl. Crystallogr. 2006, 39, 57–63. [Google Scholar] [CrossRef]
- Brünger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Coot, K.C. Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).