Association of Polymorphisms of MASP1/3, COLEC10, and COLEC11 Genes with 3MC Syndrome



Abstract
1. Introduction
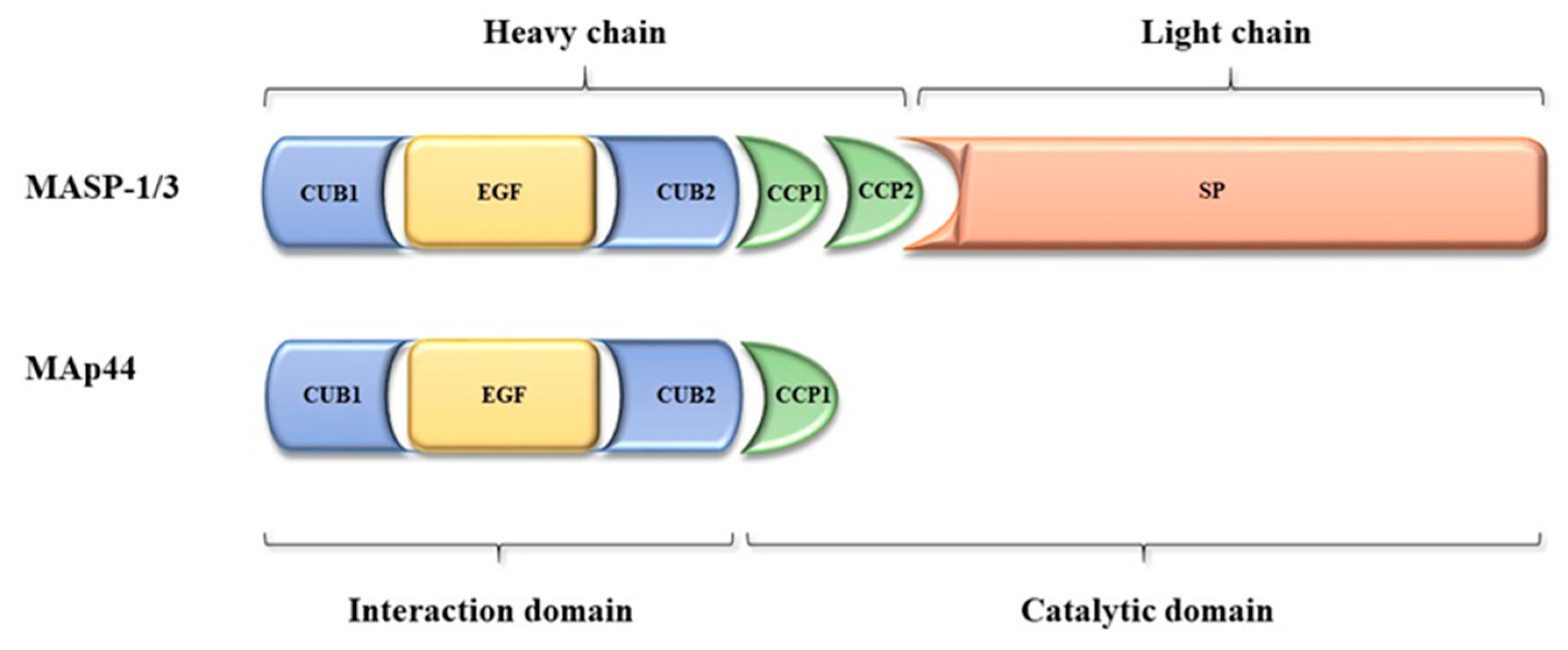
2. The MASP1/3 Gene and Its Products—MASP-1, MASP-3, and MAp44
2.1. Gene and Protein Structure
2.2. Tissue Expression of the MASP-1, MASP-3, and MAp44
2.3. Biological Activities of the MASP-1, MASP-3, and MAp44
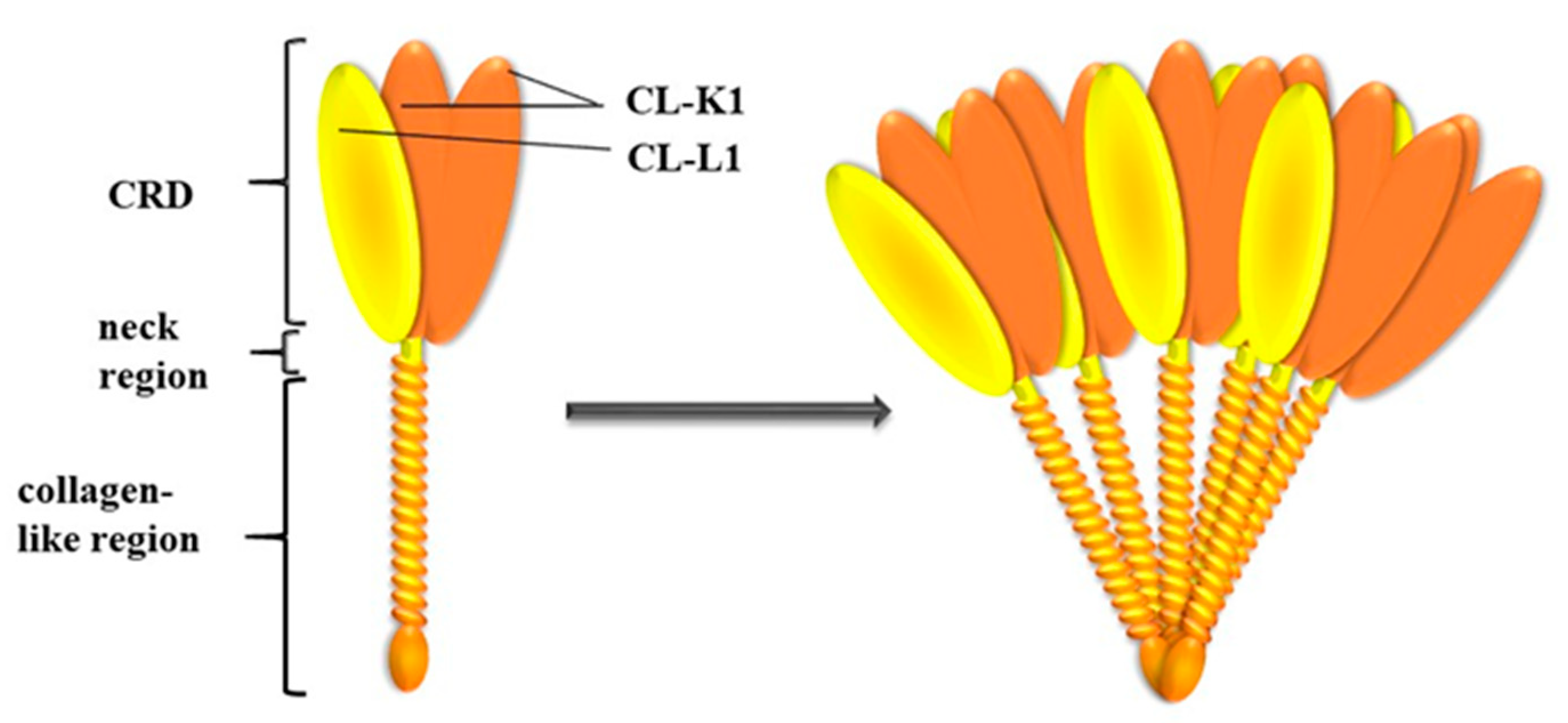
3. COLEC10 and COLEC11 Genes and Their Products, CL-L1 and CL-K1
3.1. Genes and Protein Structures
3.2. Tissue Expression of the Collectin-Liver 1 and Collectin-Kidney 1
3.3. Biological Activity of Collectin-Liver 1 and Collectin-Kidney 1
4. Associations of COLEC10, COLEC1,1 and MASP1/3 Gene Mutations with 3MC Syndrome
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3MC | Malpuech, Michels, Mingarelli, Carnevale syndrome |
| MASP | Mannose-binding lectin-associated serine proteases |
| MAp44 | Mannose-binding lectin-associated protein, 44kDa |
| SP | Serine protease |
| BMP | Bone morphogenic protein |
| SNP | Single nucleotide polymorphism |
| MBL | Mannose-binding lectin |
| PRM | Pattern recognition molecule |
| TAFI | Thrombin-activatable fibrinolysis inhibitor |
| IL | Interleukin |
| MODS | Multiorgan dysfunction |
| SIRS | Systemic inflammatory response syndrome |
| LCOS | Low cardiac output syndrome |
| CRD | Carbohydrate-recognition domain |
| CL-L1 | Collectin liver-1 (collectin-10) |
| CL-K1 | Collectin kidney-1 (collectin-11) |
| SP-A | Surfactant protein A |
| SP-D | Surfactant protein D |
| LPS | Lipopolysaccharide |
| HIV | Human immunodeficiency virus |
| D-Man | D-MANNOSE |
| D-GlcNAc | N-ACETYL-D-GLUCOSAMINE |
| D-Gal | D-GALACTOSE |
| L-Fuc | L-FUCOSE |
| D-Fuc | D-FUCOSE |
| D-ManNAc | N-ACETYL-D-MANNOSAMINE |
| CRC | Colorectal cancer |
| MSCs | Mesenchymal stem cells |
References
- Mingarelli, R.; Scanderbeg, A.C.; Dallapiccola, B. Two sisters with a syndrome of ocular, skeletal, and abdominal abnormalities (OSA syndrome). J. Med. Genet. 1996, 33, 884–886. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Malpuech, G.; Deméocq, F.; Palcoux, J.B.; Vanlieferinghen, P.; Opitz, J.M. A previously undescribed autosomal recessive multiple congenital anomalies/mental retardation (MCA/MR) syndrome with growth failure, lip/palate cleft(s), and urogenital anomalies. Am. J. Med. Genet. 1983, 16, 475–480. [Google Scholar] [CrossRef]
- Michels, V.V.; Hittner, H.M.; Beaudet, A.L. A clefting syndrome with ocular anterior chamber defect and lid anomalies. J. Pediatr. 1978, 93, 444–446. [Google Scholar] [CrossRef]
- Carnevale, F.; Krajewska, G.; Fischetto, R.; Greco, M.G.; Bonvino, A. Ptosis of eyelids, strabismus, diastasis recti, hip defect, cryptorchidism, and developmental delay in two sibs. Am. J. Med. Genet. 1989, 33, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Sirmaci, A.; Walsh, T.; Akay, H.; Spiliopoulos, M.; Şakalar, Y.B.; Hasanefendioğlu-Bayrak, A.; Duman, D.; Farooq, A.; King, M.C.; Tekin, D. MASP1 Mutations in Patients with Facial, Umbilical, Coccygeal, and Auditory Findings of Carnevale, Malpuech, OSA, and Michels Syndromes. Am. J. Hum. Genet. 2010, 87, 679–686. [Google Scholar] [CrossRef]
