Genetics in Cartilage Lesions: Basic Science and Therapy Approaches

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Genetic Basis of Cartilage Pathology
3. Diagnostic and Therapeutic Biomarkers
3.1. Metalloproteinases
3.2. Growth Factors
3.3. Inflammatory Factors
4. Types of Genetic Changes
4.1. Single Nucleotide Polymorphisms
4.2. Epigenetic Changes
5. Gene Therapy Approaches
5.1. Marrow Stimulation
5.2. Autologous Chondrocyte Implantation
5.3. Muscle and Fat Grafts
5.4. Scaffolds
5.5. Mesenchymal Stem Cells
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Fox, A.J.S.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sports Heal. A Multidiscip. Approach 2009, 1, 461–468. [Google Scholar] [CrossRef]
- Cudic, M.; Fields, G.B. Extracellular proteases as targets for drug development. Curr. Protein Pept. Sci. 2009, 10, 297–307. [Google Scholar] [CrossRef]
- Gill, S.E.; Parks, W.C. Metalloproteinases and their inhibitors: Regulators of wound healing. Int. J. Biochem. Cell Biol. 2008, 40, 1334–1347. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 1–19. [Google Scholar] [CrossRef]
- Osiecka-Iwan, A.; Hyc, A.; Radomska-Lesniewska, D.M.; Rymarczyk, A.; Skopinski, P. Antigenic and immunogenic properties of chondrocytes. Implications for chondrocyte therapeutic transplantation and pathogenesis of inflammatory and degenerative joint diseases. Cent Eur. J. Immunol. 2018, 43, 209–219. [Google Scholar] [CrossRef]
- Musumeci, G.; Aiello, F.; Szychlinska, M.; Di Rosa, M.; Castrogiovanni, P.; Mobasheri, A. Osteoarthritis in the XXIst Century: Risk Factors and Behaviours that Influence Disease Onset and Progression. Int. J. Mol. Sci. 2015, 16, 6093–6112. [Google Scholar] [CrossRef]
- Dwivedi, S.; Purohit, P.; Misra, R.; Pareek, P.; Goel, A.; Khattri, S.; Pant, K.K.; Misra, S.; Sharma, P. Diseases and Molecular Diagnostics: A Step Closer to Precision Medicine. Indian J. Clin. Biochem. 2017, 32, 374–398. [Google Scholar] [CrossRef]
- Gasperskaja, E.; Kučinskas, V. The most common technologies and tools for functional genome analysis. Acta Med. Litu. 2017, 24, 1–11. [Google Scholar] [CrossRef]
- Peffers, M.; Liu, X.; Clegg, P. Transcriptomic signatures in cartilage ageing. Arthritis Res. Ther. 2013, 15. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-Throughput Sequencing Technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef]
- Coutinho de Almeida, R.; Ramos, Y.F.M.; Mahfouz, A.; den Hollander, W.; Lakenberg, N.; Houtman, E.; van Hoolwerff, M.; Suchiman, H.E.D.; Rodríguez Ruiz, A.; Slagboom, P.E.; et al. RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage. Ann. Rheum. Dis. 2019, 78, 270–277. [Google Scholar] [CrossRef]
- Lotz, M.; Martel-Pelletier, J.; Christiansen, C.; Brandi, M.L.; Bruyere, O.; Chapurlat, R.; Collette, J.; Cooper, C.; Giacovelli, G.; Kanis, J.A.; et al. Value of biomarkers in osteoarthritis: Current status and perspectives. Ann. Rheum. Dis. 2013, 72, 1756–1763. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Sharma, A.R.; Chakraborty, C.; Saibaba, B.; Ahn, M.E.; Lee, S.S. Review of Prospects of Biological Fluid Biomarkers in Osteoarthritis. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Furumatsu, T.; Matsumoto-Ogawa, E.; Tanaka, T.; Lu, Z.; Ozaki, T. ROCK inhibition enhances aggrecan deposition and suppresses matrix metalloproteinase-3 production in human articular chondrocytes. Connect Tissue Res. 2014, 55, 89–95. [Google Scholar] [CrossRef]
- Long, D.; Blake, S.; Song, X.Y.; Lark, M.; Loeser, R.F. Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res. 2008, 10, R23. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, D.K.; Shin, H.D.; Lee, H.J.; Jo, H.S.; Jeong, J.H.; Choi, Y.L.; Lee, C.J.; Hwang, S.C. Apigenin Regulates Interleukin-1beta-Induced Production of Matrix Metalloproteinase Both in the Knee Joint of Rat and in Primary Cultured Articular Chondrocytes. Biomol. Ther. (Seoul) 2016, 24, 163–170. [Google Scholar] [CrossRef]
