Extracellular Vesicles-Based Drug Delivery Systems: A New Challenge and the Exemplum of Malignant Pleural Mesothelioma

 ,
,  ,
,  ,
, 


Abstract
1. Introduction
2. Strategies in Cancer Targeted Therapy
2.1. Nanoparticles
2.1.1. Liposomes
2.1.2. Polymer-Conjugated Drugs for Selective Delivery
2.2. Small Molecules, Peptides and Antibody in Targeted Therapy
2.3. ADC
2.4. Reconfigurable Organisms as Drug Carriers
3. EVs
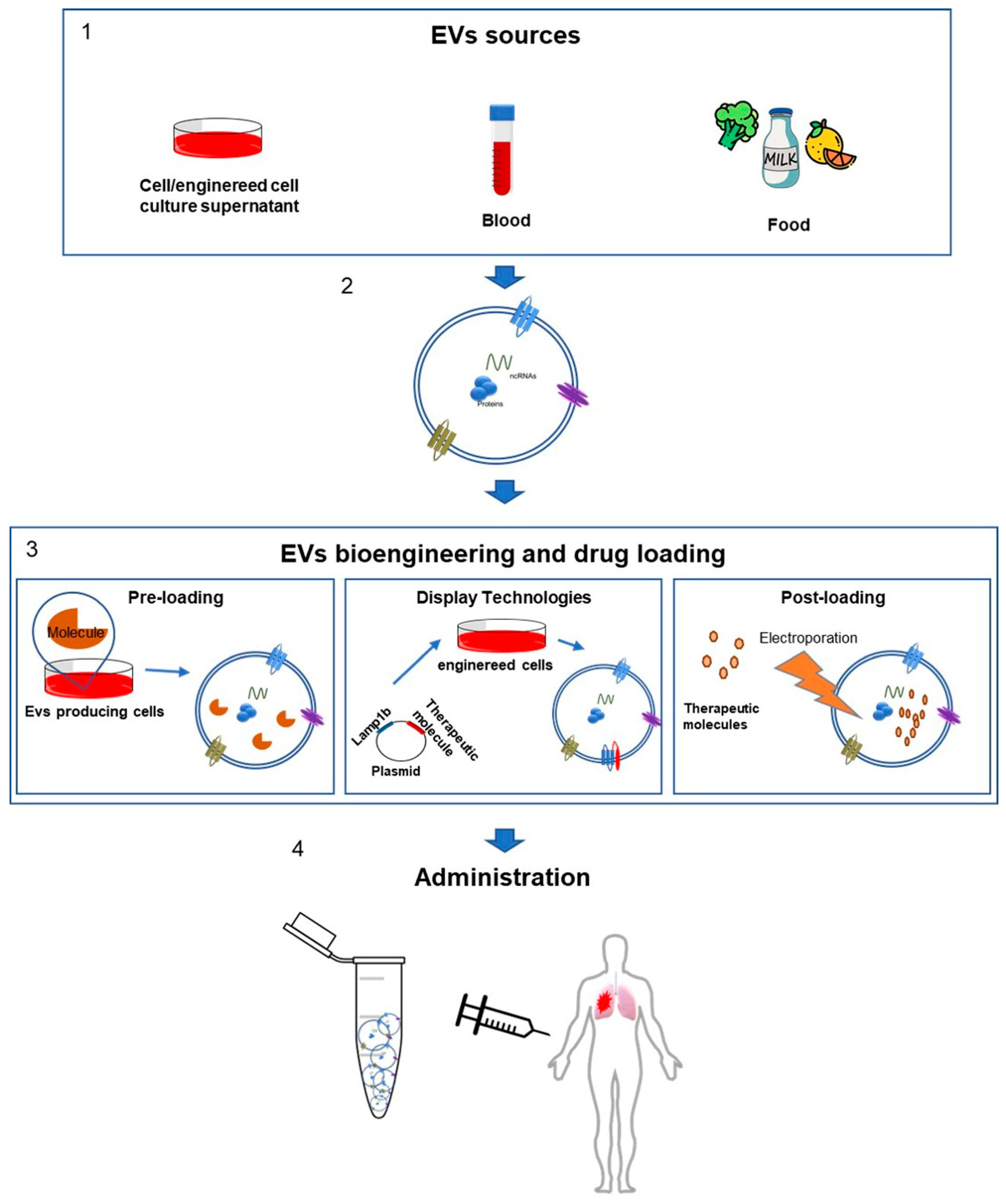
EV Bioengineering and Drug Loading
4. Malignant Pleural Mesothelioma: The Lack of Efficient Therapeutic Strategies and Possible Drug Delivery Strategies
5. Future Perspective: The Challenge in the Exploitation of EVs in MPM Therapy
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 7 May 2020).
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Odisio, E.G.; Marom, E.M.; Shroff, G.S.; Wu, C.C.; Benveniste, A.P.A.; Truong, M.T.; Benveniste, M.F. Malignant Pleural Mesothelioma: Diagnosis, Staging, Pitfalls and Follow-up. Semin. Ultrasound Ct Mr. 2017, 38, 559–570. [Google Scholar] [CrossRef]
- Pini, A. Anticorpi e peptidi, 1st ed.; ARACNE: Rome, Italy, 2015; Available online: http://www.aracneeditrice.it/aracneweb/index.php/pubblicazione.html?item=9788854840607 (accessed on 7 May 2020).
- Tan, S.Y.; Grimes, S. Paul Ehrlich (1854-1915): Man with the magic bullet. Singap. Med. J. 2010, 51, 842–843. [Google Scholar]
- Di, C.; Zhang, Q.; Wang, Y.; Wang, F.; Chen, Y.; Gan, L.; Zhou, R.; Sun, C.; Li, H.; Zhang, X.; et al. Exosomes as drug carriers for clinical application. Artif. Cells Nanomed. Biotechnol. 2018, 46, S564–S570. [Google Scholar] [CrossRef] [PubMed]
- Campanella, C.; Caruso Bavisotto, C.; Logozzi, M.; Marino Gammazza, A.; Mizzoni, D.; Cappello, F.; Fais, S. On the Choice of the Extracellular Vesicles for Therapeutic Purposes. Int. J. Mol. Sci. 2019, 20, 236. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist. Updat. 2015, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Luciani, F.; Molinari, A.; Lozupone, F.; Calcabrini, A.; Lugini, L.; Stringaro, A.; Puddu, P.; Arancia, G.; Cianfriglia, M.; Fais, S. P-glycoprotein-actin association through ERM family proteins: A role in P-glycoprotein function in human cells of lymphoid origin. Blood 2002, 99, 641–648. [Google Scholar] [CrossRef]
- Pillai, S.R.; Damaghi, M.; Marunaka, Y.; Spugnini, E.P.; Fais, S.; Gillies, R.J. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. 2019, 38, 205–222. [Google Scholar] [CrossRef]
- Luciani, F.; Spada, M.; De Milito, A.; Molinari, A.; Rivoltini, L.; Montinaro, A.; Marra, M.; Lugini, L.; Logozzi, M.; Lozupone, F.; et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J. Natl. Cancer Inst. 2004, 96, 1702–1713. [Google Scholar] [CrossRef]
- Azzarito, T.; Venturi, G.; Cesolini, A.; Fais, S. Lansoprazole induces sensitivity to suboptimal doses of paclitaxel in human melanoma. Cancer Lett. 2015, 356, 697–703. [Google Scholar] [CrossRef]
- Lugini, L.; Sciamanna, I.; Federici, C.; Iessi, E.; Spugnini, E.P.; Fais, S. Antitumor effect of combination of the inhibitors of two new oncotargets: Proton pumps and reverse transcriptase. Oncotarget 2017, 8, 4147–4155. [Google Scholar] [CrossRef] [PubMed]
- Federici, C.; Lugini, L.; Marino, M.L.; Carta, F.; Iessi, E.; Azzarito, T.; Supuran, C.T.; Fais, S. Lansoprazole and carbonic anhydrase IX inhibitors sinergize against human melanoma cells. J. Enzym. Inhib. Med. Chem. 2016, 31, 119–125. [Google Scholar] [CrossRef]
- Ferrari, S.; Perut, F.; Fagioli, F.; Brach Del Prever, A.; Meazza, C.; Parafioriti, A.; Picci, P.; Gambarotti, M.; Avnet, S.; Baldini, N.; et al. Proton pump inhibitor chemosensitization in human osteosarcoma: From the bench to the patients’ bed. J. Transl. Med. 2013, 11, 268. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.Y.; Zhang, J.; Wang, J.L.; Sun, S.; Wang, Z.H.; Wang, L.P.; Zhang, Q.L.; Lv, F.F.; Cao, E.Y.; Shao, Z.M.; et al. Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer. J. Exp. Clin. Cancer Res. 2015, 3, 109. [Google Scholar] [CrossRef] [PubMed]
- Falcone, R.; Roberto, M.; D’Antonio, C.; Romiti, A.; Milano, A.; Onesti, C.E.; Marchetti, P.; Fais, S. High-doses of proton pump inhibitors in refractory gastro-intestinal cancer: A case series and the state of art. Dig. Liver Dis. 2016, 48, 1503–1505. [Google Scholar] [CrossRef]
- Yun, Y.H.; Lee, B.K.; Park, K. Controlled Drug Delivery: Historical perspective for the next generation. J. Control. Release 2015, 219, 2–7. [Google Scholar] [CrossRef]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Itty Ipe, B.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Mirza, A.Z.; Siddiqui, F.A. Nanomedicine and drug delivery: A mini review. Int. Nano. Lett. 2014, 4, 94. [Google Scholar] [CrossRef]
- Azandaryani, A.H.; Kashanian, S.; Jamshidnejad-Tosaramandani, T. Recent Insights into Effective Nanomaterials and Biomacromolecules Conjugation in Advanced Drug Targeting. Curr. Pharm. Biotechnol. 2019, 20, 526–541. [Google Scholar] [CrossRef]
- Nazir, S.; Hussain, T.; Ayub, A.; Rashid, U.; MacRobert, A.J. Nanomaterials in combating cancer: Therapeutic applications and developments. Nanomedicine 2014, 10, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.R.; Qiao, P. Drug Delivery in Cancer Therapy, Quo Vadis? Mol. Pharm. 2018, 15, 3603–3616. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Aime, S.; Hennink, W.E.; Storm, G.; Kiessling, F. Theranostic nanomedicine. Acc. Chem. Res. 2011, 44, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Baetke, S.C.; Lammers, T.; Kiessling, F. Applications of nanoparticles for diagnosis and therapy of cancer. Br. J. Radiol. 2015, 88. [Google Scholar] [CrossRef]