- Rooryck, C.; Díaz-Font, A.; Osborn, D.P.; Chabchoub, E.; Hernandez-Hernandez, V.; Shamseldin, H.; Kenny, J.; Waters, A.; Jenkins, D.; Al Kaissi, A.; et al. Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat. Genet. 2011, 43, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Munye, M.M.; Diaz-Font, A.; Ocaka, L.; Henriksen, M.L.; Lees, M.; Brady, A.; Jenkins, D.; Morton, J.; Hansen, S.W.K.; Bacchelli, C.; et al. COLEC10 is mutated in 3MC patients and regulates early craniofacial development. PLoS Genet. 2017, 13, e1006679. [Google Scholar] [CrossRef]
- Degn, S.E.; Jensenius, J.C.; Thiel, S. Disease-Causing Mutations in Genes of the Complement System. Am. J. Hum. Genet. 2011, 88, 689–705. [Google Scholar] [CrossRef]
- Ammitzbøll, C.G.; Steffensen, R.; Nielsen, H.J.; Thiel, S.; Stengaard-Pedersen, K.; Bøgsted, M.; Jensenius, J.C. Polymorphisms in the MASP1 Gene Are Associated with Serum Levels of MASP-1, MASP-3, and MAp44. PLoS ONE 2013, 8, e73317. [Google Scholar] [CrossRef]
- Degn, S.E.; Hansen, A.G.; Steffensen, R.; Jacobsen, C.; Jensenius, J.C.; Thiel, S. MAp44, a Human Protein Associated with Pattern Recognition Molecules of the Complement System and Regulating the Lectin Pathway of Complement Activation. J. Immunol. 2009, 183, 7371–7378. [Google Scholar] [CrossRef]
- Fujita, T. Evolution of the lectin—Complement pathway and its role in innate immunity. Nat. Rev. Immunol. 2002, 2, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Endo, Y.; Fujita, T. Structural and Functional Overview of the Lectin Complement Pathway: Its Molecular Basis and Physiological Implication. Arch. Immunol. Ther. Exp. 2013, 61, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Gregory, L.A.; Thielens, N.M.; Matsushita, M.; Sorensen, R.; Arlaud, G.J.; Fontecilla-Camps, J.C.; Gaboriaud, C. The X-ray Structure of Human Mannan-binding Lectin-associated Protein 19 (MAp19) and Its Interaction Site with Mannan-binding Lectin and L-ficolin. J. Biol. Chem. 2004, 279, 29391–29397. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Beckmann, G. The CUB Domain. A Widespread Module in Developmentally Regulated Proteins. J. Mol. Biol. 1993, 23, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Bally, I.; Rossi, V.; Thielens, N.M.; Gaboriaud, C.; Arlaud, G.J. Functional Role of the Linker between the Complement Control Protein Modules of Complement Protease C1s. J. Immunol. 2005, 175, 4536–4542. [Google Scholar] [CrossRef] [PubMed]
- Gaboriaud, C.; Gupta, R.K.; Martin, L.; Lacroix, M.; Serre, L.; Teillet, F.; Arlaud, G.J.; Rossi, V.; Thielens, N.M. The Serine Protease Domain of MASP-3: Enzymatic Properties and Crystal Structure in Complex with Ecotin. PLoS ONE 2013, 8, e67962. [Google Scholar] [CrossRef] [PubMed]
- Lynch, N.J.; Stover, C.M.; Sandrini, S.M.; Marston, D.; Presanis, J.S.; Schwaeble, W.J. Composition of the Lectin Pathway of Complement in Gallus gallus: Absence of Mannan-Binding Lectin-Associated Serine Protease-1 in Birds. J. Immunol. 2005, 174, 4998–5006. [Google Scholar] [CrossRef]
- Skjoedt, M.-O.; Hummelshoj, T.; Palarasah, Y.; Hein, E.; Munthe-Fog, L.; Koch, C.; Skjodt, K.; Garred, P. Serum concentration and interaction properties of MBL/ficolin associated protein-1. Immunobiology 2011, 216, 625–632. [Google Scholar] [CrossRef]
