Cold Press Pomegranate Seed Oil Attenuates Dietary-Obesity Induced Hepatic Steatosis and Fibrosis through Antioxidant and Mitochondrial Pathways in Obese Mice

 ,
,  ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
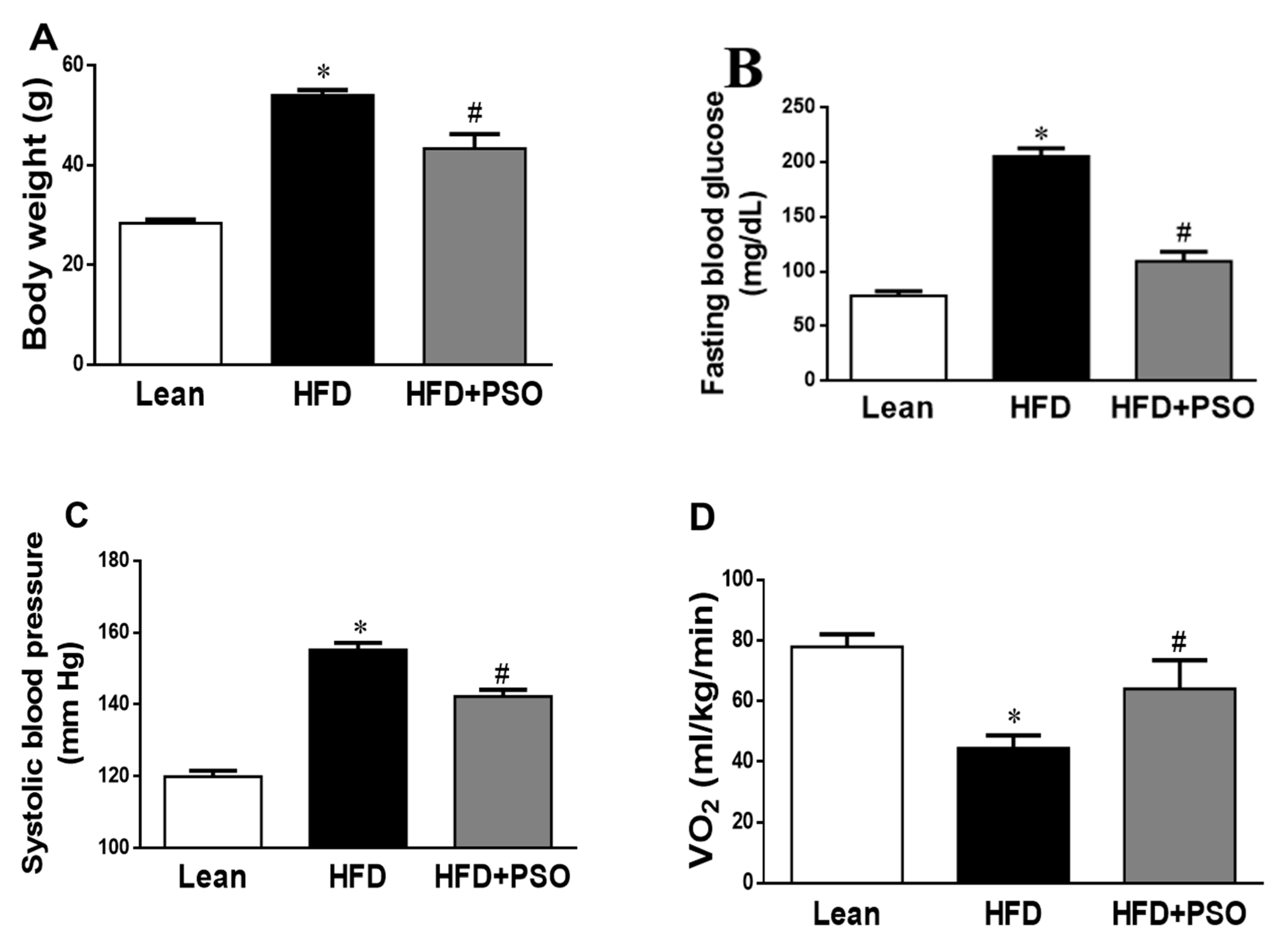
2.1. Effects of PSO on Physiological Parameters
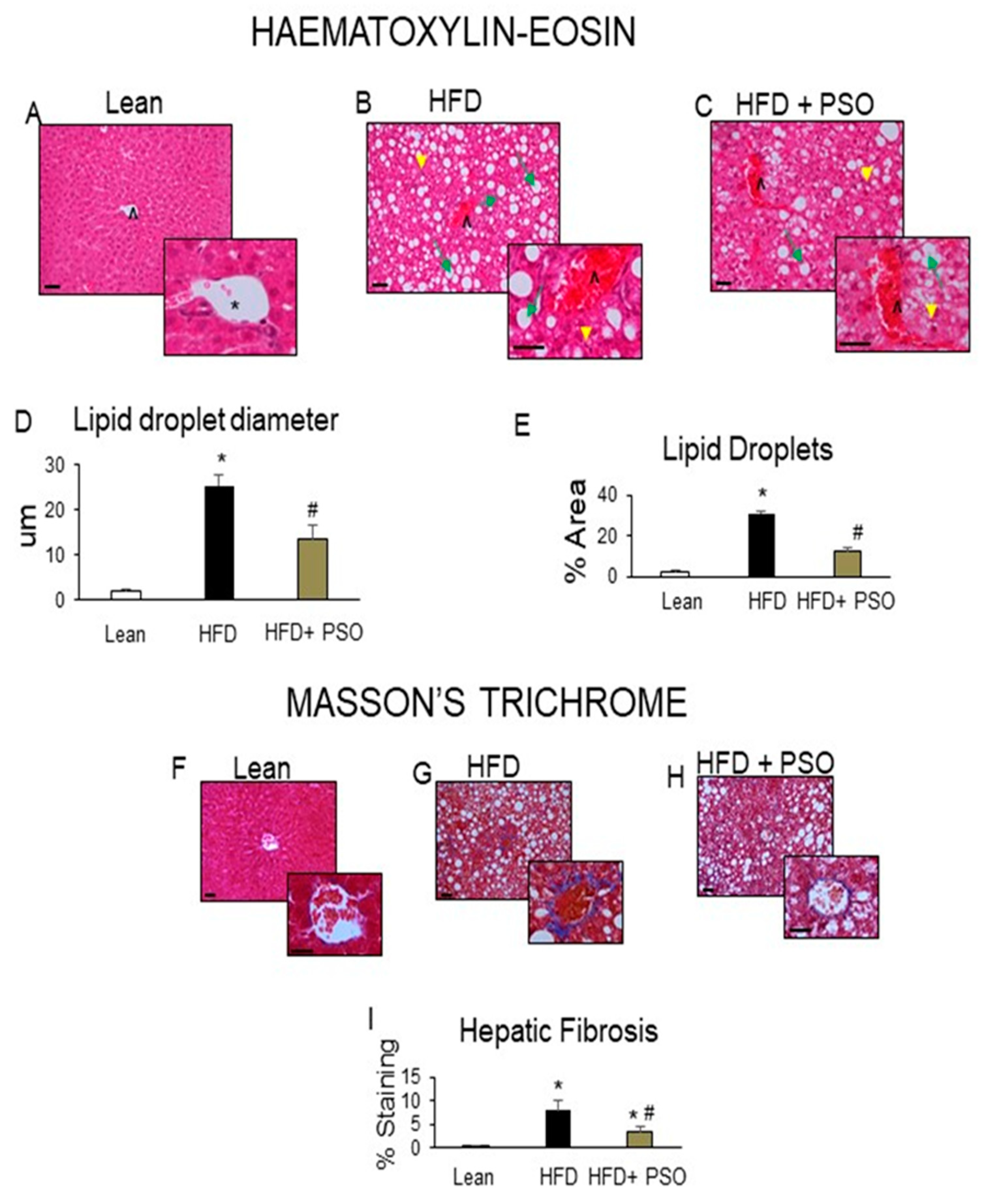
2.2. PSO Decreases Hepatic Steatosis and Fibrosis
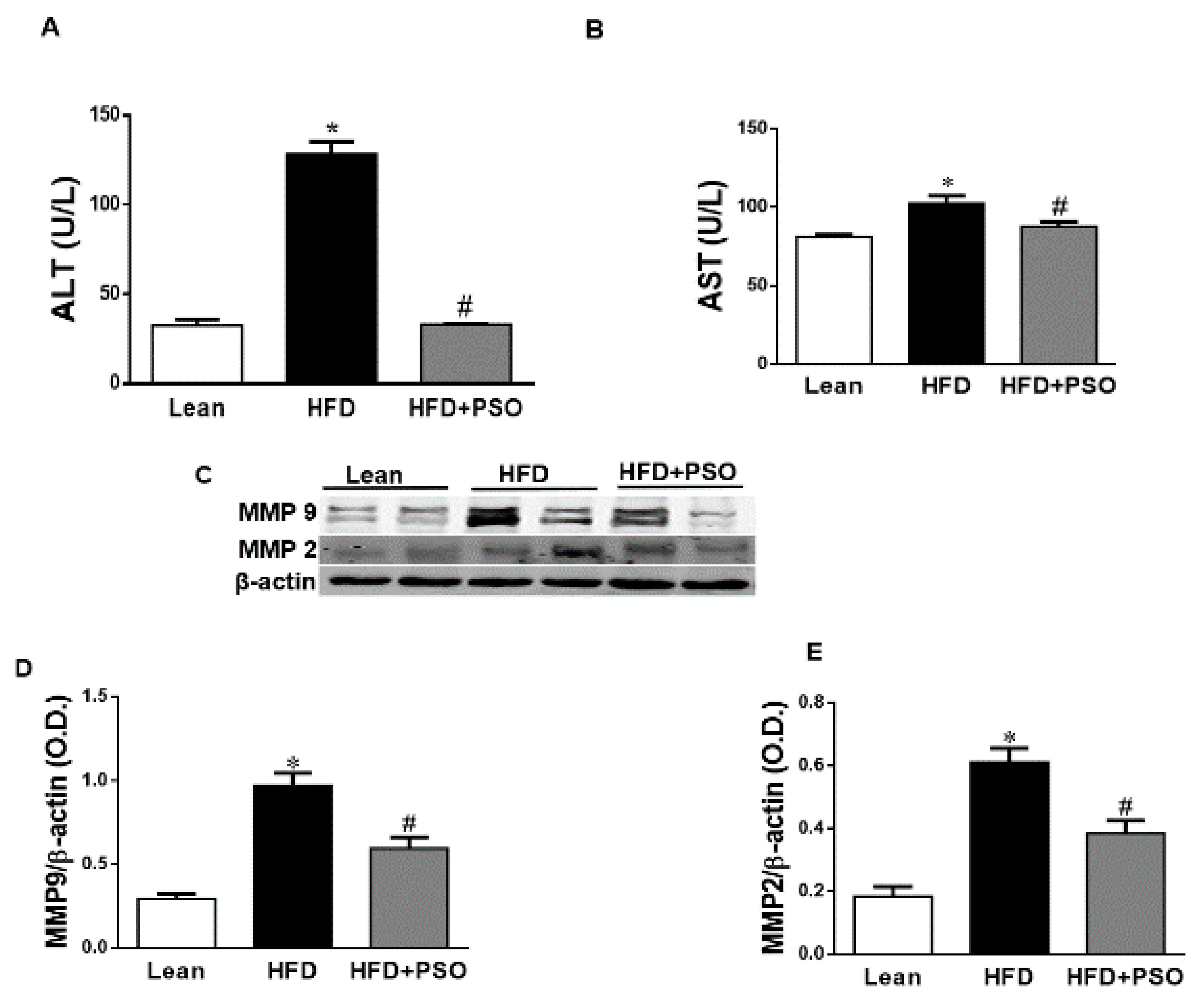
2.3. PSO Decreases Fibrotic Markers and Lowers Serum ALT and AST Levels in HFD-Fed Mice
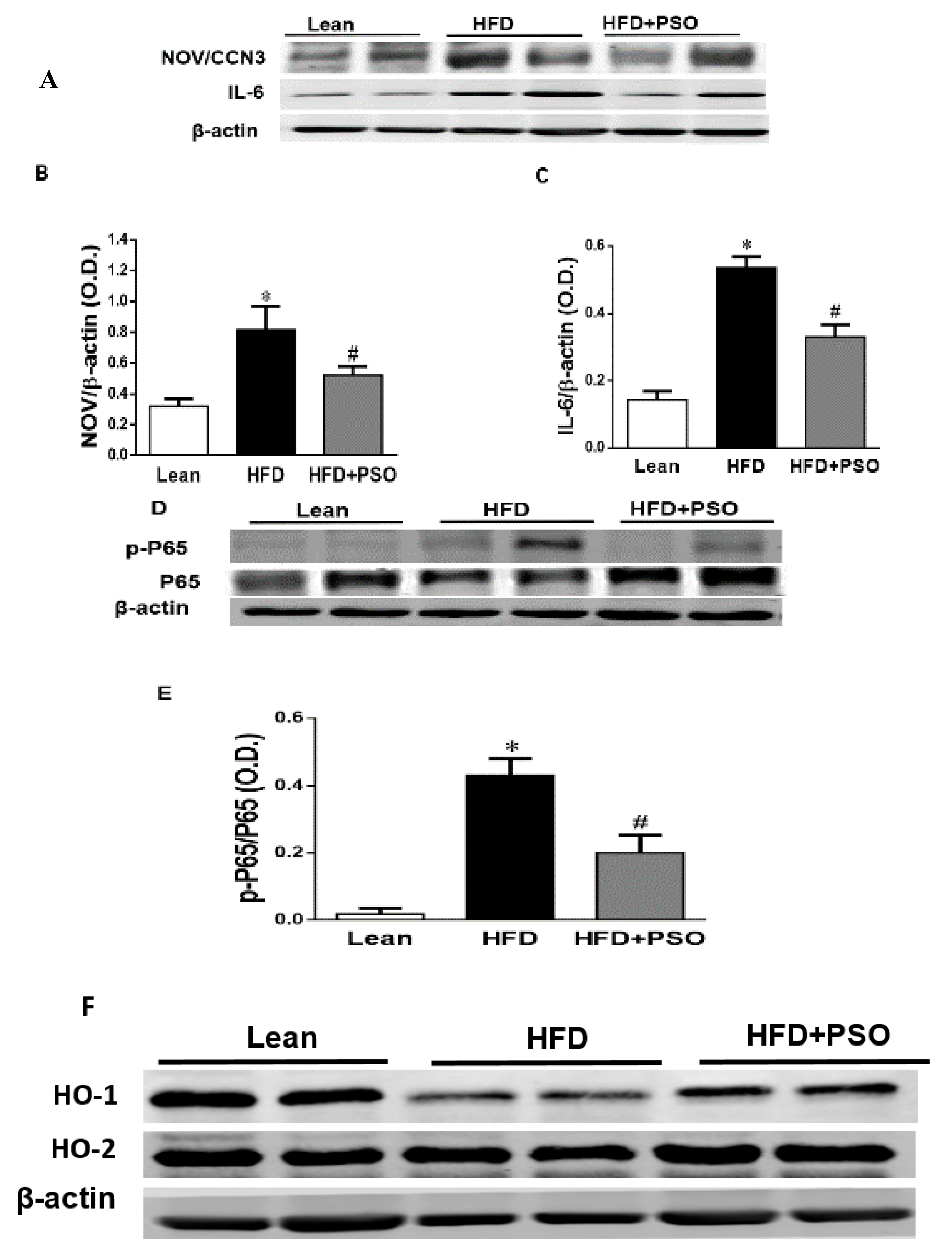
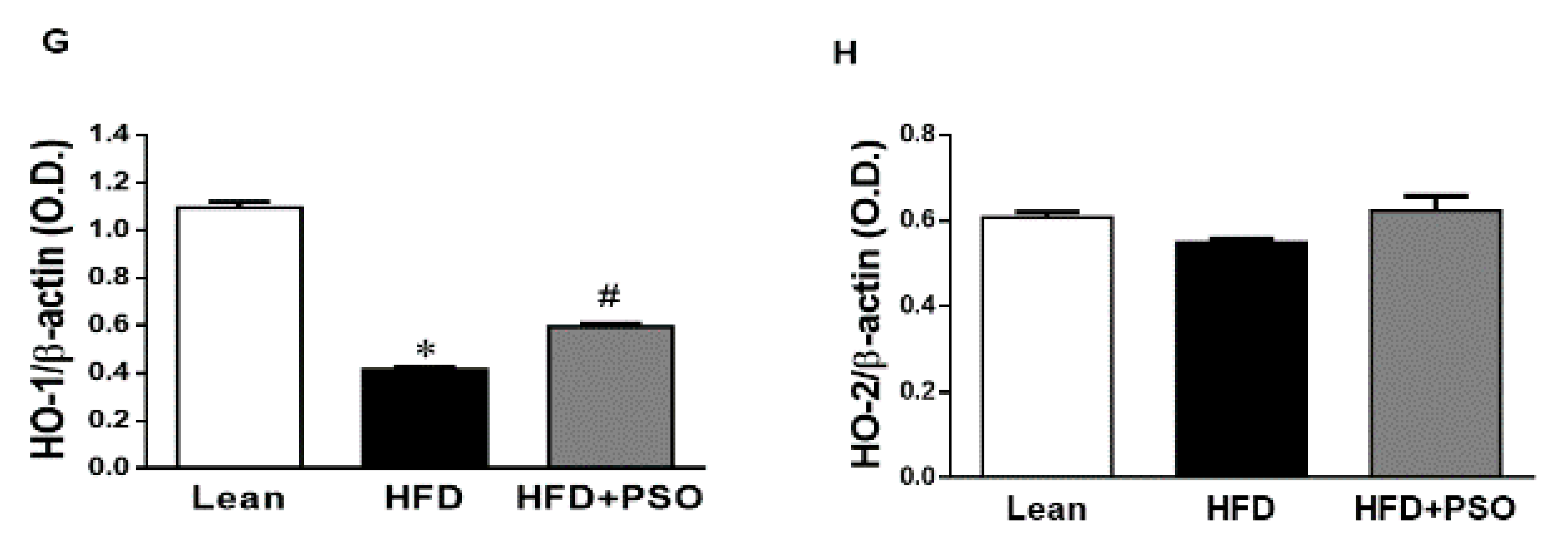
2.4. Effect PSO on Hepatic Inflammatory Proteins and Heme Oxygenase (HO)
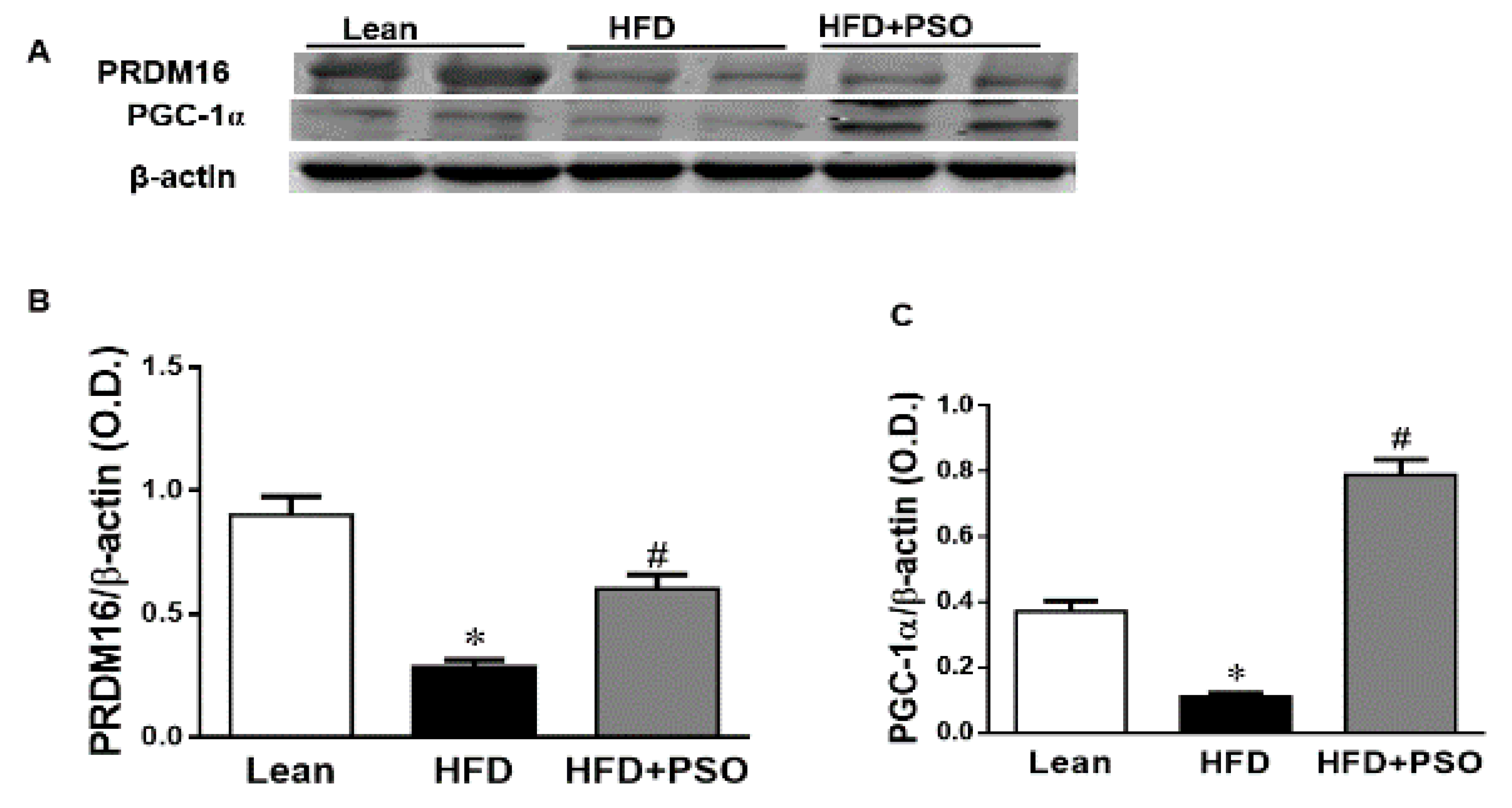
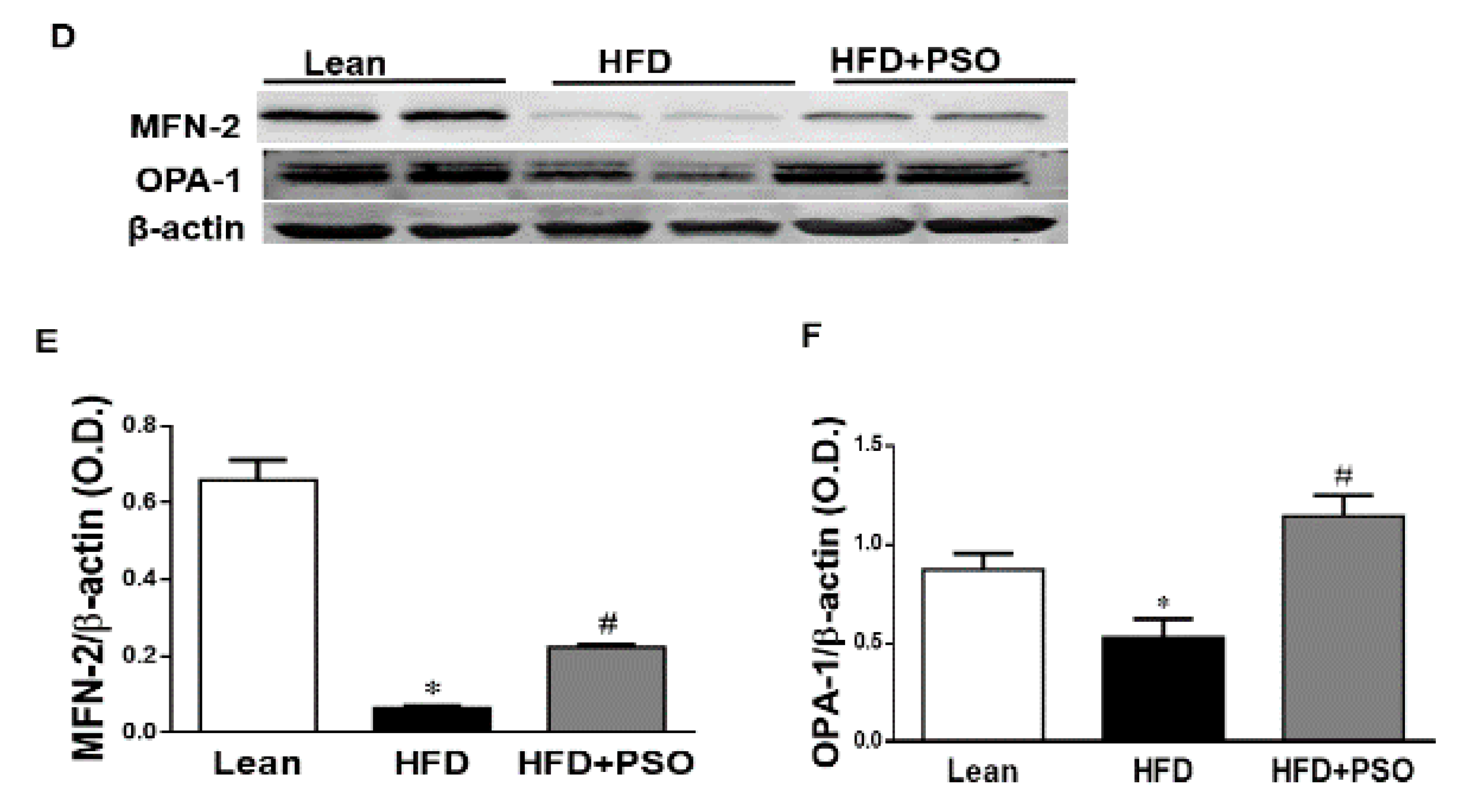
2.5. Effect PSO on Mitochondrial Function and Lipid Metabolism
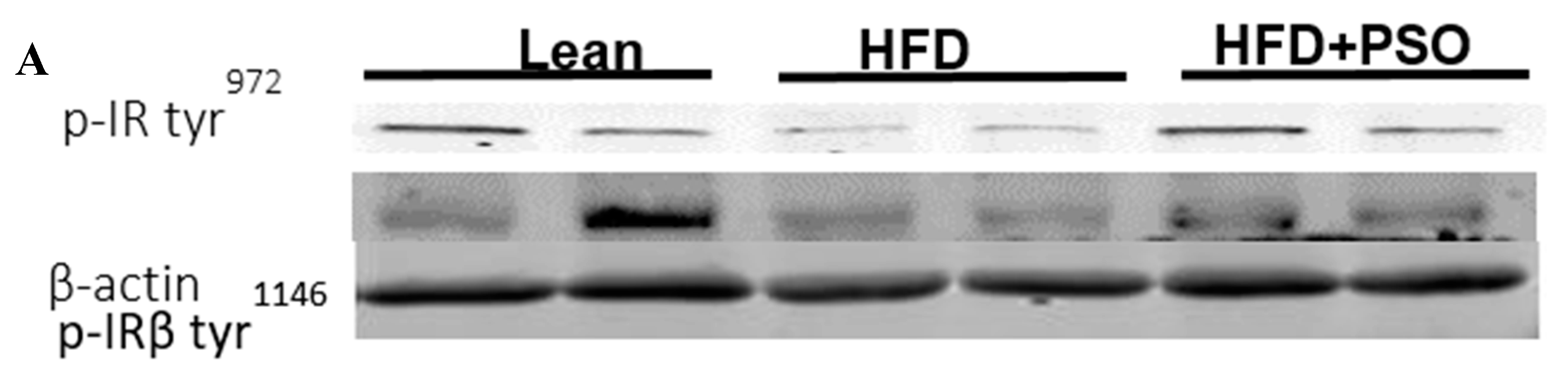
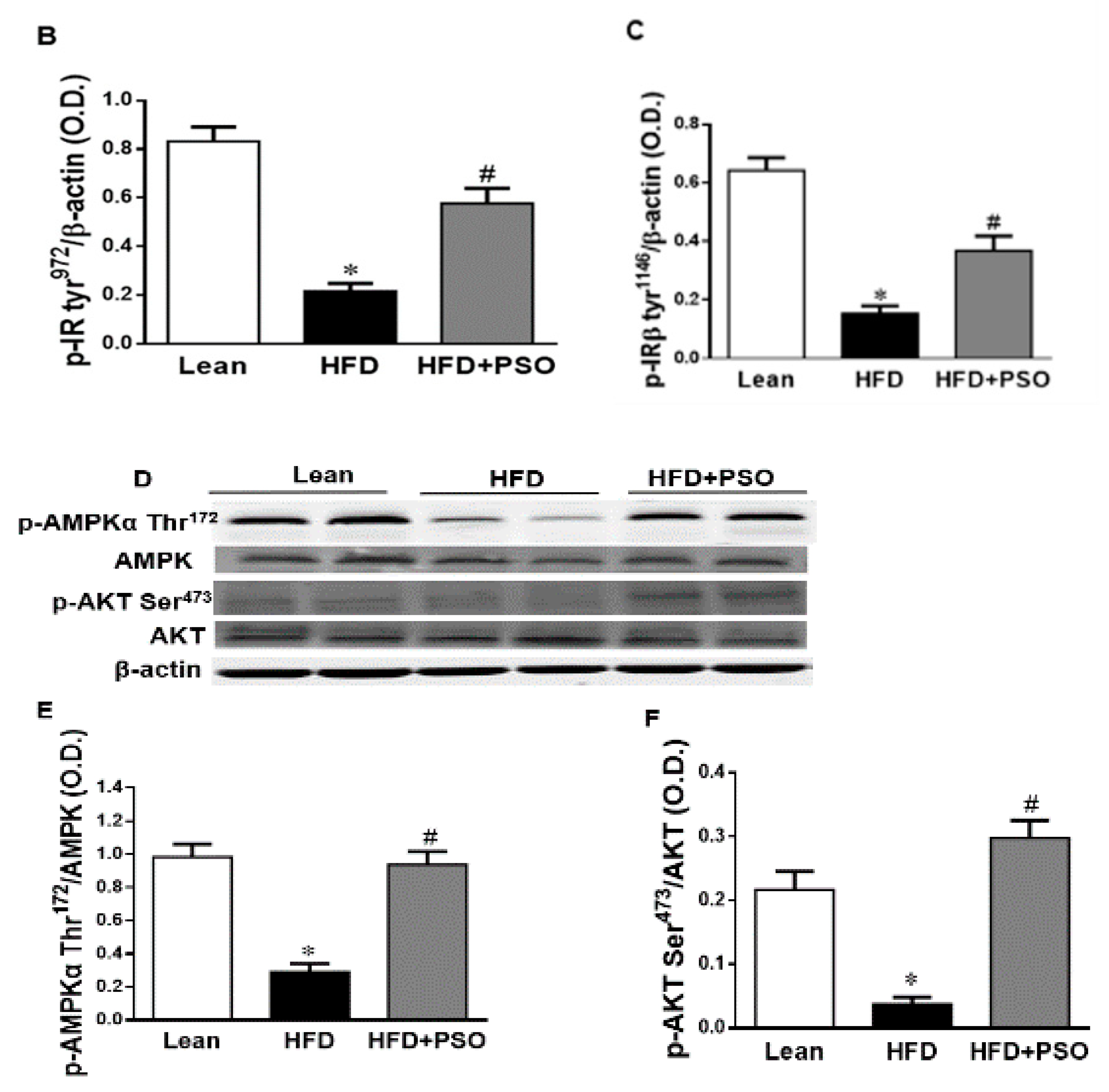
2.6. Effect PSO on Hepatic Insulin Resistance
3. Discussions
4. Materials and Methods
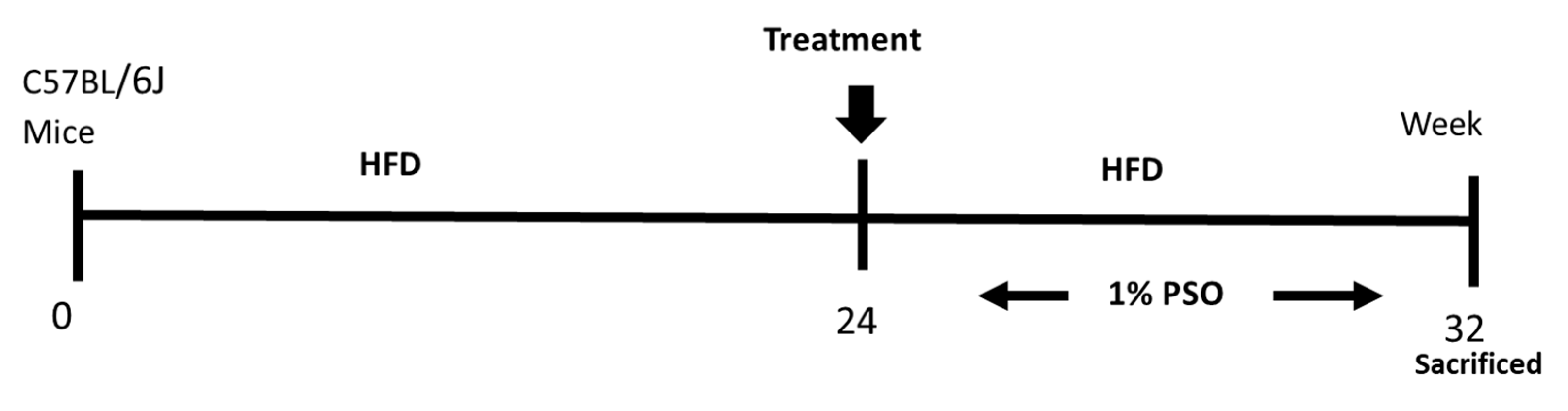
4.1. Animal Protocols and Measurement of Body Weight
4.2. Determination of Blood Glucose, Blood Pressure, and VO2
4.3. Western Blot Analysis and Histological Evaluation
4.4. Measurement of Serum Aspartate Aminotransferase (AST) and (ALT)
4.5. Statistical Analysis
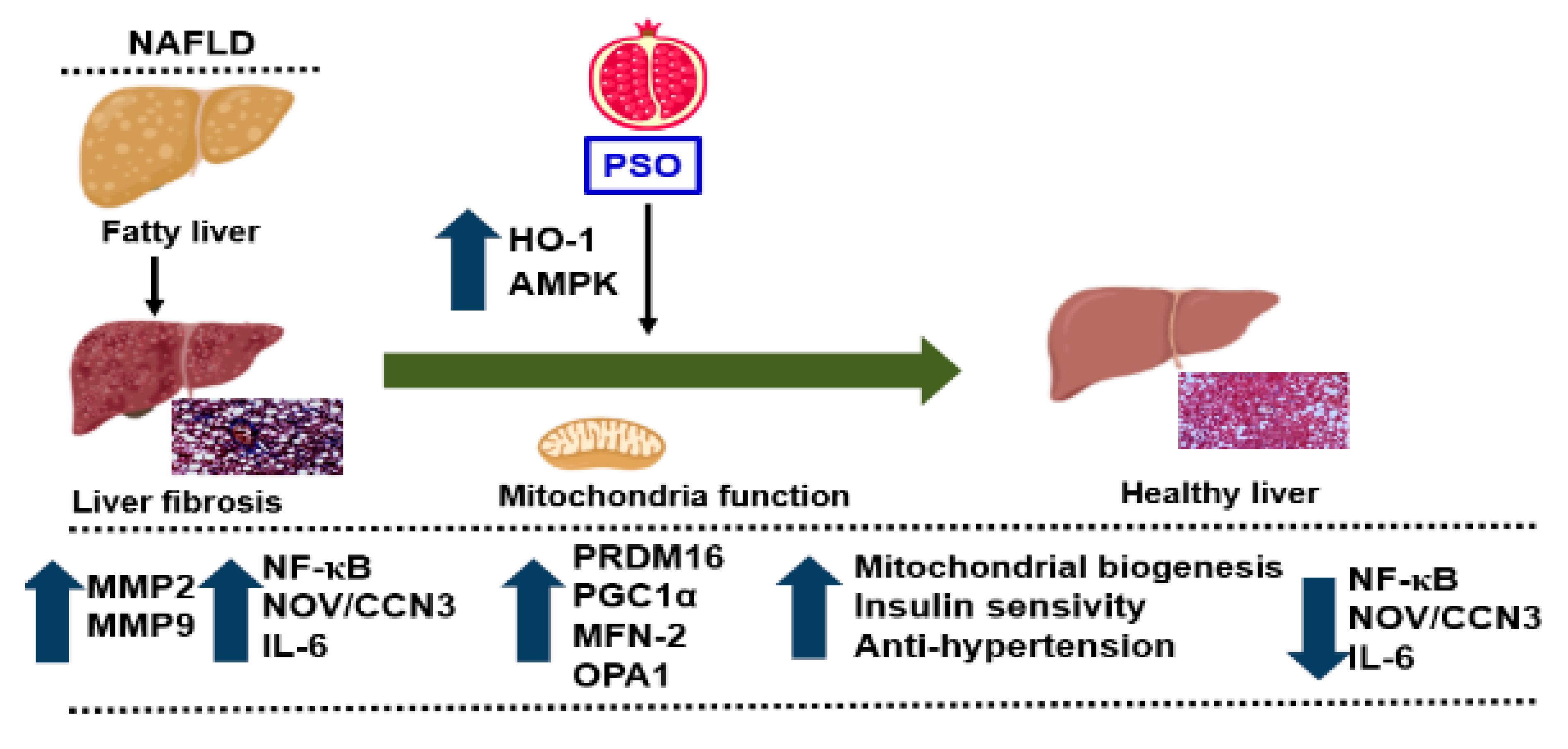
5. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PSO | pomegranate seed oil |
| HO-1 | heme oxygenase-1 |
| FA | fatty acid |
| FFA | free fatty acid |
| FAS | fatty acid synthase |
| FGF21 | fibroblast growth factor 21 |
| HF | high fat |
| HFD | high fat diet |
| IR | insulin resistance |
| DM | diabetes mellitus |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-α |
| SIRT3 | sirtuin3 |
| DRP1 | dynamin-related protein 1 |
| Fis1 | mitochondrial fission 1protein |
| OPA1 | optic atrophy 1 protein |
| Mfn 1 | mitochondrial fusion protein mitofusin 1 |
| Mfn 2 | mitochondrial fusion protein mitofusin 2 |
| UCP1 | uncoupling protein 1 |
| MnSOD | mitochondrial superoxide dismutase |
| PRDM16 | PR domain containing 16 |
| VO2 | oxygen consumption |
| Drp1 | dynamin-related protein 1 |
| ROS | reactive oxygen species |
| ALT | alanine aminotransferase |
| AST | asparagine aminotransferase |
| MMP | matrix metalloproteinase |
References