- Prasadam, I.; Crawford, R.; Xiao, Y. Aggravation of ADAMTS and matrix metalloproteinase production and role of ERK1/2 pathway in the interaction of osteoarthritic subchondral bone osteoblasts and articular cartilage chondrocytes -- possible pathogenic role in osteoarthritis. J. Rheumatol. 2012, 39, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Tetlow, L.C.; Woolley, D.E. Histamine stimulates matrix metalloproteinase-3 and -13 production by human articular chondrocytes in vitro. Ann. Rheum. Dis. 2002, 61, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Yammani, R.R.; Carlson, C.S.; Bresnick, A.R.; Loeser, R.F. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: Role of the receptor for advanced glycation end products. Arthritis Rheum. 2006, 54, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, S.; Su, H.; Cheng, J. Isoliquiritigenin Inhibits IL-1beta-Induced Production of Matrix Metalloproteinase in Articular Chondrocytes. Mol. Ther. Methods Clin. Dev. 2018, 9, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Cole, A.; Murphy, G.; Bienias, J.L.; Im, H.J.; Loeser, R.F., Jr. Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, D.; Yuan, Y.; Min, J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res. Ther. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, N.G.; Shi, Z.H.; Tang, Y.P.; Wang, Z.J.; Song, S.L.; Qian, L.H.; Qian, D.W.; Duan, J.A. New Hope for the Treatment of Osteoarthritis Through Selective Inhibition of MMP-13. Curr. Med. Chem. 2011, 18, 977–1001. [Google Scholar] [CrossRef]
- Lee, A.S.; Ellman, M.B.; Yan, D.; Kroin, J.S.; Cole, B.J.; van Wijnen, A.J.; Im, H.J. A current review of molecular mechanisms regarding osteoarthritis and pain. Gene 2013, 527, 440–447. [Google Scholar] [CrossRef]
- Goldring, M.B.; Berenbaum, F. The regulation of chondrocyte function by proinflammatory mediators: Prostaglandins and nitric oxide. Clin. Orthop. Relat. Res. 2004, S37–S46. [Google Scholar] [CrossRef]
- Wang, X.; Hunter, D.; Xu, J.; Ding, C. Metabolic triggered inflammation in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 22–30. [Google Scholar] [CrossRef]
- Arai, Y.; Kubo, T.; Kobayashi, K.; Takeshita, K.; Takahashi, K.; Ikeda, T.; Imanishi, J.; Takigawa, M.; Hirasawa, Y. Adenovirus vector-mediated gene transduction to chondrocytes: In vitro evaluation of therapeutic efficacy of transforming growth factor-beta 1 and heat shock protein 70 gene transduction. J. Rheumatol. 1997, 24, 1787–1795. [Google Scholar]
- Alaaeddine, N.; Di Battista, J.A.; Pelletier, J.-P.; Kiansa, K.; Cloutier, J.-M.; Martel-Pelletier, J. Differential Effects of Il-8, Lif (Pro-Inflammatory) and Il-11 (Anti-Inflammatory) on Tnf-α-Induced Pge2release and on Signalling Pathways in Human Oa Synovial Fibroblasts. Cytokine 1999, 11, 1020–1030. [Google Scholar] [CrossRef]
- Fleischmann, R.M.; Schechtman, J.; Bennett, R.; Handel, M.L.; Burmester, G.-R.; Tesser, J.; Modafferi, D.; Poulakos, J.; Sun, G. Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: A large, international, multicenter, placebo-controlled trial. Arthritis Rheum. 2003, 48, 927–934. [Google Scholar] [CrossRef]
- Kafienah, W.; Al-Fayez, F.; Hollander, A.P.; Barker, M.D. Inhibition of cartilage degradation: A combined tissue engineering and gene therapy approach. Arthritis Rheum. 2003, 48, 709–718. [Google Scholar] [CrossRef]
- Lotz, M. Cytokines in Cartilage Injury and Repair. Clin. Orthop. Relat. Res. 2001, 391, S108–S115. [Google Scholar] [CrossRef]
- Robbins, P.D.; Evans, C.H.; Chernajovsky, Y. Gene therapy for arthritis. Gene Ther. 2003, 10, 902–911. [Google Scholar] [CrossRef]
- Rudolphi, K.; Gerwin, N.; Verzijl, N.; van der Kraan, P.; van den Berg, W. Pralnacasan, an inhibitor of interleukin-1β converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthr. Cartil. 2003, 11, 738–746. [Google Scholar] [CrossRef]
- Neumann, E.; Judex, M.; Kullmann, F.; Grifka, J.; Robbins, P.D.; Pap, T.; Gay, R.E.; Evans, C.H.; Gay, S.; Schölmerich, J.; et al. Inhibition of cartilage destruction by double gene transfer of IL-1Ra and IL-10 involves the activin pathway. Gene Ther. 2002, 9, 1508–1519. [Google Scholar] [CrossRef]
- Nishimura, R.; Hata, K.; Takahata, Y.; Murakami, T.; Nakamura, E.; Ohkawa, M.; Ruengsinpinya, L. Role of Signal Transduction Pathways and Transcription Factors in Cartilage and Joint Diseases. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Kitoh, H.; Sugiura, F.; Ishiguro, N. Chondrogenesis enhanced by overexpression of sox9 gene in mouse bone marrow-derived mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2003, 301, 338–343. [Google Scholar] [CrossRef]
- Tew, S.R.; Li, Y.; Pothacharoen, P.; Tweats, L.M.; Hawkins, R.E.; Hardingham, T.E. Retroviral transduction with SOX9 enhances re-expression of the chondrocyte phenotype in passaged osteoarthritic human articular chondrocytes. Osteoarthr. Cartil. 2005, 13, 80–89. [Google Scholar] [CrossRef]