- Lam, P.L.; Wong, W.Y.; Bian, Z.; Chui, C.H.; Gambari, R. Recent advances in green nanoparticulate systems for drug delivery: Efficient delivery and safety concern. Nanomed. (Lond) 2017, 12, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Far, J.; Abdel-Haq, M.; Gruber, M.; Abu Ammar, A. Developing Biodegradable Nanoparticles Loaded with Mometasone Furoate for Potential Nasal Drug Delivery. ACS Omega 2020, 5, 7432–7439. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Gubernator, J. Active methods of drug loading into liposomes: Recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 8, 565–580. [Google Scholar] [CrossRef]
- Hua, S.; Wu, S.Y. The use of lipid-based nanocarriers for targeted pain therapies. Front. Pharm. 2013, 4, 143. [Google Scholar] [CrossRef]
- Monteiro, N.; Martins, A.; Reis, R.L.; Neves, N.M. Liposomes in tissue engineering and regenerative medicine. J. R. Soc. Interface 2014, 11. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Heidarli, E.; Dadashzadeh, S.; Haeri, A. State of the Art of Stimuli-Responsive Liposomes for Cancer Therapy. Iran. J. Pharm. Res. 2017, 16, 1273–1304. [Google Scholar] [PubMed]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, S.; Hu, L.; Peng, B.; Liu, Y.; Luo, X.; Liu, X.; Song, Y.; Deng, Y. Polysialic acid-polyethylene glycol conjugate-modified liposomes as a targeted drug delivery system for epirubicin to enhance anticancer efficiency. Drug Deliv. Transl. Res. 2018, 8, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Guo, X.; Tu, L.; Zou, Q.; Li, Q.; Tang, C.; Chen, B.; Xu, Y.; Wu, C. Lactoferrin-modified PEGylated liposomes loaded with doxorubicin for targeting delivery to hepatocellular carcinoma. Int. J. Nanomed. 2015, 10, 5123–5137. [Google Scholar] [CrossRef]
- Soe, Z.C.; Thapa, R.K.; Ou, W.; Gautam, M.; Nguyen, H.T.; Jin, S.G.; Ku, S.K.; Oh, K.T.; Choi, H.; Yong, C.S.; et al. Folate receptor-mediated celastrol and irinotecan combination delivery using liposomes for effective chemotherapy. Colloids Surf. B. Biointerfaces 2018, 170, 718–728. [Google Scholar] [CrossRef]
- Jhaveri, A.; Deshpande, P.; Pattni, B.; Torchilin, V. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J. Control. Release 2018, 277, 89–101. [Google Scholar] [CrossRef]
- Toh, M.R.; Chiu, G.N.C. Liposomes as sterile preparations and limitations of sterilization techniques in liposomal manufacturing. Asian J. Pharm. Sci. 2013, 8, 88–95. [Google Scholar] [CrossRef]
- Gibis, M.; Rahn, N.; Weiss, J. Physical and oxidative stability of uncoated and chitosan-coated liposomes containing grape seed extract. Pharmaceutics 2013, 5, 421–433. [Google Scholar] [CrossRef]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Ishida, T.; Harashima, H.; Kiwada, H. Liposome clearance. Biosci. Rep. 2002, 22, 197–224. [Google Scholar] [CrossRef] [PubMed]
- Ringsdorf, H. Structure and properties of pharmacologically135-active polymers. J. Polym. Sci. Polym. Symp. 1975, 51, 153. [Google Scholar]
- Harding, N.G. Amethopterin linked covalently to water-soluble macromolecules. Ann. N. Y. Acad. Sci. 1971, 186, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Tam, P. Synthetic peptide vaccine design: Synthesis and properties of a high-density multiple antigenic peptide system. Proc. Natl. Acad. Sci. USA 1988, 85, 5409–5413. [Google Scholar] [CrossRef]
- Posnett, D.N.; McGrath, H.; Tam, J.P. A novel method for producing anti-peptide antibodies. Production of site-specific antibodies to the T cell antigen receptor beta-chain. J. Biol. Chem. 1988, 263, 1719–1725. [Google Scholar]
- Brunetti, J.; Falciani, C.; Lelli, B.; Minervini, A.; Ravenni, N.; Depau, L.; Siena, G.; Tenori, E.; Menichetti, S.; Pini, A.; et al. Neurotensin branched peptide as a tumor-targeting agent for human bladder cancer. BioMed Res. Int. 2015, 2015, 173507. [Google Scholar] [CrossRef]
- Brunetti, J.; Depau, L.; Falciani, C.; Gentile, M.; Mandarini, E.; Riolo, G.; Lupetti, P.; Pini, A.; Bracci, L. Insights into the role of sulfated glycans in cancer cell adhesion and migration through use of branched peptide probe. Sci. Rep. 2016, 6, 27174. [Google Scholar] [CrossRef]
- Bracci, L.; Falciani, C.; Lelli, B.; Lozzi, L.; Runci, Y.; Pini, A.; De Montis, M.G.; Tagliamonte, A.; Neri, P. Synthetic Peptides in the Form of Dendrimers Become Resistant to Protease Activity. J. Biol. Chem. 2003, 278, 46590–46595. [Google Scholar] [CrossRef]
- Brunetti, J.; Pillozzi, S.; Falciani, C.; Depau, L.; Tenori, E.; Scali, S.; Lozzi, L.; Pini, A.; Arcangeli, A.; Menichetti, S.; et al. Tumor-selective peptide-carrier delivery of Paclitaxel increases in vivo activity of the drug. Sci. Rep. 2015, 5, 17736. [Google Scholar] [CrossRef]
- Brunetti, J.; Piantini, S.; Fragai, M.; Scali, S.; Cipriani, G.; Depau, L.; Pini, L.; Falciani, C.; Menichetti, S.; Bracci, L. A New NT4 Peptide-Based Drug Delivery System for Cancer Treatment. Molecules 2020, 25, 1088. [Google Scholar] [CrossRef]