- Andolfi, C.; Fisichella, P.M. Epidemiology of Obesity and Associated Comorbidities. J. Laparoendosc. Adv. Surg. Tech. A 2018, 28, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Artham, S.M.; Lavie, C.J.; Milani, R.V.; Ventura, H.O. Obesity and hypertension, heart failure, and coronary heart disease-risk factor, paradox, and recommendations for weight loss. Ochsner J. 2009, 9, 124–132. [Google Scholar]
- Singh, S.P.; Greenberg, M.; Glick, Y.; Bellner, L.; Favero, G.; Rezzani, R.; Rodella, L.F.; Agostinucci, K.; Shapiro, J.I.; Abraham, N.G. Adipocyte Specific HO-1 Gene Therapy is Effective in Antioxidant Treatment of Insulin Resistance and Vascular Function in an Obese Mice Model. Antioxidants (Basel) 2020, 9, 40. [Google Scholar] [CrossRef]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef]
- Qureshi, K.; Abrams, G.A. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 3540–3553. [Google Scholar] [CrossRef]
- Drummond, G.S.; Baum, J.; Greenberg, M.; Lewis, D.; Abraham, N.G. HO-1 Overexpression and Underexpression: Clinical Implications. Arch. Biochem. Biophys. 2019, 673, 108073. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Sanchez, A.; Madrigal-Santillan, E.; Bautista, M.; Esquivel-Soto, J.; Morales-Gonzalez, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sanchez-Rivera, G.; Valadez-Vega, C.; Morales-Gonzalez, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Holvoet, P. Relations between metabolic syndrome, oxidative stress and inflammation and cardiovascular disease. Verh. K. Acad. Geneeskd. Belg. 2008, 70, 193–219. [Google Scholar]
- Singh, S.; McClung, J.; Thompson, E.; Glick, Y.; Greenberg, M.; Acosta-Baez, G.; Edris, B.; Shapiro, J.; Abraham, N.G. Cardioprotective heme oxygenase-1-PGC-1α signaling in epicardial fat attenuates cardiovascular risk in humans as in obese mice. Obesity (Silver Spring) 2019, 27, 1634–1643. [Google Scholar] [CrossRef]
- Abraham, N.G.; Junge, J.M.; Drummond, G.S. Translational Significance of Heme Oxygenase in Obesity and Metabolic Syndrome. Trends Pharmacol. Sci. 2016, 37, 17–36. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; London, I.M. Hemin enhances the differentiation of mouse 3T3 cells to adipocytes. Cell 1981, 26 Pt 1, 117–122. [Google Scholar] [CrossRef]