- Troldborg, A.; Hansen, A.; Hansen, S.W.K.; Jensenius, J.C.; Stengaard-Pedersen, K.; Thiel, S. Lectin complement pathway proteins in healthy individuals. Clin. Exp. Immunol. 2017, 188, 138–147. [Google Scholar] [CrossRef]
- Dobó, J.; Pál, G.; Cervenák, L.; Gál, P. The emerging roles of mannose-binding lectin-associated serine proteases (MASPs) in the lectin pathway of complement and beyond. Immunol. Rev. 2016, 274, 98–111. [Google Scholar] [CrossRef]
- Krarup, A.; Gulla, K.C.; Gál, P.; Hajela, K.; Sim, R. The action of MBL-associated serine protease 1 (MASP1) on factor XIII and fibrinogen. Biochim. Biophys. Acta BBA Proteins Proteom. 2008, 1784, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Jenny, L.; Dobó, J.; Gal, P.; Schroeder, V. MASP-1 of the complement system promotes clotting via prothrombin activation. Mol. Immunol. 2015, 65, 398–405. [Google Scholar] [CrossRef]
- Krarup, A.; Wallis, R.; Presanis, J.S.; Gal, P.; Sim, R. Simultaneous Activation of Complement and Coagulation by MBL-Associated Serine Protease 2. PLoS ONE 2007, 2, e623. [Google Scholar] [CrossRef] [PubMed]
- Gulla, K.C.; Gupta, K.; Krarup, A.; Gal, P.; Schwaeble, W.J.; Sim, R.; O’Connor, C.D.; Hajela, K. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology 2009, 129, 482–495. [Google Scholar] [CrossRef]
- Hess, K.; Ajjan, R.; Phoenix, F.; Dobó, J.; Gál, P.; Schroeder, V. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS ONE 2012, 7, e35690. [Google Scholar] [CrossRef] [PubMed]
- Dobó, J.; Major, B.; Kékesi, K.A.; Szabo, I.; Megyeri, M.; Hajela, K.; Juhász, G.; Závodszky, P.; Gál, P. Cleavage of Kininogen and Subsequent Bradykinin Release by the Complement Component: Mannose-Binding Lectin-Associated Serine Protease (MASP)-1. PLoS ONE 2011, 6, e20036. [Google Scholar] [CrossRef] [PubMed]
- Megyeri, M.; Jani, P.K.; Kajdácsi, E.; Dobó, J.; Schwaner, E.; Major, B.; Rigó, J.; Závodszky, P.; Thiel, S.; Cervenak, L.; et al. Serum MASP-1 in complex with MBL activates endothelial cells. Mol. Immunol. 2014, 59, 39–45. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jani, P.K.; Schwaner, E.; Kajdácsi, E.; Debreczeni, M.; Ungai-Salánki, R.; Dobó, J.; Doleschall, Z.; Rigó, J.; Geiszt, M.; Szabó, B.; et al. Complement MASP-1 enhances adhesion between endothelial cells and neutrophils by up-regulating E-selectin expression. Mol. Immunol. 2016, 75, 38–47. [Google Scholar] [CrossRef]
- Jani, P.K.; Kajdácsi, E.; Megyeri, M.; Dobó, J.; Doleschall, Z.; Futosi, K.; Timár, C.I.; Mocsai, A.; Makó, V.; Gal, P.; et al. MASP-1 Induces a Unique Cytokine Pattern in Endothelial Cells: A Novel Link between Complement System and Neutrophil Granulocytes. PLoS ONE 2014, 9, e87104. [Google Scholar] [CrossRef][Green Version]
- Cortesio, C.L.; Jiang, W. Mannan-binding lectin-associated serine protease 3 cleaves synthetic peptides and insulin-like growth factor-binding protein 5. Arch. Biochem. Biophys. 2006, 449, 164–170. [Google Scholar] [CrossRef]