- Morales-Avila, E.; Ferro-Flores, G.; Ocampo-García, B.E.; De León-Rodríguez, L.M.; Santos-Cuevas, C.L.; García-Becerra, R.; Medina, L.A.; Gómez-Oliván, L. Multimeric system of 99mTc-labeled gold nanoparticles conjugated to c[RGDfK(C)] for molecular imaging of tumor α(v)β(3) expression. Bioconjug. Chem. 2011, 22, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Shatz, W.; Hass, P.E.; Peer, N.; Paluch, M.T.; Blanchette, C.; Han, G.; Sandoval, W.; Morando, A.; Loyet, K.M.; Bantseev, V.; et al. Identification and characterization of an octameric PEG-protein conjugate system for intravitreal long-acting delivery to the back of the eye. PLoS ONE 2019, 14, e0218613. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharm. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Kerr, W.G.; Chisholm, J.D. The Next Generation of Immunotherapy for Cancer: Small Molecules Could Make Big Waves. J. Immunol. 2019, 202, 11–19. [Google Scholar] [CrossRef]
- Zhong, S.; Jeong, J.H.; Chen, Z.; Chen, Z.; Luo, J.L. Targeting Tumor Microenvironment by Small-Molecule Inhibitors. Transl. Oncol. 2020, 13, 57–69. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Houghton, A.N.; Scheinberg, D.A. Monoclonal antibodies: Potential applications to the treatment of cancer. Semin. Oncol. 1986, 13, 165–179. [Google Scholar]
- Shuptrine, C.W.; Surana, R.; Weiner, L.M. Monoclonal antibodies for the treatment of cancer. Semin. Cancer Biol. 2012, 22, 3–13. [Google Scholar] [CrossRef]
- Izquierdo-Sánchez, V.; Muñiz-Hernández, S.; Vázquez-Becerra, H.; Pacheco-Yepez, J.; Romero-Piña, M.E.; Arrieta, O.; Medina, L.A. Biodistribution and Tumor Uptake of 67Ga-Nimotuzumab in a Malignant Pleural Mesothelioma Xenograft. Molecules 2018, 23, 3138. [Google Scholar] [CrossRef]
- Liu, J.K.H. The history of monoclonal antibody development—Progress, remaining challenges and future innovations. Ann. Med. Surg. 2014, 3, 113–116. [Google Scholar] [CrossRef]
- Tiller, K.E.; Tessier, P.M. Advances in Antibody Design. Annu. Rev. Biomed. Eng. 2015, 17, 191–216. [Google Scholar] [CrossRef]
- Buss, N.A.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharm. 2012, 12, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef] [PubMed]
- Kubczak, M.; Rogalińska, M. Ewolucja przeciwciał monoklonalnych w leczeniu chorób nowotworowych [Evolution of monoclonal antibodies in cancer treatment]. Postepy. Biochem. 2016, 62, 518–525. [Google Scholar] [PubMed]
- Peters, C.; Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Buratta, S.; Logozzi, M.; Mitro, N.; Sagini, K.; Di Raimo, R.; Caruso, D.; Fais, S.; Emiliani, C. Lipidomic analysis of cancer cells cultivated at acidic pH reveals phospholipid fatty acids remodelling associated with transcriptional reprogramming. J. Enzyme Inhib. Med. Chem. 2020, 35, 963–973. [Google Scholar] [CrossRef]
- Ahmadzada, T.; Kao, S.; Reid, G.; Clarke, S.; Grau, G.E.; Hosseini-Beheshti, E. Extracellular vesicles as biomarkers in malignant pleural mesothelioma: A review. Crit. Rev. Oncol. Hematol. 2020, 150, 102949. [Google Scholar] [CrossRef]
- Kazazi-Hyseni, F.; Beijnen, J.H.; Schellens, J.H. Bevacizumab. Oncologist 2010, 15, 819–825. [Google Scholar] [CrossRef]
- The European Medicines Agency. European Public Assessment Report. Avastin Product Information. Avastin-H-C-582-II-23. August 2008. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/avastin (accessed on 7 May 2020).
- Kaminski, M.S.; Tuck, M.; Estes, J.; Kolstad, A.; Ross, C.W.; Zasadny, K.; Regan, D.; Kison, P.; Fisher, S.; Kroll, S.; et al. 131I-tositumomab therapy as initial treatment for follicular lymphoma. N. Engl. J. Med. 2005, 352, 441–449. [Google Scholar] [CrossRef]
- Borgheai, H.; Schilder, R.J. Safety and efficacy of radioimmunotherapy with yttrium 90 ibritumomab tiuxetan (Zevalin). Semin. Nucl. Med. 2004, 34, 4–9. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, E.G.; Kim, K.M. Strategies and advancement in antibody-drug conjugate, Optimization for Targeted Cancer Therapeutics. Biomol. Ther. 2015, 23, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Wadleigh, M.; Richardson, P.G.; Zahrieh, D.; Lee, S.J.; Cutler, C.; Ho, V.; Alyea, E.P.; Antin, J.H.; Stone, R.M.; Soiffer, R.J.; et al. Prior gemtuzumab ozogamicin exposure significantly increases the risk of veno-occlusive disease in patients who undergo myeloablative allogeneic stem cell transplantation. Blood 2003, 102, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Rashid, Z.; Shokri, F.; Abbasi, A.; Khoobi, M.; Zarnani, A.H. Surface modification and bioconjugation of anti-CD4 monoclonal antibody to magnetic nanoparticles as a highly efficient affinity adsorbent for positive selection of peripheral blood T CD4+ lymphocytes [published online ahead of print, 2020 June 1]. Int. J. Biol. Macromol. 2020. [Google Scholar] [CrossRef]
- Catapano, A.L.; Pirillo, A.; Norata, G.D. New Pharmacological Approaches to Target PCSK9. Curr. Atheroscler. Rep. 2020, 22, 24. [Google Scholar] [CrossRef]
- Kriegman, S.; Blackiston, D.; Levin, M.; Bongard, J. A scalable pipeline for designing reconfigurable organisms. Proc. Natl. Acad. Sci. USA 2020, 117, 1853–1859. [Google Scholar] [CrossRef]
- Chargaff, E.; West, R. The biological significance of the thromboplastic protein of blood. J. Biol. Chem. 1946, 166, 189–197. [Google Scholar]
- Wolf, P. The nature and significance of platelet products in human plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef]