- Abraham, N.G. Therapeutic applications of human heme oxygenase gene transfer and gene therapy. Curr. Pharm. Des. 2003, 9, 2513–2524. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Tsenovoy, P.L.; McClung, J.; Drummond, G.S. Heme oxygenase: A target gene for anti-diabetic and obesity. Curr. Pharm. Des. 2008, 14, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Martinerie, C.; Garcia, M.; Do, T.T.; Antoine, B.; Moldes, M.; Dorothee, G.; Kazazian, C.; Auclair, M.; Buyse, M.; Ledent, T.; et al. NOV/CCN3: A New Adipocytokine Involved in Obesity-Associated Insulin Resistance. Diabetes 2016, 65, 2502–2515. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Singh, S.P.; McClung, J.; Joseph, G.; Vanella, L.; Barbagallo, I.; Jiang, H.; Falck, J.R.; Arad, M.; Shapiro, J.I.; et al. EET Intervention on Wnt1, NOV and HO-1 Signaling Prevents Obesity-Induced Cardiomyopathy in Obese Mice. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H368–H380. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Jamaluddin, M.; Wang, S.; Boldogh, I.; Tian, B.; Brasier, A.R. TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal. 2007, 19, 1419–1433. [Google Scholar] [CrossRef]
- Maury, E.; Brichard, S.M. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol. Cell. Endocrinol. 2010, 314, 1–16. [Google Scholar] [CrossRef]
- Peterson, S.J.; Shapiro, J.I.; Thompson, E.; Singh, S.; Liu, L.; Weingarten, J.A.; O’Hanlon, K.; Bialczak, A.; Bhesania, S.R.; Abraham, N.G. Oxidized HDL, Adipokines, and Endothelial Dysfunction: A Potential Biomarker Profile for Cardiovascular Risk in Women with Obesity. Obesity (Silver Spring) 2019, 27, 87–93. [Google Scholar] [CrossRef]
- Uemura, S.; Matsushita, H.; Li, W.; Glassford, A.J.; Asagami, T.; Lee, K.H.; Harrison, D.G.; Tsao, P.S. Diabetes mellitus enhances vascular matrix metalloproteinase activity: Role of oxidative stress. Circ. Res. 2001, 88, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Vasko, R.; Xavier, S.; Chen, J.; Lin, C.H.; Ratliff, B.; Rabadi, M.; Maizel, J.; Tanokuchi, R.; Zhang, F.; Cao, J.; et al. Endothelial Sirtuin 1 Deficiency Perpetrates Nephrosclerosis through Downregulation of Matrix Metalloproteinase-14: Relevance to Fibrosis of Vascular Senescence. J. Am. Soc. Nephrol. 2014, 25, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Sinfield, J.K.; Das, A.; O’Regan, D.J.; Ball, S.G.; Porter, K.E.; Turner, N.A. p38 MAPK alpha mediates cytokine-induced IL-6 and MMP-3 expression in human cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2013, 430, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Theret, N.; Lehti, K.; Musso, O.; Clement, B. MMP2 activation by collagen I and concanavalin A in cultured human hepatic stellate cells. Hepatology 1999, 30, 462–468. [Google Scholar] [CrossRef]