- Li, Y.; Tew, S.R.; Russell, A.M.; Gonzalez, K.R.; Hardingham, T.E.; Hawkins, R.E. Transduction of Passaged Human Articular Chondrocytes with Adenoviral, Retroviral, and Lentiviral Vectors and the Effects of Enhanced Expression of SOX9. Tissue Eng. 2004, 10, 575–584. [Google Scholar] [CrossRef]
- Cucchiarini, M.; Thurn, T.; Weimer, A.; Kohn, D.; Terwilliger, E.F.; Madry, H. Restoration of the extracellular matrix in human osteoarthritic articular cartilage by overexpression of the transcription factorSOX9. Arthritis Rheum. 2007, 56, 158–167. [Google Scholar] [CrossRef]
- Minina, E.; Wenzel, H.M.; Kreschel, C.; Karp, S.; Gaffield, W.; McMahon, A.P.; Vortkamp, A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 2001, 128, 4523–4534. [Google Scholar] [PubMed]
- Grande, D.A.; Mason, J.; Light, E.; Dines, D. Stem cells as platforms for delivery of genes to enhance cartilage repair. J. Bone Jt Surg. Am. 2003, 85-A (Suppl. S2), 111–116. [Google Scholar] [CrossRef] [PubMed]
- Tavella, S.; Biticchi, R.; Schito, A.; Minina, E.; Di Martino, D.; Pagano, A.; Vortkamp, A.; Horton, W.A.; Cancedda, R.; Garofalo, S. Targeted expression of SHH affects chondrocyte differentiation, growth plate organization, and Sox9 expression. J. Bone Min. Res. 2004, 19, 1678–1688. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A. Interaction of growth factors regulating chondrocyte differentiation in the developing embryo. Osteoarthr. Cartil. 2001, 9 (Suppl. SA), S109–S117. [Google Scholar] [PubMed]
- Scharstuhl, A.; Diepens, R.; Lensen, J.; Vitters, E.; van Beuningen, H.; van der Kraan, P.; van den Berg, W. Adenoviral overexpression of Smad-7 and Smad-6 differentially regulates TGF-beta-mediated chondrocyte proliferation and proteoglycan synthesis. Osteoarthr. Cartil. 2003, 11, 773–782. [Google Scholar] [CrossRef]
- Scharstuhl, A.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Reduction of osteophyte formation and synovial thickening by adenoviral overexpression of transforming growth factor beta/bone morphogenetic protein inhibitors during experimental osteoarthritis. Arthritis Rheum. 2003, 48, 3442–3451. [Google Scholar] [CrossRef]
- D’Lima, D.D.; Hashimoto, S.; Chen, P.C.; Lotz, M.K.; Colwell, C.W. Cartilage Injury Induces Chondrocyte Apoptosis. J. Bone Jt. Surg. Am. 2001, 83, 19–21. [Google Scholar] [CrossRef]
- Yao, Q.; Wang, S.; Gambotto, A.; Glorioso, J.C.; Evans, C.H.; Robbins, P.D.; Ghivizzani, S.C.; Oligino, T.J. Intra-articular adenoviral-mediated gene transfer of trail induces apoptosis of arthritic rabbit synovium. Gene Ther. 2003, 10, 1055–1060. [Google Scholar] [CrossRef][Green Version]
- Lotz, M.; Hashimoto, S.; Kühn, K. Mechanisms of chondrocyte apoptosis. Osteoarthr. Cartil. 1999, 7, 389–391. [Google Scholar] [CrossRef]
- Yao, Q.; Glorioso, J.C.; Evans, C.H.; Robbins, P.D.; Kovesdi, I.; Oligino, T.J.; Ghivizzani, S.C. Adenoviral mediated delivery of FAS ligand to arthritic joints causes extensive apoptosis in the synovial lining. J. Gene Med. 2000, 2, 210–219. [Google Scholar] [CrossRef]
- Kobayashi, H.; Hirata, M.; Saito, T.; Itoh, S.; Chung, U.I.; Kawaguchi, H. Transcriptional induction of ADAMTS5 protein by nuclear factor-kappaB (NF-kappaB) family member RelA/p65 in chondrocytes during osteoarthritis development. J. Biol. Chem. 2013, 288, 28620–28629. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef]
- Akiyama, H. Interactions between Sox9 and -catenin control chondrocyte differentiation. Genes Dev. 2004, 18, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Loughlin, J.; Dowling, B.; Chapman, K.; Marcelline, L.; Mustafa, Z.; Southam, L.; Ferreira, A.; Ciesielski, C.; Carson, D.A.; Corr, M. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc. Natl. Acad. Sci. USA 2004, 101, 9757–9762. [Google Scholar] [CrossRef] [PubMed]
- Terashima, A.; Takayanagi, H. Overview of Osteoimmunology. Calcif. Tissue Int. 2018, 102, 503–511. [Google Scholar] [CrossRef]
- Romas, E.; Gillespie, M.T.; Martin, T.J. Involvement of receptor activator of NFkappaB ligand and tumor necrosis factor-alpha in bone destruction in rheumatoid arthritis. Bone 2002, 30, 340–346. [Google Scholar] [CrossRef]
- Babaei, M.; Javadian, Y.; Narimani, H.; Ranaei, M.; Heidari, B.; Basereh, H.; Gholinia, H.; Firouzjahi, A. Correlation between systemic markers of inflammation and local synovitis in knee osteoarthritis. Casp. J. Intern Med. 2019, 10, 383–387. [Google Scholar] [CrossRef]
- Nishimura, R.; Hata, K.; Nakamura, E.; Murakami, T.; Takahata, Y. Transcriptional network systems in cartilage development and disease. Histochem. Cell Biol. 2018, 149, 353–363. [Google Scholar] [CrossRef]