- Hargett, L.A.; Bauer, N.N. On the origin of microparticles: From “platelet dust” to mediators of intercellular communication. Pulm. Circ. 2013, 3, 329–340. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan., M.L.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Milosevic, J.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Zhu, Y.L.; Zhou, Y.Y.; Liang, G.F.; Wang, Y.Y.; Hu, F.H.; Xiao, Z.D. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [PubMed]
- Caruso Bavisotto, C.; Marino Gammazza, A.; Rappa, F.; Fucarino, A.; Pitruzzella, A.; David, S.; Campanella, C. Exosomes: Can doctors still ignore their existence? EuroMediterr. Biomed. J. 2013, 8, 136–139. [Google Scholar]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated HNRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Kırbaş, O.K.; Bozkurt, B.T.; Asutay, A.B.; Mat, B.; Ozdemir, B.; Ozturkoglu, D.; Olmez, H.; Islek, Z.; Sahin, F.; Tasli, P.N. Optimized Isolation of Extracellular Vesicles From Various Organic Sources Using Aqueous Two-Phase System. Sci. Rep. 2019, 9, 19159. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Konoshenko, M.Y.; Lekchnov, E.A.; Vlassov, A.V.; Laktionov, P.P. Isolation of Extracellular Vesicles: General Methodologies and Latest Trends. Biomed. Res. Int. 2018, 2018, 8545347. [Google Scholar] [CrossRef]
- Guerreiro, E.M.; Vestad, B.; Steffensen, L.A.; Aass, H.; Saeed, M.; Øvstebø, R.; Costea, D.E.; Galtung, H.K.; Søland, T.M. Efficient extracellular vesicle isolation by combining cell media modifications, ultrafiltration, and size-exclusion chromatography. PLoS ONE 2018, 13, e0204276. [Google Scholar] [CrossRef]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef]
- Cappello, F.; Logozzi, M.; Campanella, C.; Caruso Bavisotto, C.; Marcilla, A.; Properzi, F.; Fais, S. Exosome levels in human body fluids: A tumor marker by themselves? Eur. J. Pharm. Sci. 2017, 96, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Franchi, L.; Nunez, G.; Dubyak, G.R. Nonclassical IL-1 secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J. Immunol. 2007, 179, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Phoonsawat, W.; Aoki-Yoshida, A.; Tsuruta, T.; Sonoyama, K. Adiponectin is partially associated with exosomes in mouse serum. Biochem. Biophys. Res. Commun. 2014, 448, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, R.; Kemper, S.; Charrier, A.; Brigstock, D.R. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: Role of exosomes in horizontal transfer of Twist1. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G491–G499. [Google Scholar] [CrossRef]
- Campanella, C.; Bucchieri, F.; Merendino, A.M.; Fucarino, A.; Burgio, G.; Corona, D.F.; Barbieri, G.; David, S.; Farina, F.; Zummo, G.; et al. The odyssey of Hsp60 from tumor cells to other destinations includes plasma membrane-associated stages and Golgi and exosomal protein-trafficking modalities. PLoS ONE 2012, 7, e42008. [Google Scholar] [CrossRef]
- Campanella, C.; Rappa, F.; Sciumè, C.; Marino Gammazza, A.; Barone, R.; Bucchieri, F.; David, S.; Curcurù, G.; Caruso Bavisotto, C.; Pitruzzella, A.; et al. Heat shock protein 60 levels in tissue and circulating exosomes in human large bowel cancer before and after ablative surgery. Cancer 2015, 121, 3230–3239. [Google Scholar] [CrossRef]
- Caruso Bavisotto, C.; Graziano, F.; Rappa, F.; Marino Gammazza, A.; Logozzi, M.; Fais, S.; Maugeri, R.; Bucchieri, F.; Conway de Macario, E.; Macario, A.J.L.; et al. Exosomal Chaperones and miRNAs in Gliomagenesis: State-of-Art and Theranostics Perspectives. Int. J. Mol. Sci. 2018, 19, 2626. [Google Scholar] [CrossRef]
- Caruso Bavisotto, C.; Cappello, F.; Macario, A.J.L.; Conway de Macario, E.; Logozzi, M.; Fais, S.; Campanella, C. Exosomal HSP60: A potentially useful biomarker for diagnosis, assessing prognosis, and monitoring response to treatment. Expert Rev. Mol. Diagn. 2017, 17, 815–822. [Google Scholar] [CrossRef]
- Campanella, C.; D’Anneo, A.; Marino Gammazza, A.; Caruso Bavisotto, C.; Barone, R.; Emanuele, S.; Lo Cascio, F.; Mocciaro, E.; Fais, S.; Conway De Macario, E.; et al. The histone deacetylase inhibitor SAHA induces HSP60 nitration and its extracellular release by exosomal vesicles in human lung-derived carcinoma cells. Oncotarget 2016, 7, 28849–28867. [Google Scholar] [CrossRef]
- Caruso Bavisotto, C.; Scalia, F.; Marino Gammazza, A.; Carlisi, D.; Bucchieri, F.; Conway de Macario, E.; Macario, A.; Cappello, F.; Campanella, C. Extracellular Vesicle-Mediated Cell-Cell Communication in the Nervous System: Focus on Neurological Diseases. Int. J. Mol. Sci. 2019, 20, 434. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Febbraio, M.A. Exosome-dependent trafficking of Hsp70: A novel secretory pathway for cellular stress proteins. J. Biol. Chem. 2005, 280, 23349–23355. [Google Scholar] [CrossRef] [PubMed]
- Bausero, M.A.; Gastpar, R.; Multhoff, G.; Asea, A. Alternative mechanism by which IFN-gamma enhances tumor recognition: Active release of heat shock protein 72. J. Immunol. 2005, 175, 2900–2912. [Google Scholar] [CrossRef] [PubMed]
- Vega, V.L.; Rodríguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A.A. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [PubMed]
- Vesiclepedia: Browse Results. Available online: http://microvesicles.org/browse_results?org_name=Homosapiens&cont_type=&tissue=&gene_symbol=&ves_type= (accessed on 28 July 2020).
- ExoCarta: Browse Results. Available online: http://exocarta.org/browse_results?org_name=Homosapiens&cont_type=&tissue=&gene_symbol= (accessed on 28 July 2020).