- Son, Y.; Lee, J.H.; Chung, H.T.; Pae, H.O. Therapeutic roles of heme oxygenase-1 in metabolic diseases: Curcumin and resveratrol analogues as possible inducers of heme oxygenase-1. Oxid. Med. Cell. Longev. 2013, 2013, 639541. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Akhov, L.; Selvaraj, G.; Wang, M.; Alam, J. Regulation of heme oxygenase-1 expression by demethoxy curcuminoids through Nrf2 by a PI3-kinase/Akt-mediated pathway in mouse beta-cells. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E645–E655. [Google Scholar] [CrossRef]
- Chi, T.C.; Chen, W.P.; Chi, T.L.; Kuo, T.F.; Lee, S.S.; Cheng, J.T.; Su, M.J. Phosphatidylinositol-3-kinase is involved in the antihyperglycemic effect induced by resveratrol in streptozotocin-induced diabetic rats. Life Sci. 2007, 80, 1713–1720. [Google Scholar] [CrossRef]
- Shen, H.H.; Peterson, S.J.; Bellner, L.; Choudhary, A.; Levy, L.; Gancz, L.; Sasson, A.; Trainer, J.; Rezzani, R.; Resnick, A.; et al. Cold-Pressed Nigella Sativa Oil Standardized to 3% Thymoquinone Potentiates Omega-3 Protection against Obesity-Induced Oxidative Stress, Inflammation, and Markers of Insulin Resistance Accompanied with Conversion of White to Beige Fat in Mice. Antioxidants (Basel) 2020, 9, 489. [Google Scholar] [CrossRef]
- Singh, S.; Das, S.S.; Singh, G.; Schuff, C.; de Lampasona, M.P.; Catalan, C.A. Composition, in vitro antioxidant and antimicrobial activities of essential oil and oleoresins obtained from black cumin seeds (Nigella sativa L.). Biomed. Res. Int. 2014, 2014, 918209. [Google Scholar] [CrossRef]
- Baranano, D.E.; Wolosker, H.; Bae, B.I.; Barrow, R.K.; Snyder, S.H.; Ferris, C.D. A mammalian iron ATPase induced by iron. J. Biol. Chem 2000, 275, 15166–15173. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M. Pomegranate for your cardiovascular health. Rambam Maimonides Med. J. 2013, 4, e0013. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ozen, C.; Abu-Reidah, I.M.; Chigurupati, S.; Patra, J.K.; Horbanczuk, J.O.; Jozwik, A.; Tzvetkov, N.T.; Uhrin, P.; Atanasov, A.G. Vasculoprotective Effects of Pomegranate (Punica granatum L.). Front. Pharmacol. 2018, 9, 544. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.A. Composition of pomegranate juice. J. AOAC Int. 2012, 95, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Puri, N.; Favero, G.; Stevens, S.; Meadows, C.; Abraham, N.G.; Rezzani, R.; Ansinelli, H.; Lebovics, E.; Shapiro, J.I. Fructose Mediated Non-Alcoholic Fatty Liver Is Attenuated by HO-1-SIRT1 Module in Murine Hepatocytes and Mice Fed a High Fructose Diet. PLoS ONE 2015, 10, e0128648. [Google Scholar] [CrossRef]