- Skjoedt, M.-O.; Roversi, P.; Hummelshøj, T.; Palarasah, Y.; Rosbjerg, A.; Johnson, S.; Lea, S.M.; Garred, P. Crystal Structure and Functional Characterization of the Complement Regulator Mannose-binding Lectin (MBL)/Ficolin-associated Protein-1 (MAP-1). J. Biol. Chem. 2012, 287, 32913–32921. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.A.; Skov, L.L.; Kjaer-Sorensen, K.; Hansen, A.G.; Hansen, S.W.K.; Dagnæs-Hansen, F.; Jensenius, J.C.; Oxvig, C.; Thiel, S.; Degn, S.E. Endogenous Natural Complement Inhibitor Regulates Cardiac Development. J. Immunol. 2017, 198, 3118–3126. [Google Scholar] [CrossRef] [PubMed]
- Mendes, H.W.; Boldt, A.B.W.; Stahlke, E.V.R.S.; Jensenius, J.C.; Thiel, S.; Messias-Reason, I.J.T. Adding MASP1 to the lectin pathway—Leprosy association puzzle: Hints from gene polymorphisms and protein levels. PLoS Negl. Trop. Dis. 2020, 14, e0007534. [Google Scholar] [CrossRef]
- Larsen, J.B.; Laursen, M.A.; Hvas, C.L.; Larsen, K.M.; Thiel, S.; Hvas, A.-M. Reduced Mannose-Binding Lectin-Associated Serine Protease (MASP)-1 is Associated with Disturbed Coagulation in Septic Shock. Thromb. Haemost. 2019, 119, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Michalski, M.; Pągowska-Klimek, I.; Thiel, S.; Świerzko, A.S.; Hansen, A.G.; Jensenius, J.C.; Cedzynski, M. Factors involved in initiation and regulation of complement lectin pathway influence postoperative outcome after pediatric cardiac surgery involving cardiopulmonary bypass. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Fanelli, G.; Cordero, A.G.; Gardner, P.J.; Peng, Q.; Fernando, M.; Kloc, M.; Farrar, C.A.; Naeem, A.; Garred, P.; Ali, R.R.; et al. Human stem cell-derived retinal epithelial cells activate complement via collectin 11 in response to stress. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Henriksen, M.L.; Brandt, J.; Andrieu, J.-P.; Nielsen, C.; Jensen, P.H.; Holmskov, U.; Jørgensen, T.; Palarasah, Y.; Thielens, N.M.; Hansen, S.W.K. Heteromeric Complexes of Native Collectin Kidney 1 and Collectin Liver 1 Are Found in the Circulation with MASPs and Activate the Complement System. J. Immunol. 2013, 191, 6117–6127. [Google Scholar] [CrossRef]
- Hansen, S.; Selman, L.; Palaniyar, N.; Ziegler, K.; Brandt, J.; Kliem, A.; Jonasson, M.; Skjoedt, M.-O.; Nielsen, O.; Hartshorn, K.; et al. Collectin 11 (CL-11, CL-K1) Is a MASP-1/3–Associated Plasma Collectin with Microbial-Binding Activity. J. Immunol. 2010, 185, 6096–6104. [Google Scholar] [CrossRef]
- Selman, L.; Henriksen, M.; Brandt, J.; Palarasah, Y.; Waters, A.; Beales, P.; Holmskov, U.; Jørgensen, T.J.D.; Nielsen, C.; Skjodt, K.; et al. An enzyme-linked immunosorbent assay (ELISA) for quantification of human collectin 11 (CL-11, CL-K1). J. Immunol. Methods 2012, 375, 182–188. [Google Scholar] [CrossRef]
- Bayarri-Olmos, R.; Hansen, S.W.K.; Henriksen, M.L.; Storm, L.; Thiel, S.; Garred, P.; Munthe-Fog, L. Genetic Variation of COLEC10 and COLEC11 and Association with Serum Levels of Collectin Liver 1 (CL-L1) and Collectin Kidney 1 (CL-K1). PLoS ONE 2015, 10, e0114883. [Google Scholar] [CrossRef]
- Hansen, S.W.K.; Aagaard, J.B.; Bjerrum, K.B.; Hejbøl, E.K.; Nielsen, O.; Schrøder, H.D.; Skjoedt, K.; Sørensen, A.L.; Graversen, J.H.; Henriksen, M.L. CL-L1 and CL-K1 Exhibit Widespread Tissue Distribution With High and Co-Localized Expression in Secretory Epithelia and Mucosa. Front. Immunol. 2018, 9, 1757. [Google Scholar] [CrossRef] [PubMed]