- Stoorvogel, W.; Kleijmeer, M.J.; Geuze, H.J.; Raposo, G. The biogenesis and functions of exosomes. Traffic 2002, 3, 321–330. [Google Scholar] [CrossRef]
- Song, Y.H.; Warncke, C.; Choi, S.J.; Choi, S.; Chiou, A.E.; Ling, L.; Liu, H.Y.; Daniel, S.; Antonyak, M.A.; Cerione, R.A.; et al. Breast cancer-derived extracellular vesicles stimulate myofibroblast differentiation and pro-angiogenic behavior of adipose stem cells. Matrix Biol. 2017, 61, 190–205. [Google Scholar] [CrossRef]
- Lindoso, R.S.; Collino, F.; Camussi, G. Extracellular vesicles derived from renal cancer stem cells induce a pro-tumorigenic phenotype in mesenchymal stromal cells. Oncotarget 2015, 6, 7959–7969. [Google Scholar] [CrossRef]
- Webber, J.; Spary, L.; Sanders, A.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D.; et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34, 290–302. [Google Scholar] [CrossRef]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef]
- Goh, C.Y.; Wyse, C.; Ho, M.; O’Beirne, E.; Howard, J.; Lindsay, S.; Kelly, P.; Higgins, M.; McCann, A. Exosomes in triple negative breast cancer: Garbage disposals or Trojan horses? Cancer Lett. 2020, 473, 90–97. [Google Scholar] [CrossRef]
- Kikuchi, S.; Yoshioka, Y.; Prieto-Vila, M.; Ochiya, T. Involvement of Extracellular Vesicles in Vascular-Related Functions in Cancer Progression and Metastasis. Int. J. Mol. Sci. 2019, 20, 2584. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, O.; Fontana, S.; Monteleone, F.; Taverna, S.; Di Bella, M.A.; Di Vizio, D.; Alessandro, R. Exosomes from metastatic cancer cells transfer amoeboid phenotype to non-metastatic cells and increase endothelial permeability: Their emerging role in tumor heterogeneity. Sci. Rep. 2017, 7, 4711. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 124, 3748–3757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef]
- Conigliaro, A.; Costa, V.; Lo Dico, A.; Saieva, L.; Buccheri, S.; Dieli, F.; Manno, M.; Raccosta, S.; Mancone, C.; Tripodi, M.; et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol. Cancer 2015, 14, 155. [Google Scholar] [CrossRef]
- Dickman, C.T.D.; Lawson, J.; Jabalee, J.; MacLellan, S.A.; LePard, N.E.; Bennewith, K.L.; Garnis, C. Selective extracellular vesicle exclusion of miR-142-3p by oral cancer cells promotes both internal and extracellular malignant phenotypes. Oncotarget 2017, 8, 15252–15266. [Google Scholar] [CrossRef]
- Lawson, J.; Dickman, C.; MacLellan, S.; Towle, R.; Jabalee, J.; Lam, S.; Garnis, C. Selective secretion of microRNAs from lung cancer cells via extracellular vesicles promotes CAMK1D-mediated tube formation in endothelial cells. Oncotarget 2017, 8, 83913–83924. [Google Scholar] [CrossRef]
- Zhuang, G.; Wu, X.; Jiang, Z.; Kasman, I.; Yao, J.; Guan, Y.; Oeh, J.; Modrusan, Z.; Bais, C.; Sampath, D.; et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J. 2012, 31, 3513–3523. [Google Scholar] [CrossRef]
- Zhou, M.; Chen, J.; Zhou, L.; Chen, W.; Ding, G.; Cao, L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol. 2014, 292, 65–69. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Marton, A.; Vizler, C.; Kusz, E.; Temesfoi, V.; Szathmary, Z.; Nagy, K.; Szegletes, Z.; Varo, G.; Siklos, L.; Katona, R.L.; et al. Melanoma cell-derived exosomes alter macrophage and dendritic cell functions in vitro. Immunol. Lett. 2012, 148, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Zhou, W.; Liu, L.; Fong, M.Y.; Champer, J.; Van Haute, D.; Chin, A.R.; Ren, X.; Gugiu, B.G.; Meng, Z.; et al. Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-κB. Sci. Rep. 2015, 4, 5750. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, E.L.; Lindsay, S.; Halasz, M.; Gubbins, L.C.; Weiner-Gorzel, K.; Guang, M.; McGoldrick, A.; Collins, E.; Henry, M.; Blanco-Fernández, A.; et al. Protein and chemotherapy profiling of extracellular vesicles harvested from therapeutic induced senescent triple negative breast cancer cells. Oncogenesis 2017, 6, e388. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Théry, C. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012, 72, 4920–4930. [Google Scholar] [CrossRef]
- Kalimuthu, S.; Gangadaran, P.; Rajendran, R.L.; Zhu, L.; Oh, J.M.; Lee, H.W.; Gopal, A.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; et al. A New Approach for Loading Anticancer Drugs into Mesenchymal Stem Cell-Derived Exosome Mimetics for Cancer Therapy. Front. Pharmacol. 2018, 9, 1116. [Google Scholar] [CrossRef]
- Gomari, H.; Forouzandeh Moghadam, M.; Soleimani, M. Targeted cancer therapy using engineered exosome as a natural drug delivery vehicle. Onco. Targets Ther. 2018, 11, 5753–5762. [Google Scholar] [CrossRef]
- Jiskoot, W.; van Schie, R.M.F.; Carstens, M.G.; Schellekens, H. Immunological Risk of Injectable Drug Delivery Systems. Pharm. Res. 2009, 26, 1303–1314. [Google Scholar] [CrossRef]
- Harding, C.V.; Heuser, J.E.; Stahl, P.D. Exosomes: Looking back three decades and into the future. J. Cell Biol. 2013, 200, 367–371. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- Armstrong, J.P.; Holme, M.N.; Stevens, M.M. Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics. ACS Nano 2017, 11, 69–83. [Google Scholar] [CrossRef]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef] [PubMed]
- Iessi, E.; Logozzi, M.; Lugini, L.; Azzarito, T.; Federici, C.; Spugnini, E.P.; Mizzoni, D.; Di Raimo, R.; Angelini, D.F.; Battistini, L.; et al. Acridine Orange/exosomes increase the delivery and the effectiveness of Acridine Orange in human melanoma cells: A new prototype for theranostics of tumors. J. Enzyme Inhib. Med. Chem. 2017, 32, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Kusuzaki, K.; Matsubara, T.; Murata, H.; Logozzi, M.; Iessi, E.; Di Raimo, R.; Carta, F.; Supuran, C.T.; Fais, S. Natural extracellular nanovesicles and photodynamic molecules: Is there a future for drug delivery? J. Enzyme Inhib. Med. Chem. 2017, 32, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Logozzi, M.; Mizzoni, D.; Bocca, B.; Di Raimo, R.; Petrucci, F.; Caimi, S.; Alimonti, A.; Falchi, M.; Cappello, F.; Campanella, C.; et al. Human primary macrophages scavenge AuNPs and eliminate it through exosomes. A natural shuttling for nanomaterials. Eur. J. Pharm. Biopharm. 2019, 137, 23–36. [Google Scholar] [CrossRef]
- Fais, S.; Logozzi, M.; Lugini, L.; Federici, C.; Azzarito, T.; Zarovni, N.; Chiesi, A. Exosomes: The ideal nanovectors for biodelivery. Biol. Chem. 2013, 394, 1–15. [Google Scholar] [CrossRef]
- Fais, S.; O’Driscoll, L.; Borras, F.E.; Buzas, E.; Camussi, G.; Cappello, F.; Carvalho, J.; Cordeiro da Silva, A.; Del Portillo, H.; El Andaloussi, S.; et al. Evidence-Based Clinical Use of Nanoscale Extracellular Vesicles in Nanomedicine. ACS Nano. 2016, 10, 3886–3899. [Google Scholar] [CrossRef]
- Logozzi, M.; Mizzoni, D.; Capasso, C.; Del Prete, S.; Di Raimo, R.; Falchi, M.; Angelini, D.F.; Sciarra, A.; Maggi, M.; Supuran, C.T.; et al. Plasmatic exosomes from prostate cancer patients show increased carbonic anhydrase IX expression and activity and low pH. J. Enzyme Inhib. Med. Chem. 2020, 35, 280–288. [Google Scholar] [CrossRef]
- Logozzi, M.; Capasso, C.; Di Raimo, R.; Del Prete, S.; Mizzoni, D.; Falchi, M.; Supuran, C.T.; Fais, S. Prostate cancer cells and exosomes in acidic condition show increased carbonic anhydrase IX expression and activity. J. Enzyme Inhib. Med. Chem. 2019, 34, 272–278. [Google Scholar] [CrossRef]