- Rose, B.J.; Kooyman, D.L. A Tale of Two Joints: The Role of Matrix Metalloproteases in Cartilage Biology. Dis. Markers 2016, 2016, 4895050. [Google Scholar] [CrossRef]
- Yang, C.Y.; Chanalaris, A.; Troeberg, L. ADAMTS and ADAM metalloproteinases in osteoarthritis–looking beyond the ‘usual suspects’. Osteoarth. Cart 2017, 25, 1000–1009. [Google Scholar] [CrossRef]
- Kevorkian, L.; Young, D.A.; Darrah, C.; Donell, S.T.; Shepstone, L.; Porter, S.; Brockbank, S.M.; Edwards, D.R.; Parker, A.E.; Clark, I.M. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004, 50, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Lee, M.H. What are the roles of metalloproteinases in cartilage and bone damage? Ann. Rheum. Dis. 2005, 64 (Suppl. S4), iv44–iv47. [Google Scholar] [CrossRef]
- Young, D.A.; Barter, M.J.; Wilkinson, D.J. Recent advances in understanding the regulation of metalloproteinases. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Huang, W.; Dehnade, F.; Ahmad, M.; Zafarullah, M. Induction of matrix metalloproteinase-13 gene expression by TNF-alpha is mediated by MAP kinases, AP-1, and NF-kappaB transcription factors in articular chondrocytes. Exp. Cell Res. 2003, 288, 208–217. [Google Scholar] [CrossRef]
- Vincenti, M.P.; Brinckerhoff, C.E. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: Integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002, 4, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Burrage, P.S.; Mix, K.S.; Brinckerhoff, C.E. Matrix metalloproteinases: Role in arthritis. Front Biosci. 2006, 11, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, E.; Malemud, C.J. Prospects for treating osteoarthritis: Enzyme–protein interactions regulating matrix metalloproteinase activity. Ther. Adv. Chronic Dis. 2012, 3, 219–229. [Google Scholar] [CrossRef]
- Moore, C.S.; Crocker, S.J. An alternate perspective on the roles of TIMPs and MMPs in pathology. Am. J. Pathol. 2012, 180, 12–16. [Google Scholar] [CrossRef]
- Clutterbuck, A.L.; Asplin, K.E.; Harris, P.; Allaway, D.; Mobasheri, A. Targeting matrix metalloproteinases in inflammatory conditions. Curr. Drug Targets 2009, 10, 1245–1254. [Google Scholar] [CrossRef]
- Murphy, G.; Knauper, V.; Atkinson, S.; Butler, G.; English, W.; Hutton, M.; Stracke, J.; Clark, I. Matrix metalloproteinases in arthritic disease. Arthritis Res. 2002, 4 (Suppl. S3), S39–S49. [Google Scholar] [CrossRef]
- Ye, S.; Henney, A.M. Detecting polymorphisms in MMP genes. Methods Mol. Biol. 2001, 151, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Honsawek, S.; Malila, S.; Yuktanandana, P.; Tanavalee, A.; Deepaisarnsakul, B.; Parvizi, J. Association of MMP-3 (-1612 5A/6A) polymorphism with knee osteoarthritis in Thai population. Rheumatol. Int. 2013, 33, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Xu, P.; Jin, T.; Wang, J.; Fan, D.; Hao, Z.; Ji, Y.; Jing, S.; Han, C.; Du, J.; et al. MMP-3 gene polymorphisms are associated with increased risk of osteoarthritis in Chinese men. Oncotarget 2017, 8, 79491–79497. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Zhao, J.; Bao, N. The effect of single nucleotide polymorphism on susceptibility of osteoarthritis: Recent progression and implications. Ann. Jt. 2017, 2, 52. [Google Scholar] [CrossRef]
- Salminen, H.J.; Saamanen, A.M.; Vankemmelbeke, M.N.; Auho, P.K.; Perala, M.P.; Vuorio, E.I. Differential expression patterns of matrix metalloproteinases and their inhibitors during development of osteoarthritis in a transgenic mouse model. Ann. Rheum. Dis. 2002, 61, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Cheon, E.J.; Lee, M.H.; Kim, H.A. MicroRNA-127-5p Regulates Matrix Metalloproteinase 13 Expression and Interleukin-1β-Induced Catabolic Effects in Human Chondrocytes. Arthritis Rheum. 2013, 65, 3141–3152. [Google Scholar] [CrossRef]
- Akhtar, N.; Rasheed, Z.; Ramamurthy, S.; Anbazhagan, A.N.; Voss, F.R.; Haqqi, T.M. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010, 62, 1361–1371. [Google Scholar] [CrossRef]
- Miyaki, S.; Sato, T.; Inoue, A.; Otsuki, S.; Ito, Y.; Yokoyama, S.; Kato, Y.; Takemoto, F.; Nakasa, T.; Yamashita, S.; et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010, 24, 1173–1185. [Google Scholar] [CrossRef]
- Si, H.B.; Zeng, Y.; Liu, S.Y.; Zhou, Z.K.; Chen, Y.N.; Cheng, J.Q.; Lu, Y.R.; Shen, B. Intra-articular injection of microRNA-140 (miRNA-140) alleviates osteoarthritis (OA) progression by modulating extracellular matrix (ECM) homeostasis in rats. Osteoarthr. Cartil. 2017, 25, 1698–1707. [Google Scholar] [CrossRef]