- Keshi, H.; Sakamoto, T.; Kawai, T.; Ohtani, K.; Katoh, T.; Jang, S.; Motomura, W.; Yoshizaki, T.; Fukuda, M.; Koyama, S.; et al. Identification and characterization of a novel human collectin CL-K1. Microbiol. Immunol. 2006, 50, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Suzuki, Y.; Eda, S.; Kawai, T.; Kase, T.; Yamazaki, H.; Shimada, T.; Keshi, H.; Sakai, Y.; Fukuoh, A.; et al. Molecular Cloning of a Novel Human Collectin from Liver (CL-L1). J. Biol. Chem. 1999, 274, 13681–13689. [Google Scholar] [CrossRef] [PubMed]
- Axelgaard, E.; Jensen, L.; Dyrlund, T.F.; Nielsen, H.J.; Enghild, J.J.; Thie, L.S.; Jensenius, J.C. Investigations on collectin-liver 1 (CL-L1 or CL-10). J. Biol. Chem. 2013, 288, 23407–23420. [Google Scholar]
- Świerzko, A.S.; Michalski, M.; Sokołowska, A.; Nowicki, M.; Eppa, Ł.; Szala-Poździej, A.; Mitrus, I.; Szmigielska-Kapłon, A.; Sobczyk-Kruszelnicka, M.; Michalak, K.; et al. The Role of Complement Activating Collectins and Associated Serine Proteases in Patients With Hematological Malignancies, Receiving High-Dose Chemotherapy, and Autologous Hematopoietic Stem Cell Transplantations (Auto-HSCT). Front. Immunol. 2018, 9, 2153. [Google Scholar] [CrossRef]
- Troegeler, A.; Lugo, G.; Hansen, S.W.K.; Rasolofo, V.; Henriksen, M.L.; Mori, K.; Ohtani, K.; Duval, C.; Mercier, I.; Bénard, A.; et al. Collectin CL-LK Is a Novel Soluble Pattern Recognition Receptor for Mycobacterium tuberculosis. PLoS ONE 2015, 10, e0132692. [Google Scholar] [CrossRef]
- Girija, U.V.; Furze, C.M.; Gingras, A.R.; Yoshizaki, T.; Ohtani, K.; Marshall, J.E.; Wallis, A.K.; Schwaeble, W.J.; El-Mezgueldi, M.; Mitchell, D.A.; et al. Molecular basis of sugar recognition by collectin-K1 and the effects of mutations associated with 3MC syndrome. BMC Biol. 2015, 13, 1–14. [Google Scholar] [CrossRef]
- Hansen, S.W.K. Lung Surfactant Protein D (SP-D) and the Molecular Diverted Descendants: Conglutinin, CL-43 and CL-46. Immunobiology 2002, 205, 498–517. [Google Scholar] [CrossRef]
- Henriksen, M.L.; Brandt, J.; Iyer, S.S.; Thielens, N.M.; Hansen, S. Characterization of the interaction between collectin 11 (CL-11, CL-K1) and nucleic acids. Mol. Immunol. 2013, 56, 757–767. [Google Scholar] [CrossRef]
- Selman, L.; Hansen, S. Structure and function of collectin liver 1 (CL-L1) and collectin 11 (CL-11, CL-K1). Immunobiology 2012, 217, 851–863. [Google Scholar] [CrossRef]
- Smedbråten, J.; Sagedal, S.; Åsberg, A.; Hartmann, A.; Rollag, H.; Mjøen, G.; Fagerland, M.W.; Hansen, S.; Mollnes, T.E.; Thiel, S. Collectin Liver 1 and Collectin Kidney 1 of the Lectin Complement Pathway Are Associated With Mortality After Kidney Transplantation. Am. J. Transplant. 2016, 17, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Troldborg, A.; Thiel, S.; Trendelenburg, M.; Friebus-Kardash, J.; Nehring, J.; Steffensen, R.; Hansen, S.; Laska, M.J.; Deleuran, B.; Jensenius, J.C.; et al. The Lectin Pathway of Complement Activation in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2018, 45, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Storm, L.; The Danish Study Group on Early Detection of Colorectal Cancer; Christensen, I.J.; Jensenius, J.C.; Nielsen, H.J.; Thiel, S. Evaluation of complement proteins as screening markers for colorectal cancer. Cancer Immunol. Immunother. 2014, 64, 41–50. [Google Scholar] [CrossRef]