- González-Ortiz, M.; Martínez-Abundis, E.; Espinel-Bermúdez, M.C.; Pérez-Rubio, K.G. Effect of pomegranate juice on insulin secretion and sensitivity in patients with obesity. Ann. Nutr. Metab. 2011, 58, 220–223. [Google Scholar] [CrossRef]
- Vroegrijk, I.O.; van Diepen, J.A.; van den Berg, S.; Westbroek, I.; Keizer, H.; Gambelli, L.; Hontecillas, R.; Bassaganya-Riera, J.; Zondag, G.C.; Romijn, J.A.; et al. Pomegranate seed oil, a rich source of punicic acid, prevents diet-induced obesity and insulin resistance in mice. Food Chem. Toxicol. 2011, 49, 1426–1430. [Google Scholar] [CrossRef]
- Moazzen, H.; Alizadeh, M. Effects of Pomegranate Juice on Cardiovascular Risk Factors in Patients with Metabolic Syndrome: A Double-Blinded, Randomized Crossover Controlled Trial. Plant Foods Hum. Nutr. 2017, 72, 126–133. [Google Scholar] [CrossRef]
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of nonalcoholic fatty liver disease in the United States: The Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Epidemiol. 2013, 178, 38–45. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Cusi, K.; Sanyal, A.J.; Zhang, S.; Hartman, M.L.; Bue-Valleskey, J.M.; Hoogwerf, B.J.; Haupt, A. Non-alcoholic fatty liver disease (NAFLD) prevalence and its metabolic associations in patients with type 1 diabetes and type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1630–1634. [Google Scholar] [CrossRef]
- Noureddin, M.; Sanyal, A.J. Pathogenesis of NASH: The Impact of Multiple Pathways. Curr. Hepatol. Rep. 2018, 17, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M. Weight loss as a treatment for nonalcoholic fatty liver disease. J. Clin. Gastroenterol. 2006, 40, S39–S43. [Google Scholar]
- Petroni, M.L.; Brodosi, L.; Barbanti, F.A.; di Domizio, S.; Petta, S.; Marchesini, G. Lifestyle changes for the treatment of nonalcoholic fatty liver disease—A 2015-19 update. Curr. Pharm. Des. 2020, 26, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Reginato, E.; Pippi, R.; Aiello, C.; Sbroma Tomaro, E.; Ranucci, C.; Buratta, L.; Bini, V.; Marchesini, G.; De Feo, P.; Fanelli, C. Effect of Short Term Intensive Lifestyle Intervention on Hepatic Steatosis Indexes in Adults with Obesity and/or Type 2 Diabetes. J. Clin. Med. 2019, 8, 851. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Grant, I.; Meissner, A.; Kappas, A.; Abraham, N. Ablation of adipose-HO-1 expression increases white fat over beige fat through inhibition of mitochondrial fusion and of PGC1alpha in female mice. Horm. Mol. Biol. Clin. Investig. 2017, 31. [Google Scholar] [CrossRef]
- Alcala, M.; Calderon-Dominguez, M.; Bustos, E.; Ramos, P.; Casals, N.; Serra, D.; Viana, M.; Herrero, L. Increased inflammation, oxidative stress and mitochondrial respiration in brown adipose tissue from obese mice. Sci. Rep. 2017, 7, 16082. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef]
- Sacerdoti, D.; Singh, S.P.; Schragenheim, J.; Bellner, L.; Vanella, L.; Raffaele, M.; Meissner, A.; Grant, I.; Favero, G.; Rezzani, R.; et al. Development of NASH in Obese Mice is Confounded by Adipose Tissue Increase in Inflammatory NOV and Oxidative Stress. Int. J. Hepatol. 2018, 2018, 3484107. [Google Scholar] [CrossRef]
- Spahis, S.; Delvin, E.; Borys, J.M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef]
- Kleiner, S.; Mepani, R.J.; Laznik, D.; Ye, L.; Jurczak, M.J.; Jornayvaz, F.R.; Estall, J.L.; Chatterjee, B.D.; Shulman, G.I.; Spiegelman, B.M. Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proc. Natl. Acad. Sci. USA 2012, 109, 9635–9640. [Google Scholar] [CrossRef]
- Singh, S.P.; Schragenheim, J.; Cao, J.; Falck, J.R.; Abraham, N.G.; Bellner, L. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat. 2016, 125, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Rosell, M.; Diaz-Delfin, J.; Olmos, Y.; Monsalve, M.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor alpha (PPARalpha) induces PPARgamma coactivator 1alpha (PGC-1alpha) gene expression and contributes to thermogenic activation of brown fat: Involvement of PRDM16. J. Biol. Chem. 2011, 286, 43112–43122. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.V.; Fuhrmann, N. Dominant optic atrophy, OPA1, and mitochondrial quality control: Understanding mitochondrial network dynamics. Mol. Neurodegener. 2013, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Munoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef]