- Logozzi, M.; Mizzoni, D.; Angelini, D.F.; Di Raimo, R.; Falchi, M.; Battistini, L.; Fais, S. Microenvironmental pH and Exosome Levels Interplay in Human Cancer Cell Lines of Different Histotypes. Cancers 2018, 10, 370. [Google Scholar] [CrossRef]
- Meng, W.; He, C.; Hao, Y.; Wang, L.; Li, L.; Zhu, G. Prospects and challenges of extracellular vesicle-based drug delivery system: Considering cell source. Drug Deliv. 2020, 27, 585–598. [Google Scholar] [CrossRef]
- Xin, H.; Li, Y.; Buller, B.; Katakowski, M.; Zhang, Y.; Wang, X.; Shang, X.; Zhang, Z.G.; Chopp, M. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 2012, 30, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Y.; Zhao, B.; Niu, X.; Hu, B.; Li, Q.; Zhang, J.; Ding, J.; Chen, Y.; Wang, Y. Comparison of exosomes secreted by induced pluripotent stem cell-derived mesenchymal stem cells and synovial membrane-derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res. Ther. 2017, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chu, W.C.; Lai, R.C.; Lim, S.K.; Hui, J.H.; Toh, W.S. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthr. Cartil. 2016, 24, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Miyaki, S.; Ishitobi, H.; Ogura, T.; Kato, Y.; Kamei, N.; Miyado, K.; Higashi, Y.; Ochi, M. Mesenchymal Stem Cell-Derived Exosomes Promote Fracture Healing in a Mouse Model. Stem Cells Transl. Med. 2016, 5, 1620–1630. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Li, H.; Chen, C.; Hu, B.; Niu, X.; Li, Q.; Zhao, B.; Xie, Z.; Wang, Y. Exosomes/tricalcium phosphate combination scaffolds can enhance bone regeneration by activating the PI3K/Akt signaling pathway. Stem Cell Res. 2016, 7, 136. [Google Scholar] [CrossRef]
- Ibrahim, A.G.-E.; Cheng, K.; Marbán, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef]
- Das, C.K.; Jena, B.C.; Banerjee, I.; Das, S.; Parekh, A.; Bhutia, S.K.; Mandal, M. Exosome as a Novel Shuttle for Delivery of Therapeutics across Biological Barriers. Mol. Pharm. 2019, 16, 24–40. [Google Scholar] [CrossRef]
- Pullan, J.E.; Confeld, M.I.; Osborn, J.K.; Kim, J.; Sarkar, K.; Mallik, S. Exosomes as Drug Carriers for Cancer Therapy. Mol. Pharm. 2019, 16, 1789–1798. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Fliervoet, L.A.L.; van der Meel, R.; Fens, M.; Heijnen, H.; van Bergen En Henegouwen, P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Fridman, E.; Yaari, Z.; Milman, N.; Schroeder, A.; Ben David, G.; Shlomi, T.; Gil, Z. Transfer of miRNA in Macrophage-Derived Exosomes Induces Drug Resistance in Pancreatic Adenocarcinoma. Cancer Res. 2018, 78, 5287–5299. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Agrahari, V.; Agrahari, V.; Burnouf, P.A.; Chew, C.H.; Burnouf, T. Extracellular microvesicles as new industrial therapeutic frontiers. Trends Biotechnol. 2019, 37, 707–729. [Google Scholar] [CrossRef]
- Burger, D.; Viñas, J.L.; Akbari, S.; Dehak, H.; Knoll, W.; Gutsol, A.; Carter, A.; Touyz, R.M.; Allan, D.S.; Burns, K.D. Human endothelial colony-forming cells protect against acute kidney injury: Role of exosomes. Am. J. Pathol. 2015, 185, 2309–2323. [Google Scholar] [CrossRef] [PubMed]
- Record, M. Exosome-like nanoparticles from food: Protective nanoshuttles for bioactive cargo. Mol. Ther. 2013, 21, 1294–1296. [Google Scholar] [CrossRef]
- Izumi, H.; Tsuda, M.; Sato, Y.; Kosaka, N.; Ochiya, T.; Iwamoto, H.; Namba, K.; Takeda, Y. Bovine milk exosomes contain microRNA and mRNA and are taken up by human macrophages. J. Dairy Sci. 2015, 98, 2920–2933. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-C.; Tao, S.-C.; Yin, W.-J.; Qi, X.; Yuan, T.; Zhang, C.-Q. Exosomes derived from platelet-rich plasma promote the re-epithelization of chronic cutaneous wounds via activation of YAP in a diabetic rat model. Theranostics 2017, 7, 81–96. [Google Scholar] [CrossRef]
- Burnouf, T.; Goubran, H.A.; Chou, M.L.; Devos, D.; Radosevic, M. Platelet microparticles: Detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Rev. 2014, 28, 155–166. [Google Scholar] [CrossRef]
- Tao, S.C.; Guo, S.C.; Zhang, C.Q. Platelet-derived extracellular vesicles: An emerging therapeutic approach. Int. J. Biol. Sci. 2017, 13, 828–834. [Google Scholar] [CrossRef]
- Burnouf, T.; Agrahari, V.; Agrahari, V. Extracellular Vesicles as Nanomedicine: Hopes and Hurdles in Clinical Translation. Int. J. Nanomed. 2019, 14, 8847–8859. [Google Scholar] [CrossRef]
- Abrami, L.; Brandi, L.; Moayeri, M.; Brown, M.J.; Krantz, B.A.; Leppla, S.H.; van der Goot, F.G. Hijacking multivesicular bodies enables long-term and exosome-mediated long-distance action of anthrax toxin. Cell Rep. 2013, 5, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Pieters, B.C.; Arntz, O.J.; Bennink, M.B.; Broeren, M.G.; van Caam, A.P.; Koenders, M.I.; van Lent, P.L.; van den Berg, W.B.; de Vries, M.; van der Kraan, P.M.; et al. Commercial cow milk contains physically stable extracellular vesicles expressing immunoregulatory TGF-β. PLoS ONE 2015, 10, e0121123. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Shimizu, K.; Yamauchi, M.; Takase, H.; Ugawa, S.; Okada, A.; Inoshima, Y. Acidification effects on isolation of extracellular vesicles from bovine milk. PLoS ONE 2019, 14, e0222613. [Google Scholar] [CrossRef] [PubMed]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials-an ISEV position paper. J. Extracell. Vesicles 2015, 4, 31–41. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Hagiwara, K.; Takeshita, F.; Ochiya, T. Competitive interactions of cancer cells and normal cells via secretory micrornas. J. Biol. Chem. 2012, 287, 1397–1405. [Google Scholar] [CrossRef]
- Somiya, M.; Yoshioka, Y.; Ochiya, T. Drug delivery application of extracellular vesicles; insight into production, drug loading, targeting, and pharmacokinetics. Aims Bioeng. 2017, 4, 73–92. [Google Scholar] [CrossRef]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef]
- Hagiwara, K.; Katsuda, T.; Gailhouste, L.; Kosaka, N.; Ochiya, T. Commitment of annexin A2 in recruitment of micrornas into extracellular vesicles. FEBS Lett. 2015, 589, 4071–4078. [Google Scholar] [CrossRef]
- Shurtleff, M.J.; Temoche-Diaz, M.M.; Karfilis, K.V.; Ri, S.; Schekman, R. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife 2016, 5, e19276. [Google Scholar] [CrossRef]
- García-Manrique, P.; Matos, M.; Gutiérrez, G.; Pazos, C.; Blanco-López, M.C. Therapeutic biomaterials based on extracellular vesicles: Classification of bio-engineering and mimetic preparation routes. J. Extracell. Vesicles 2018, 7, 1422676. [Google Scholar]
- Delcayre, A.; Estelles, A.; Sperinde, J.; Roulon, T.; Paz, P.; Aguilar, B.; Villanueva, J.; Khine, S.; Le Pecq, J.B. Exosome display technology: Applications to the development of new diagnostics and therapeutics. Blood Cells Mol. Dis. 2005, 35, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- El-Say, K.M. Maximizing the encapsulation efficiency and the bioavailability of controlled-release cetirizine microspheres using Draper-Lin small composite design. Drug Des. Dev. 2016, 10, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Chen, A.Z.; Weng, L.J.; Chen, M.Y.; Xie, X.L. Effect of drug-loading methods on drug load, encapsulation efficiency and release properties of alginate/poly-L-arginine/chitosan ternary complex microcapsules. Macromol. Biosci. 2004, 4, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 2013, 172, 229–238. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 655–664. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Pinho, M.J.; Zuzarte, M.; Oliveira, C.; Pereira, P.; Sluijter, J.P.; Gomes, C.; Girao, H. Presence of CX43 in extracellular vesicles reduces the cardiotoxicity of the anti-tumour therapeutic approach with doxorubicin. J. Extracell. Vesicles 2016, 5, 1–12. [Google Scholar] [CrossRef]