- van Meurs, J.; van Lent, P.; Stoop, R.; Holthuysen, A.; Singer, I.; Bayne, E.; Mudgett, J.; Poole, R.; Billinghurst, C.; van der Kraan, P.; et al. Cleavage of aggrecan at the Asn341-Phe342 site coincides with the initiation of collagen damage in murine antigen-induced arthritis: A pivotal role for stromelysin 1 in matrix metalloproteinase activity. Arthritis Rheum. 1999, 42, 2074–2084. [Google Scholar] [CrossRef]
- van Meurs, J.; van Lent, P.; Holthuysen, A.; Lambrou, D.; Bayne, E.; Singer, I.; van den Berg, W. Active matrix metalloproteinases are present in cartilage during immune complex-mediated arthritis: A pivotal role for stromelysin-1 in cartilage destruction. J. Immunol. 1999, 163, 5633–5639. [Google Scholar]
- Leong, D.J.; Gu, X.I.; Li, Y.; Lee, J.Y.; Laudier, D.M.; Majeska, R.J.; Schaffler, M.B.; Cardoso, L.; Sun, H.B. Matrix metalloproteinase-3 in articular cartilage is upregulated by joint immobilization and suppressed by passive joint motion. Matrix Biol. 2010, 29, 420–426. [Google Scholar] [CrossRef]
- Elliott, S.; Cawston, T. The clinical potential of matrix metalloproteinase inhibitors in the rheumatic disorders. Drugs Aging 2001, 18, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.G.; Andrews, R.C.; Beaudet, B.; Bickett, D.M.; Boncek, V.; Brodie, T.A.; Clark, R.L.; Crumrine, R.C.; Leenitzer, M.A.; McDougald, D.L.; et al. Inhibition of tumor necrosis factor-alpha (TNF-alpha) production and arthritis in the rat by GW3333, a dual inhibitor of TNF-alpha-converting enzyme and matrix metalloproteinases. J. Pharm. Exp. Ther. 2001, 298, 900–908. [Google Scholar]
- Borden, P.; Heller, R.A. Transcriptional control of matrix metalloproteinases and the tissue inhibitors of matrix metalloproteinases. Crit. Rev. Eukaryot Gene Expr. 1997, 7, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Guibert, M.; Cailotto, F.; Gasser, A.; Presle, N.; Mainard, D.; Netter, P.; Kempf, H.; Jouzeau, J.Y.; Reboul, P. Fibroblast Growth Factor 23 drives MMP13 expression in human osteoarthritic chondrocytes in a Klotho-independent manner. Osteoarthr. Cartil. 2016, 24, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Tuan, R.S.; Chen, A.F.; Klatt, B.A. Cartilage regeneration. J. Am. Acad. Orthop. Surg. 2013, 21, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Fortier, L.A.; Barker, J.U.; Strauss, E.J.; McCarrel, T.M.; Cole, B.J. The role of growth factors in cartilage repair. Clin. Orthop. Relat. Res. 2011, 469, 2706–2715. [Google Scholar] [CrossRef]
- Chia, S.-L.; Sawaji, Y.; Burleigh, A.; McLean, C.; Inglis, J.; Saklatvala, J.; Vincent, T. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheum. 2009, 60, 2019–2027. [Google Scholar] [CrossRef]
- Goodrich, L.R.; Hidaka, C.; Robbins, P.D.; Evans, C.H.; Nixon, A.J. Genetic modification of chondrocytes with insulin-like growth factor-1 enhances cartilage healing in an equine model. J. Bone Jt. Surg. Br. Vol. 2007, 89-B, 672–685. [Google Scholar] [CrossRef]
- Longo, U.G.; Petrillo, S.; Franceschetti, E.; Berton, A.; Maffulli, N.; Denaro, V. Stem Cells and Gene Therapy for Cartilage Repair. Stem Cells Int. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.B.; Chen, E.H.; Lynch, S.E. A review of the effects of insulin-like growth factor and platelet derived growth factor on in vivo cartilage healing and repair. Osteoarthr. Cartil. 2006, 14, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Sawaji, Y.; Hynes, J.; Vincent, T.; Saklatvala, J. Fibroblast growth factor 2 inhibits induction of aggrecanase activity in human articular cartilage. Arthritis Rheum. 2008, 58, 3498–3509. [Google Scholar] [CrossRef]
- Fortier, L.A.; Balkman, C.E.; Sandell, L.J.; Ratcliffe, A.; Nixon, A.J. Insulin-like growth factor-I gene expression patterns during spontaneous repair of acute articular cartilage injury. J. Orthop. Res. 2001, 19, 720–728. [Google Scholar] [CrossRef]
- Morisset, S.; Frisbie, D.D.; Robbins, P.D.; Nixon, A.J.; McIlwraith, C.W. IL-1ra/IGF-1 Gene Therapy Modulates Repair of Microfractured Chondral Defects. Clin. Orthop. Relat. Res. 2007, 462, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hunter, D.J.; Jin, X.; Ding, C. The importance of synovial inflammation in osteoarthritis: Current evidence from imaging assessments and clinical trials. Osteoarthr. Cartil. 2018, 26, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M.N.M. The crucial roles of inflammatory mediators in inflammation: A review. Vet World 2018, 11, 627–635. [Google Scholar] [CrossRef]