- Farrar, C.A.; Tran, D.; Li, K.; Wu, W.; Peng, Q.; Schwaeble, W.; Zhou, W.; Sacks, S.H. Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J. Clin. Investig. 2016, 126, 1911–1925. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Liu, C.; Farrar, C.A.; Ma, L.; Dong, X.; Sacks, S.H.; Li, K.; Zhou, W. Collectin-11 Promotes the Development of Renal Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2017, 29, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.R.K.G.; Pandit, H.; Warty, N.; Madan, T. Endometriotic mesenchymal stem cells exhibit a distinct immune phenotype. Int. Immunol. 2014, 27, 195–204. [Google Scholar] [CrossRef]
- Atik, T.; Koparir, A.; Bademci, G.; Foster, J.; Altunoglu, U.; Mutlu, G.Y.; Bowdin, S.; Elcioglu, N.; Tayfun, G.A.; Atik, S.S.; et al. Novel MASP1 mutations are associated with an expanded phenotype in 3MC1 syndrome. Orphanet J. Rare Dis. 2015, 10, 128. [Google Scholar] [CrossRef]
- Urquhart, J.E.; Roberts, R.; De Silva, D.; Shalev, S.; Chervinsky, E.; Nampoothiri, S.; Sznajer, Y.; Revencu, N.; Gunasekera, R.; Suri, M.; et al. Exploring the genetic basis of 3MC syndrome: Findings in 12 further families. Am. J. Med Genet. Part A 2016, 170, 1216–1224. [Google Scholar] [CrossRef]
- Pihl, R.; Jensen, L.; Hansen, A.G.; Thøgersen, I.B.; Andres, S.; Dagnæs-Hansen, F.; Oexle, K.; Enghild, J.J.; Thiel, S. Analysis of Factor D Isoforms in Malpuech-Michels-Mingarelli-Carnevale Patients Highlights the Role of MASP-3 as a Maturase in the Alternative Pathway of Complement. J. Immunol. 2017, 199, 2158–2170. [Google Scholar] [CrossRef]
- Graul-Neumann, L.M.; Mensah, M.A.; Klopocki, E.; Uebe, S.; Ekici, A.B.; Thiel, C.T.; Reis, A.; Zweier, C. Biallelic intragenic deletion in MASP1 in an adult female with 3MC syndrome. Eur. J. Med. Genet. 2018, 61, 363–368. [Google Scholar] [CrossRef]
- Basdemirci, M.; Sen, A.; Ceylaner, S. Novel mutation in MASP1 gene in a new family with 3MC syndrome. Clin. Dysmorphol. 2019, 28, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Çakmaklı, S.; Kandur, Y. 3MC syndrome: A case report. Arch. Clin. Exp. Med. 2019, 4, 107–109. [Google Scholar] [CrossRef]
- Gardner, O.K.; Haynes, K.; Schweitzer, D.; Magee, W.P.; Urata, M.M.; Sanchez-Lara, P.A.; Johns, A. Familial Recurrence of 3MC Syndrome in Consanguineous Families: A Clinical and Molecular Diagnostic Approach with review of the Literature. Cleft Palate Craniofacial J. 2017, 54, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Minoux, M.; Rijli, F.M. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development 2010, 137, 2605–2621. [Google Scholar] [CrossRef]
- Carmona-Fontaine, C.; Theveneau, E.; Tzekou, A.; Tada, M.; Woods, M.; Page, K.M.; Parsons, M.; Lambris, J.D.; Mayor, R. Complement Fragment C3a Controls Mutual Cell Attraction during Collective Cell Migration. Dev. Cell 2011, 21, 1026–1037. [Google Scholar] [CrossRef]
- Cai, G.-Z.; Griffin, G.L.; Senior, R.M.; Longmore, W.J.; Moxley, M.A. Recombinant SP-D carbohydrate recognition domain is a chemoattractant for human neutrophils. Am. J. Physiol. Content 1999, 276, L131–L136. [Google Scholar] [CrossRef]