- Schragenheim, J.; Bellner, L.; Cao, J.; Singh, S.P.; Bamshad, D.; McClung, J.A.; Maayan, O.; Meissner, A.; Grant, I.; Stier, C.T., Jr.; et al. EET enhances renal function in obese mice resulting in restoration of HO-1-Mfn1/2 signaling, and decrease in hypertension through inhibition of sodium chloride co-transporter. Prostaglandins Other Lipid Mediat. 2018, 137, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, E.W.; Peterson, S.J.; Kothari, J.; Alex, R.; Shapiro, J.I.; Abraham, N.G. Genetic Polymorphisms Complicate COVID-19 Therapy: Pivotal Role of HO-1 in Cytokine Storm. Antioxidants (Basel) 2020, 9, 636. [Google Scholar] [CrossRef]
- Cao, J.; Peterson, S.J.; Sodhi, K.; Vanella, L.; Barbagallo, I.; Rodella, L.F.; Schwartzman, M.L.; Abraham, N.G.; Kappas, A. Heme oxygenase gene targeting to adipocytes attenuates adiposity and vascular dysfunction in mice fed a high-fat diet. Hypertension 2012, 60, 467–475. [Google Scholar] [CrossRef]
- Burgess, A.; Li, M.; Vanella, L.; Kim, D.H.; Rezzani, R.; Rodella, L.; Sodhi, K.; Canestraro, M.; Martasek, P.; Peterson, S.J.; et al. Adipocyte heme oxygenase-1 induction attenuates metabolic syndrome in both male and female obese mice. Hypertension 2010, 56, 1124–1130. [Google Scholar] [CrossRef]
- Csongradi, E.; Docarmo, J.M.; Dubinion, J.H.; Vera, T.; Stec, D.E. Chronic HO-1 induction with cobalt protoporphyrin (CoPP) treatment increases oxygen consumption, activity, heat production and lowers body weight in obese melanocortin-4 receptor-deficient mice. Int. J. Obes. (Lond.) 2012, 36, 244–253. [Google Scholar] [CrossRef]
- Vanella, L.; Kim, D.H.; Asprinio, D.; Peterson, S.J.; Barbagallo, I.; Vanella, A.; Goldstein, D.; Ikehara, S.; Kappas, A.; Abraham, N.G. HO-1 expression increases mesenchymal stem cell-derived osteoblasts but decreases adipocyte lineage. Bone 2010, 46, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Sodhi, K.; Meadows, C.; Fedorova, L.; Puri, N.; Kim, D.H.; Peterson, S.J.; Shapiro, J.; Abraham, N.G.; Kappas, A. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity (Silver. Spring) 2014, 22, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, H.V.; Zehra, M.; Pillai, S.S.; Puri, N.; Shapiro, J.I.; Abraham, N.G.; Sodhi, K. Beneficial Role of HO-1-SIRT1 Axis in Attenuating Angiotensin II-Induced Adipocyte Dysfunction. Int. J. Mol. Sci. 2019, 20, 3205. [Google Scholar] [CrossRef] [PubMed]
- Waldman, M.; Arad, M.; Abraham, N.G.; Hochhauser, E. The PGC-1alpha -HO-1 axis, a powerful antioxidative pathway with potential to attenuate diabetic cardiomyopathy. Antioxid. Redox Signal. 2020, 32, 1273–1290. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, H.; Liu, J. Akt activation: A potential strategy to ameliorate insulin resistance. Diabetes Res. Clin. Pract. 2019, 156, 107092. [Google Scholar] [CrossRef]