- Scanzello, C.R.; McKeon, B.; Swaim, B.H.; DiCarlo, E.; Asomugha, E.U.; Kanda, V.; Nair, A.; Lee, D.M.; Richmond, J.C.; Katz, J.N.; et al. Synovial inflammation in patients undergoing arthroscopic meniscectomy: Molecular characterization and relationship to symptoms. Arthritis Rheum. 2011, 63, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Henrotin, Y.; Biesalski, H.K.; Shakibaei, M. Scientific evidence and rationale for the development of curcumin and resveratrol as nutraceutricals for joint health. Int. J. Mol. Sci. 2012, 13, 4202–4232. [Google Scholar] [CrossRef] [PubMed]
- Madson, K.L.; Moore, T.L.; Lawrence, J.M., 3rd; Osborn, T.G. Cytokine levels in serum and synovial fluid of patients with juvenile rheumatoid arthritis. J. Rheumatol. 1994, 21, 2359–2363. [Google Scholar] [PubMed]
- Perry, M.G.; Jessop, D.S.; Hunt, L.P.; Sharif, M.; Kirwan, J.R. Overnight variations in IL-6 in synovial fluid and plasma in patients with active rheumatoid arthritis. Int. J. Clin. Rheumatol. 2010, 5, 593–600. [Google Scholar] [CrossRef]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef]
- Fingleton, B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 2036–2042. [Google Scholar] [CrossRef]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef]
- Maldonado, M.; Nam, J. The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. Biomed. Res. Int. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Goldring, M.B.; Otero, M.; Tsuchimochi, K.; Ijiri, K.; Li, Y. Defining the roles of inflammatory and anabolic cytokines in cartilage metabolism. Ann. Rheum. Dis. 2008, 67 (Suppl. S3), iii75–iii82. [Google Scholar] [CrossRef]
- Ayral, X.; Pickering, E.H.; Woodworth, T.G.; Mackillop, N.; Dougados, M. Synovitis: A potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis-results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthr. Cartil. 2005, 13, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.; Dunlop, D.D.; Peterfy, C.; Guermazi, A.; Prasad, P.; Hayes, K.W.; Song, J.; Cahue, S.; Chang, A.; Marshall, M.; et al. The relationship between specific tissue lesions and pain severity in persons with knee osteoarthritis. Osteoarthr. Cartil. 2006, 14, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Benito, M.J.; Veale, D.J.; FitzGerald, O.; van den Berg, W.B.; Bresnihan, B. Synovial tissue inflammation in early and late osteoarthritis. Ann. Rheum. Dis. 2005, 64, 1263–1267. [Google Scholar] [CrossRef]
- Tong, Z.; Liu, Y.; Chen, B.; Yan, L.; Hao, D. Association between MMP3 and TIMP3 polymorphisms and risk of osteoarthritis. Oncotarget 2017, 8, 83563–83569. [Google Scholar] [CrossRef] [PubMed]
- Nakki, A.; Rodriguez-Fontenla, C.; Gonzalez, A.; Harilainen, A.; Leino-Arjas, P.; Heliovaara, M.; Eriksson, J.G.; Tallroth, K.; Videman, T.; Kaprio, J.; et al. Association study of MMP8 gene in osteoarthritis. Connect Tissue Res. 2016, 57, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Su, S.L.; Yang, H.Y.; Lee, H.S.; Huang, G.S.; Lee, C.H.; Liu, W.S.; Wang, C.C.; Peng, Y.J.; Lai, C.H.; Chen, C.Y.; et al. Gene-gene interactions between TGF-beta/Smad3 signalling pathway polymorphisms affect susceptibility to knee osteoarthritis. BMJ Open 2015, 5, e007931. [Google Scholar] [CrossRef]
- Rodriguez-Fontenla, C.; Calaza, M.; Evangelou, E.; Valdes, A.M.; Arden, N.; Blanco, F.J.; Carr, A.; Chapman, K.; Deloukas, P.; Doherty, M.; et al. Assessment of Osteoarthritis Candidate Genes in a Meta-Analysis of Nine Genome-Wide Association Studies. Arthritis Rheumatol. 2014, 66, 940–949. [Google Scholar] [CrossRef]
- Nakki, A.; Videman, T.; Kujala, U.M.; Suhonen, M.; Mannikko, M.; Peltonen, L.; Battie, M.C.; Kaprio, J.; Saarela, J. Candidate gene association study of magnetic resonance imaging-based hip osteoarthritis (OA): Evidence for COL9A2 gene as a common predisposing factor for hip OA and lumbar disc degeneration. J. Rheumatol. 2011, 38, 747–752. [Google Scholar] [CrossRef]
- Zhu, L.; Shi, D.; Dai, J.; Qin, J.; Fan, J.; Wang, Z.; Qiu, X.; Xu, Z.; Chen, D.; Jiang, Q. Lack of evidence for association between DVWA gene polymorphisms and developmental dysplasia of the hip in Chinese Han population. Rheumatol. Int. 2011, 31, 883–887. [Google Scholar] [CrossRef]
- Valdes, A.M.; Spector, T.D.; Doherty, S.; Wheeler, M.; Hart, D.J.; Doherty, M. Association of the DVWA and GDF5 polymorphisms with osteoarthritis in UK populations. Ann. Rheum. Dis. 2009, 68, 1916–1920. [Google Scholar] [CrossRef]