- Lane, R.E.; Korbie, D.; Anderson, W.; Vaidyanathan, R.; Trau, M. Analysis of exosome purification methods using a model liposome system and tunable-resistive pulse sensing. Sci. Rep. 2015, 5, 7639. [Google Scholar] [CrossRef]
- Alanazi, F.K.; Lu, D.R.; Shakeel, F.; Haq, N. Density gradient separation of carborane-containing liposome from low density lipoprotein and detection by inductively coupled plasma spectrometry. J. Liposome Res. 2014, 24, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Miyanohara, A.; Friedmann, T. Enhanced gene transfer with fusogenic liposomes containing vesicular stomatitis virus G glycoprotein. J. Virol. 1998, 72, 6159–6163. [Google Scholar] [CrossRef] [PubMed]
- Antimisiaris, S.G.; Mourtas, S.; Marazioti, A. Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery. Pharmaceutics 2018, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Kriebardis, A.; Antonelou, M.; Stamoulis, K.; Papassideri, I. Cell-derived microparticles in stored blood products: Innocent-bystanders or effective mediators of post-transfusion reactions? Blood Transfus. 2012, 10, s25–s38. [Google Scholar] [PubMed]
- Danesh, A.; Inglis, H.C.; Jackman, R.P.; Wu, S.; Deng, X.; Muench, M.O.; Heitman, J.W.; Norris, P.J. Exosomes from red blood cell units bind to monocytes and induce proinflammatory cytokines, boosting T-cell responses in vitro. Blood 2014, 123, 687–696. [Google Scholar] [CrossRef]
- Van Balkom, B.W.M.; Gremmels, H.; Giebel, B.; Lim, S.K. Proteomic Signature of Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles. Proteomics 2019, 19, e1800163. [Google Scholar] [CrossRef]
- Murphy, D.E.; de Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Sato, Y.T.; Umezaki, K.; Sawada, S.; Mukai, S.A.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, J.; Gu, W.; Huang, Y.; Tong, Z.; Huang, L.; Tan, J. Exosome-Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs. Adv. Sci. 2018, 5, 1700611. [Google Scholar] [CrossRef] [PubMed]
- Rayamajhi, S.; Nguyen, T.D.T.; Marasini, R.; Aryal, S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019, 94, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Piffoux, M.; Silva, A.K.A.; Wilhelm, C.; Gazeau, F.; Tareste, D. Modification of Extracellular Vesicles by Fusion with Liposomes for the Design of Personalized Biogenic Drug Delivery Systems. ACS Nano 2018, 12, 6830–6842. [Google Scholar] [CrossRef] [PubMed]
- Sawada, S.I.; Sato, Y.T.; Kawasaki, R.; Yasuoka, J.I.; Mizuta, R.; Sasaki, Y.; Akiyoshi, K. Nanogel hybrid assembly for exosome intracellular delivery: Effects on endocytosis and fusion by exosome surface polymer engineering. Biomater. Sci. 2020, 8, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.C.; Gho, Y.S. Could bioengineered exosome- mimetic nanovesicles be an efficient strategy for the delivery of chemotherapeutics? Nanomedicine 2014, 9, 177–180. [Google Scholar] [CrossRef]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef]
- Avadhani, V. Mesothelioma—General. PathologyOutlines.com Website. Available online: http://www.pathologyoutlines.com/topic/pleuramesothelioma.html (accessed on 20 June 2019).
- Cappello, F.; Barnes, L. Synovial sarcoma and malignant mesothelioma of the pleura: Review, differential diagnosis and possible role of apoptosis. Pathology 2001, 33, 142–148. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to the 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef]
- Vimercati, L.; Cavone, D.; Delfino, M.C.; De Maria, L.; Caputi, A.; Ferri, G.M.; Serio, G. Asbestos exposure and malignant mesothelioma of the tunica vaginalis testis: A systematic review and the experience of the Apulia (southern Italy) mesothelioma register. Environ. Health 2019, 18, 78. [Google Scholar] [CrossRef]
- Marinaccio, A.; Consonni, D.; Mensi, C.; Mirabelli, D.; Migliore, E.; Magnani, C.; Di Marzio, D.; Gennaro, V.; Mazzoleni, G.; Girardi, P.; et al. Association between asbestos exposure and pericardial and tunica vaginalis testis malignant mesothelioma: A case-control study and epidemiological remarks. Scand. J. Work Environ. Health 2020, 7, 3895. [Google Scholar]
- Moore, A.J.; Parker, R.J.; Wiggins, J. Malignant mesothelioma. Orphanet. J. Rare Dis. 2008, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Delgermaa, V.; Takahash, K.; Park, E.K.; Le, G.V.; Hara, T.; Sorahan, T. Global mesothelioma deaths reported to the World Health Organization between 1994 and 2008. Bull. World Health Organ. 2011, 89, 716–724C. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, T.; Nelson, D.I.; Steenland, K.; Leigh, J.; Concha-Barrientos, M.; Fingerhut, M.; Prüss-Ustün, A. The global burden of disease due to occupational carcinogens. Am. J. Ind. Med. 2005, 48, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Marinaccio, A.; Corfiati, M.; Binazzi, A.; Di Marzio, D.; Bonafede, M.; Verardo, M.; Migliore, E.; Gennaro, V.; Mensi, C.; Schallemberg, G.; et al. The epidemiological surveillance of malignant mesothelioma in Italy (1993–2015): Methods, findings, and research perspectives. Epidemiol. Prev. 2020, 44, 23–30. [Google Scholar]
- Robinson, B.M. Malignant pleural mesothelioma: An epidemiological perspective. Ann. Cardiothorac. Surg. 2012, 1, 491–496. [Google Scholar]
- Bruno, R.; Alì, G.; Giannini, R.; Proietti, A.; Lucchi, M.; Chella, A.; Melfi, F.; Mussi, A.; Fontanini, G. Malignant pleural mesothelioma and mesothelial hyperplasia: A new molecular tool for the differential diagnosis. Oncotarget 2017, 8, 2758–2770. [Google Scholar] [CrossRef]
- Ikeda, K.; Tate, G.; Suzuki, T.; Kitamura, T.; Mitsuya, T. IMP3/L523S, a novel immunocytochemical marker that distinguishes benign and malignant cells: The expression profiles of IMP3/L523S in effusion cytology. Hum. Pathol. 2010, 41, 745–750. [Google Scholar] [CrossRef]
- Chang, S.; Oh, M.H.; Ji, S.Y.; Han, J.; Kim, T.J.; Eom, M.; Kwon, K.Y.; Ha, S.Y.; Choi, Y.D.; Lee, C.H.; et al. Practical utility of insulin-like growth factor II mRNA-binding protein 3, glucose transporter 1, and epithelial membrane antigen for distinguishing malignant mesotheliomas from benign mesothelial proliferations. Pathol. Int. 2014, 64, 607–612. [Google Scholar] [CrossRef]
- Husain, A.N.; Colby, T.V.; Ordóñez, N.G.; Allen, T.C.; Attanoos, R.L.; Beasley, M.B.; Butnor, K.J.; Chirieac, L.R.; Churg, A.M.; Dacic, S.; et al. Guidelines for Pathologic Diagnosis of Malignant Mesothelioma 2017 Update of the Consensus Statement From the International Mesothelioma Interest Group. Arch. Pathol. Lab. Med. 2018, 142, 89–108. [Google Scholar] [CrossRef]
- Nabeshima, K.; Matsumoto, S.; Hamasaki, M.; Hida, T.; Kamei, T.; Hiroshima, K.; Tsujimura, T.; Kawahara, K. Use of p16 FISH for differential diagnosis of mesothelioma in smear preparations. Diagn. Cytopathol. 2016, 44, 774–780. [Google Scholar] [CrossRef]