- Raffaele, M.; Bellner, L.; Singh, S.P.; Favero, G.; Rezzani, R.; Rodella, L.F.; Falck, J.R.; Abraham, N.G.; Vanella, L. Epoxyeicosatrienoic intervention improves NAFLD in leptin receptor deficient mice by an increase in PGC1alpha-HO-1-PGC1alpha-mitochondrial signaling. Exp. Cell Res. 2019, 380, 180–187. [Google Scholar] [CrossRef]
- Sacerdoti, D.; Abraham, N.G.; Oyekan, A.O.; Yang, L.; Gatta, A.; McGiff, J.C. Role of the heme oxygenases in abnormalities of the mesenteric circulation in cirrhotic rats. J. Pharmacol. Exp. Ther. 2004, 308, 636–643. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raffaele, M.; Licari, M.; Amin, S.; Alex, R.; Shen, H.-h.; Singh, S.P.; Vanella, L.; Rezzani, R.; Bonomini, F.; Peterson, S.J.; et al. Cold Press Pomegranate Seed Oil Attenuates Dietary-Obesity Induced Hepatic Steatosis and Fibrosis through Antioxidant and Mitochondrial Pathways in Obese Mice. Int. J. Mol. Sci. 2020, 21, 5469. https://doi.org/10.3390/ijms21155469
Raffaele M, Licari M, Amin S, Alex R, Shen H-h, Singh SP, Vanella L, Rezzani R, Bonomini F, Peterson SJ, et al. Cold Press Pomegranate Seed Oil Attenuates Dietary-Obesity Induced Hepatic Steatosis and Fibrosis through Antioxidant and Mitochondrial Pathways in Obese Mice. International Journal of Molecular Sciences. 2020; 21(15):5469. https://doi.org/10.3390/ijms21155469
Chicago/Turabian StyleRaffaele, Marco, Maria Licari, Sherif Amin, Ragin Alex, Hsin-hsueh Shen, Shailendra P. Singh, Luca Vanella, Rita Rezzani, Francesca Bonomini, Stephen J. Peterson, and et al. 2020. "Cold Press Pomegranate Seed Oil Attenuates Dietary-Obesity Induced Hepatic Steatosis and Fibrosis through Antioxidant and Mitochondrial Pathways in Obese Mice" International Journal of Molecular Sciences 21, no. 15: 5469. https://doi.org/10.3390/ijms21155469
APA StyleRaffaele, M., Licari, M., Amin, S., Alex, R., Shen, H.-h., Singh, S. P., Vanella, L., Rezzani, R., Bonomini, F., Peterson, S. J., Stec, D. E., & Abraham, N. G. (2020). Cold Press Pomegranate Seed Oil Attenuates Dietary-Obesity Induced Hepatic Steatosis and Fibrosis through Antioxidant and Mitochondrial Pathways in Obese Mice. International Journal of Molecular Sciences, 21(15), 5469. https://doi.org/10.3390/ijms21155469