- Meulenbelt, I.; Chapman, K.; Dieguez-Gonzalez, R.; Shi, D.; Tsezou, A.; Dai, J.; Malizos, K.N.; Kloppenburg, M.; Carr, A.; Nakajima, M.; et al. Large replication study and meta-analyses of DVWA as an osteoarthritis susceptibility locus in European and Asian populations. Hum. Mol. Genet. 2009, 18, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Baker-Lepain, J.C.; Lynch, J.A.; Parimi, N.; McCulloch, C.E.; Nevitt, M.C.; Corr, M.; Lane, N.E. Variant alleles of the Wnt antagonist FRZB are determinants of hip shape and modify the relationship between hip shape and osteoarthritis. Arthritis Rheum. 2012, 64, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, E.; Chapman, K.; Meulenbelt, I.; Karassa, F.B.; Loughlin, J.; Carr, A.; Doherty, M.; Doherty, S.; Gomez-Reino, J.J.; Gonzalez, A.; et al. Large-scale analysis of association between GDF5 and FRZB variants and osteoarthritis of the hip, knee, and hand. Arthritis Rheum. 2009, 60, 1710–1721. [Google Scholar] [CrossRef]
- Zhang, R.; Yao, J.; Xu, P.; Ji, B.; Luck, J.V.; Chin, B.; Lu, S.; Kelsoe, J.R.; Ma, J. A comprehensive meta-analysis of association between genetic variants of GDF5 and osteoarthritis of the knee, hip and hand. Inflamm. Res. 2015, 64, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Mototani, H.; Mabuchi, A.; Saito, S.; Fujioka, M.; Iida, A.; Takatori, Y.; Kotani, A.; Kubo, T.; Nakamura, K.; Sekine, A.; et al. A functional single nucleotide polymorphism in the core promoter region of CALM1 is associated with hip osteoarthritis in Japanese. Hum. Mol. Genet. 2005, 14, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Poulou, M.; Kaliakatsos, M.; Tsezou, A.; Kanavakis, E.; Malizos, K.N.; Tzetis, M. Association of the CALM1 core promoter polymorphism with knee osteoarthritis in patients of Greek origin. Genet Test 2008, 12, 263–265. [Google Scholar] [CrossRef]
- Loughlin, J.; Sinsheimer, J.S.; Carr, A.; Chapman, K. The CALM1 core promoter polymorphism is not associated with hip osteoarthritis in a United Kingdom Caucasian population. Osteoarthr. Cartil. 2006, 14, 295–298. [Google Scholar] [CrossRef]
- Shi, D.; Ni, H.; Dai, J.; Qin, J.; Xu, Y.; Zhu, L.; Yao, C.; Shao, Z.; Chen, D.; Xu, Z.; et al. Lack of association between the CALM1 core promoter polymorphism (-16C/T) and susceptibility to knee osteoarthritis in a Chinese Han population. BMC Med. Genet 2008, 9, 91. [Google Scholar] [CrossRef]
- Liu, C.; Sun, J.; Zhang, H.; Li, L. TGF beta1 gene polymorphisms correlate with the susceptibility of osteoarthritis. Int. J. Clin. Exp. Pathol. 2017, 10, 8780–8785. [Google Scholar]
- Urano, T.; Narusawa, K.; Shiraki, M.; Usui, T.; Sasaki, N.; Hosoi, T.; Ouchi, Y.; Nakamura, T.; Inoue, S. Association of a single nucleotide polymorphism in the insulin-like growth factor-1 receptor gene with spinal disc degeneration in postmenopausal Japanese women. Spine (Phila Pa 1976) 2008, 33, 1256–1261. [Google Scholar] [CrossRef]
- Claessen, K.M.; Ramautar, S.R.; Pereira, A.M.; Smit, J.W.; Biermasz, N.R.; Kloppenburg, M. Relationship between insulin-like growth factor-1 and radiographic disease in patients with primary osteoarthritis: A systematic review. Osteoarthr. Cartil. 2012, 20, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Limer, K.L.; Tosh, K.; Bujac, S.R.; McConnell, R.; Doherty, S.; Nyberg, F.; Zhang, W.; Doherty, M.; Muir, K.R.; Maciewicz, R.A. Attempt to replicate published genetic associations in a large, well-defined osteoarthritis case-control population (the GOAL study). Osteoarthr. Cartil. 2009, 17, 782–789. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meulenbelt, I.; Bijkerk, C.; Miedema, H.S.; Breedveld, F.C.; Hofman, A.; Valkenburg, H.A.; Pols, H.A.; Slagboom, P.E.; van Duijn, C.M. A genetic association study of the IGF-1 gene and radiological osteoarthritis in a population-based cohort study (the Rotterdam Study). Ann. Rheum. Dis. 1998, 57, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Keen, L.J.; Billingham, M.J.; Perry, M.J.; Elson, C.J.; Kirwan, J.R.; Sims, J.E.; Doherty, M.; Spector, T.D.; Bidwell, J.L. Extended haplotypes and linkage disequilibrium in the IL1R1-IL1A-IL1B-IL1RN gene cluster: Association with knee osteoarthritis. Genes Immun. 2004, 5, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Moxley, G.; Han, J.; Stern, A.G.; Riley, B.P. Potential influence of IL1B haplotype and IL1A-IL1B-IL1RN extended haplotype on hand osteoarthritis risk. Osteoarthr. Cartil. 2007, 15, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Solovieva, S.; Kamarainen, O.P.; Hirvonen, A.; Hamalainen, S.; Laitala, M.; Vehmas, T.; Luoma, K.; Nakki, A.; Riihimaki, H.; Ala-Kokko, L.; et al. Association between interleukin 1 gene cluster polymorphisms and bilateral distal interphalangeal osteoarthritis. J. Rheumatol. 2009, 36, 1977–1986. [Google Scholar] [CrossRef]