- Crouch, E.C.; Persson, A.; Griffin, G.L.; Chang, D.; Senior, R.M. Interactions of pulmonary surfactant protein D (SP-D) with human blood leukocytes. Am. J. Respir. Cell Mol. Biol. 1995, 12, 410–415. [Google Scholar] [CrossRef]
- Schagat, T.L.; Wofford, J.A.; Greene, K.E.; Wright, J.R. Surfactant protein A differentially regulates peripheral and inflammatory neutrophil chemotaxis. Am. J. Physiol. Cell. Mol. Physiol. 2003, 284, L140–L147. [Google Scholar] [CrossRef]
- Gorelik, A.; Sapir, T.; Haffner-Krausz, R.; Olender, T.; Woodruff, T.M.; Reiner, O. Developmental activities of the complement pathway in migrating neurons. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Ma, Y.J.; Skjoedt, M.O.; Garred, P. Collectin-11/MASP complex formation triggers activation of the lectin complement pathway—The fifth lectin pathway initiation complex. J. Innate Immun. 2013, 5, 242–250. [Google Scholar] [CrossRef]
- Takahashi, M.; Iwaki, D.; Kanno, K.; Ishida, Y.; Xiong, J.; Matsushita, M.; Endo, Y.; Miura, S.; Ishii, N.; Sugamura, K.; et al. Mannose-Binding Lectin (MBL)-Associated Serine Protease (MASP)-1 Contributes to Activation of the Lectin Complement Pathway. J. Immunol. 2008, 180, 6132–6138. [Google Scholar] [CrossRef] [PubMed]
- Banda, N.K.; Takahashi, M.; Levitt, B.; Glogowska, M.; Nicholas, J.; Takahashi, K.; Stahl, G.L.; Fujita, T.; Arend, W.P.; Holers, V.M. Essential Role of Complement Mannose-Binding Lectin-Associated Serine proteases-1/3 in the Murine Collagen Antibody-Induced Model of Inflammatory Arthritis. J. Immunol. 2010, 185, 5598–5606. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Mori, K.; Ohtani, K.; Matsuda, Y.; Roy, N.; Kim, Y.; Suzuki, Y.; Wakamiya, N. Collectin Kidney 1 Plays an Important Role in Innate Immunity against Streptococcus pneumoniae Infection. J. Innate Immun. 2017, 9, 217–228. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Family (Number of Carriers) | Origin | Nucleotide Change | Amino acid Change/Protein Change | References |
|---|---|---|---|---|
| F1 (2) | Turkey | c.2059G>A exon 12 | p.Gly687Arg MASP-3 SP | Sirmaci et al., 2010 [5] |
| F2 (1) | Turkey | c.870G>A exon 6 | p.Trp290* MASP-1/3 CUB1-EGF-CUB2-CCP1 | |
| F3 (1) | Greece | c.1489 C>T exon 12 | p.His497Tyr MASP-3 SP | Rooryck et al., 2011 [6] |
| F4 (2) | Italy | c.1888T>C exon 12 | p.Cys630Arg MASP-3 SP | |
| F5 (1) | Brazil | c.1997G>A exon 12 | p.Gly666Glu MASP-3 SP | |
| F6 (1) | Brazil | c.1997G>A exon 12 | p.Gly666Glu MASP-3 SP | |
| F7 (1) | Turkey | c.1012-2A>G intron 7 | Splice site mutation | Atik et al., 2015 [57] |
| F8 (1) | Turkey | c.891 +1G>T intron 6 | Splice site mutation | |
| F9 (1) | Turkey | c.891 +1G>T intron 6 | Splice site mutation | |
| F10 (1) | Pakistan | c.1451G>A exon 12 | p. Gly484Glu MASP-3 SP | |
| F11 (1) | Turkey | c.1657G>A exon 12 | p. Asp553Asn MASP-3 SP | |
| F12 (1) | Syria | c.1987G>T exon 12 | p. Asp663Tyr MASP-3 SP | |
| F13 (1) | Israel | c.1987G>T exon 12 | p.Asp663Tyr MASP-3 SP | Urquhart et al., 2016 [58] |
| F14 (1) | Sri Lanka | c.9G>A exon 2 | p.Trp3* MASP-1/3 CUB1-EGF-CUB2-CCP1 | |
| F14 (2) | Sri Lanka | c.760A>T exon 6 | p.Leu 254* MASP-1/3 CUB1-EGF-CUB2-CCP1 | |
| F15 (1) | India | c.547G>T exon 4 | p.Val 183Leu MASP-1/3 CUB1-EGF-CUB2-CCP1 | |