- Hwang, H.C.; Sheffield, B.S.; Rodriguez, S.; Thompson, K.; Tse, C.H.; Gown, A.M.; Churg, A. Utility of BAP1 Immunohistochemistry and p16 (CDKN2A) FISH in the Diagnosis of Malignant Mesothelioma in Effusion Cytology Specimens. Surg. Pathol. 2016, 40, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Serio, G.; Fortarezza, F.; Scattone, A.; Caporusso, C.; Punzi, A.; Cavone, D.; Pennella, A.; Marzullo, A.; Vimercati, L. Prognostic Value of Ki67 Percentage, WT-1 Expression and p16/CDKN2A Deletion in Diffuse Malignant Peritoneal Mesothelioma: A Single-Centre Cohort Study. Diagnostics 2020, 10, 386. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.; Ji, H.; Chen, M.; Robinson, B.W.; Dick, I.M.; Creaney, J.; Simpson, R.J. Secreted primary human malignant mesothelioma exosome signature reflects oncogenic cargo. Sci. Rep. 2016, 6, 32643. [Google Scholar] [CrossRef] [PubMed]
- Bard, M.P.; Hegmans, J.P.; Hemmes, A.; Luider, T.M.; Willemsen, R.; Severijnen, L.A.; van Meerbeeck, J.P.; Burgers, S.A.; Hoogsteden, H.C.; Lambrecht, B.N. Proteomic analysis of exosomes isolated from human malignant pleural effusions. Am. J. Respir. Cell Mol. Biol. 2004, 31, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Thayanithy, V.; Babatunde, V.; Dickson, E.L.; Wong, P.; Oh, S.; Ke, X.; Barlas, A.; Fujisawa, S.; Romin, Y.; Moreira, A.L.; et al. Tumor exosomes induce tunneling nanotubes in lipid raft-enriched regions of human mesothelioma cells. Exp. Cell Res. 2014, 323, 178–188. [Google Scholar] [CrossRef]
- Clayton, A.; Tabi, Z. Exosomes and the MICA-NKG2D system in cancer. Blood Cells Mol. Dis. 2005, 34, 206–213. [Google Scholar] [CrossRef]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef]
- Scherpereel, A.; Astoul, P.; Baas, P.; Berghmans, T.; Clayson, H.; de Vuyst, P.; Dienemann, H.; Galateau-Salle, F.; Hennequin, C.; Hillerdal, G.; et al. Guidelines of the European Respiratory Society and the European Society of Thoracic Surgeons for the management of malignant pleural mesothelioma. Eur. Respir. J. 2010, 35, 479–495. [Google Scholar] [CrossRef]
- Kovac, V.; Zwitter, M.; Zagar, T. Improved survival after introduction of chemotherapy for malignant pleural mesothelioma in Slovenia: Population-based survey of 444 patients. Radiol. Oncol. 2012, 46, 136–144. [Google Scholar] [CrossRef]
- Eccles, B.K.; Geldart, T.R.; Laurence, V.M.; Bradley, K.L.; Lwin, M.T. Experience of first- and subsequent-line systemic therapy in the treatment of non-small cell lung cancer. Ther. Adv. Med. Oncol. 2011, 3, 163–170. [Google Scholar] [CrossRef]
- Arrieta, Ó.; Medina, L.A.; Estrada-Lobato, E.; Hernández-Pedro, N.; Villanueva-Rodríguez, G.; Martínez-Barrera, L.; Macedo, E.O.; López-Rodríguez, V.; Motola-Kuba, D.; Corona-Cruz, J.F. First-line chemotherapy with liposomal doxorubicin plus cisplatin for patients with advanced malignant pleural mesothelioma: Phase II trial. Br. J. Cancer 2012, 106, 1027–1032. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Steele, J.P.; Nolan, L.; Gilligan, D.; Taylor, P.; Spicer, J.; Lind, M.; Mitra, S.; Shamash, J.; Phillips, M.M.; et al. Arginine Deprivation With Pegylated Arginine Deiminase in Patients with Argininosuccinate Synthetase 1-Deficient Malignant Pleural Mesothelioma: A Randomized Clinical Trial. JAMA Oncol. 2017, 3, 58–66. [Google Scholar] [CrossRef]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef]
- Jabalee, J.; Towle, R.; Garnis, C. The Role of Extracellular Vesicles in Cancer: Cargo, Function, and Therapeutic Implications. Cells 2018, 7, 93. [Google Scholar] [CrossRef]


{kind=link}
| Strategy | Advantages | Disadvantages | References |
|---|---|---|---|
| Nanoparticles | • Flexibility • High stability in vivo • Increased compound half-life • Hydrophilic and lipophilic compounds can be both loaded • Enhanced permeability of tumor vasculature facilitates nanoparticle delivery on the tumor site | • Depends on the nanoparticle type • Inorganic nanoparticles may trigger the immune-system | [19,20,21,22,23,24,25,26,27,28] |
| Liposomes | • Similarity with cell membrane • High variety of drug encapsulation • Low systemic toxicity | • Sensitivity to sterilization methods • Low stability in circulation • Low reproducibility in liposome loading and size control • Short shelf-life | [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43] |
| Polymer Conjugated Drugs | • Increased compound half-life • Increased high-dose drug tolerance • Increased specificity | • Deep knowledge of polymer–receptor molecular interactions required • Poor information about long-term side effects | [44,45,46,47,48,49,50,51,52,53,54] |
| Small molecules, peptides and antibodies | • Various anticancer effects • High cell permeability • Low systemic toxicity | • Depends on the small molecule type • Size-influenced pharmacokinetics • Short half-life | [55,56,57,58,59,60,61,62,63,64,65,66,67,68,69] |
| ADC | • High specificity • Remarkable results for AB working as single entities | • May trigger the immune system • High production costs • Some clinical trials showed no significant improvement of patient outcomes compared to canonical therapies | [4,70,71,72,73,74,75,76,77,78] |
| Reconfigurable Organisms | • Extremely configurable organisms • High drug loading efficiency • Autonomous, self-repairing system | • Machine learning is still unripe • Research in this field is still in the embryonic stage | [79] |
| EVs | • High biocompatibility due to the endogenous origin • High specificity • Can be highly modified in order to enhance or modify tissue specificity • Due to the phospholipid bilayer, they can easily fuse directly to the target’s plasma membrane • Presence of specific surface markers that preserve them from phagocytosis • Functional complexity, not easy to artificially replicate | • Generally low efficiency of EVs isolation methods • Lack of clinical evaluations • Lack of standardized isolation methods • Deepening of the understanding of intracellular production/packaging mechanisms still ongoing | [80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgio, S.; Noori, L.; Marino Gammazza, A.; Campanella, C.; Logozzi, M.; Fais, S.; Bucchieri, F.; Cappello, F.; Caruso Bavisotto, C. Extracellular Vesicles-Based Drug Delivery Systems: A New Challenge and the Exemplum of Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2020, 21, 5432. https://doi.org/10.3390/ijms21155432
Burgio S, Noori L, Marino Gammazza A, Campanella C, Logozzi M, Fais S, Bucchieri F, Cappello F, Caruso Bavisotto C. Extracellular Vesicles-Based Drug Delivery Systems: A New Challenge and the Exemplum of Malignant Pleural Mesothelioma. International Journal of Molecular Sciences. 2020; 21(15):5432. https://doi.org/10.3390/ijms21155432
Chicago/Turabian StyleBurgio, Stefano, Leila Noori, Antonella Marino Gammazza, Claudia Campanella, Mariantonia Logozzi, Stefano Fais, Fabio Bucchieri, Francesco Cappello, and Celeste Caruso Bavisotto. 2020. "Extracellular Vesicles-Based Drug Delivery Systems: A New Challenge and the Exemplum of Malignant Pleural Mesothelioma" International Journal of Molecular Sciences 21, no. 15: 5432. https://doi.org/10.3390/ijms21155432
APA StyleBurgio, S., Noori, L., Marino Gammazza, A., Campanella, C., Logozzi, M., Fais, S., Bucchieri, F., Cappello, F., & Caruso Bavisotto, C. (2020). Extracellular Vesicles-Based Drug Delivery Systems: A New Challenge and the Exemplum of Malignant Pleural Mesothelioma. International Journal of Molecular Sciences, 21(15), 5432. https://doi.org/10.3390/ijms21155432