| F16 (1) | Germany | c.1993G>A exon 12 | p.G665S MASP-3 SP | Pihl et al., 2017 [59] |
| F17 (1) | Pakistan | c.9G>A exon 2 | p. Trp3* MASP-1/3 CUB1-EGF-CUB2-CCP1 | Munye et al., 2017 [7] |
| F18 (1) | Turkey | c.1895_*1602+411del exon 12 | p.Arg637Cysfs*1 MASP-3 SP | Graul-Neumann et al., 2018 [60] |
| F19 (3) | Turkey | c.2111T>G exon 12 | p. Val704Gly MASP-1/MASP-3 CCP2 | Basdemirci et al., 2019 [61] |
| F20 (1) | Syria | c.1987G>T exon 12 | p.Asp663Tyr MASP-3 SP | Çakmaklı et al., 2019 [62] |
| Family (Number of Carriers) | Origin | Nucleotide Change | Amino Acid Change/Protein Change | References |
|---|---|---|---|---|
| F1 (2) | Tunisia | c.496T>C exon 8 | p.Ser169Pro CRD | Rooryck et al., 2011 [6] |
| F2 (2) | Bangaldesh | c.45delC exon 2 | p.Phe16SerfsX 85 N-terminal Collagen-like region | |
| F3 (2) | Afganistan | c.610G>A exon 8 | p.Gly204Ser CRD | |
| F4 (1) | Saudi Arabia | c.648_650delCTC exon 8 | p.Ser217del CRD | |
| F5 (1) | Pakistan | c.610G>A exon 8 | p.Gly204Ser CRD | |
| F6 (1) | Italy | c.300delT exon 6 | p.Gly101ValfsX 113 Neck domain | |
| F7 (1) | Italy | ex 1-3 deletion exons 1,2,3 | Predicted: complete loss of N-terminus and partial loss of the collagen-like domains N-terminal collagen-like region | |
| F8 (2) | Israel | c.627_628delGC Exon 8 | p. Ala213Leufs 5 CRD | Urquhart et al., 2016 [58] |
| F9 (2) | Pakistan | ex 8 deletion exon 8 | Predicted: complete loss of C terminus and at least partial loss of the carbohydrate-recognition domain (CRD) | Gardner et al., 2017 [63] |
| F10 (1) | Pakistan | c.309delT exon 4 | p.Gly104Valfs29 Collagen-like domain | Munye et al., 2017 [7] |
| F11 (1) | Somalia | c.G496A exon 6 | p.Ala166Thr Neck domain | |
| F12 (1) | United Arab Emirates | 89_98del ATGACGCCTG exon 2 | p.Asp30Alafs68 N-terminal domain |
| Family (Number of Ccarriers) | Origin | Nucleotide Change | Amino Acid Change/Protein Change | References |
|---|---|---|---|---|
| F1 (2) | Pakistan | c.25C>T exon 1 | p. Arg9Ter Signal peptide | Munye et al., 2017 [7] |
| c.226delA exon 3 | p.Gly77Glufs*66 Collagen-like region | |||
| F2 (1) | Pakistan | c.25C>T exon 1 | p. Arg9Ter Signal peptide | |
| c.528C>G exon 6 | p.Cys176Trp Neck domain |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajek, G.; Świerzko, A.S.; Cedzyński, M. Association of Polymorphisms of MASP1/3, COLEC10, and COLEC11 Genes with 3MC Syndrome. Int. J. Mol. Sci. 2020, 21, 5483. https://doi.org/10.3390/ijms21155483
Gajek G, Świerzko AS, Cedzyński M. Association of Polymorphisms of MASP1/3, COLEC10, and COLEC11 Genes with 3MC Syndrome. International Journal of Molecular Sciences. 2020; 21(15):5483. https://doi.org/10.3390/ijms21155483
Chicago/Turabian StyleGajek, Gabriela, Anna S. Świerzko, and Maciej Cedzyński. 2020. "Association of Polymorphisms of MASP1/3, COLEC10, and COLEC11 Genes with 3MC Syndrome" International Journal of Molecular Sciences 21, no. 15: 5483. https://doi.org/10.3390/ijms21155483
APA StyleGajek, G., Świerzko, A. S., & Cedzyński, M. (2020). Association of Polymorphisms of MASP1/3, COLEC10, and COLEC11 Genes with 3MC Syndrome. International Journal of Molecular Sciences, 21(15), 5483. https://doi.org/10.3390/ijms21155483