- Stern, A.G.; de Carvalho, M.R.; Buck, G.A.; Adler, R.A.; Rao, T.P.; Disler, D.; Moxley, G.; Network, I.N. Association of erosive hand osteoarthritis with a single nucleotide polymorphism on the gene encoding interleukin-1 beta. Osteoarthr. Cartil. 2003, 11, 394–402. [Google Scholar] [CrossRef]
- Kerkhof, H.J.; Doherty, M.; Arden, N.K.; Abramson, S.B.; Attur, M.; Bos, S.D.; Cooper, C.; Dennison, E.M.; Doherty, S.A.; Evangelou, E.; et al. Large-scale meta-analysis of interleukin-1 beta and interleukin-1 receptor antagonist polymorphisms on risk of radiographic hip and knee osteoarthritis and severity of knee osteoarthritis. Osteoarthr. Cartil. 2011, 19, 265–271. [Google Scholar] [CrossRef]
- Attur, M.; Zhou, H.; Samuels, J.; Krasnokutsky, S.; Yau, M.; Scher, J.U.; Doherty, M.; Wilson, A.G.; Bencardino, J.; Hochberg, M.; et al. Interleukin 1 receptor antagonist (IL1RN) gene variants predict radiographic severity of knee osteoarthritis and risk of incident disease. Ann. Rheum. Dis. 2020, 79, 400–407. [Google Scholar] [CrossRef]
- Pola, E.; Papaleo, P.; Pola, R.; Gaetani, E.; Tamburelli, F.C.; Aulisa, L.; Logroscino, C.A. Interleukin-6 gene polymorphism and risk of osteoarthritis of the hip: A case-control study. Osteoarthr. Cartil. 2005, 13, 1025–1028. [Google Scholar] [CrossRef]
- Day-Williams, A.G.; Southam, L.; Panoutsopoulou, K.; Rayner, N.W.; Esko, T.; Estrada, K.; Helgadottir, H.T.; Hofman, A.; Ingvarsson, T.; Jonsson, H.; et al. A variant in MCF2L is associated with osteoarthritis. Am. J. Hum. Genet 2011, 89, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.; Skelton, A.J.; Rushton, M.D.; Reynard, L.N.; Loughlin, J. Expression analysis of the osteoarthritis genetic susceptibility locus mapping to an intron of the MCF2L gene and marked by the polymorphism rs11842874. BMC Med. Genet 2015, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Doherty, S.; Muir, K.R.; Zhang, W.; Maciewicz, R.A.; Wheeler, M.; Arden, N.; Cooper, C.; Doherty, M. Genetic contribution to radiographic severity in osteoarthritis of the knee. Ann. Rheum. Dis. 2012, 71, 1537–1540. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Zhao, F.; Zhang, X.; Deng, X.; He, X. Association between the interaction of SMAD3 polymorphisms with body mass index and osteoarthritis susceptibility. Int. J. Clin. Exp. Pathol. 2015, 8, 7364–7370. [Google Scholar]
- Sharma, A.C.; Srivastava, R.N.; Srivastava, S.R.; Parmar, D.; Singh, A.; Raj, S. Association between Single Nucleotide Polymorphisms of SMAD3 and BMP5 with the Risk of Knee Osteoarthritis. J. Clin. Diagn. Res. 2017, 11, GC01–GC04. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Hart, D.J.; Jones, K.A.; Surdulescu, G.; Swarbrick, P.; Doyle, D.V.; Schafer, A.J.; Spector, T.D. Association study of candidate genes for the prevalence and progression of knee osteoarthritis. Arthritis Rheum. 2004, 50, 2497–2507. [Google Scholar] [CrossRef]
- Rodriguez-Lopez, J.; Pombo-Suarez, M.; Loughlin, J.; Tsezou, A.; Blanco, F.J.; Meulenbelt, I.; Slagboom, P.E.; Valdes, A.M.; Spector, T.D.; Gomez-Reino, J.J.; et al. Association of a nsSNP in ADAMTS14 to some osteoarthritis phenotypes. Osteoarthr. Cartil. 2009, 17, 321–327. [Google Scholar] [CrossRef]
- El Khoury, L.; Posthumus, M.; Collins, M.; Handley, C.J.; Cook, J.; Raleigh, S.M. Polymorphic variation within the ADAMTS2, ADAMTS14, ADAMTS5, ADAM12 and TIMP2 genes and the risk of Achilles tendon pathology: A genetic association study. J. Sci. Med. Sport 2013, 16, 493–498. [Google Scholar] [CrossRef]
- Poonpet, T.; Honsawek, S.; Tammachote, N.; Kanitnate, S.; Tammachote, R. ADAMTS14 gene polymorphism associated with knee osteoarthritis in Thai women. Genet Mol. Res. 2013, 12, 5301–5309. [Google Scholar] [CrossRef]
- Wang, D.D.; Gan, Y.H.; Ma, X.C.; Meng, J.H. Association between ADAMTS14 gene polymorphism and the temporomandibular joint osteoarthritis in Chinese Han females. J. Peking Univ. Heal. Sci. 2018, 50, 279–283. [Google Scholar]
- Lv, Z.T.; Liang, S.; Huang, X.J.; Cheng, P.; Zhu, W.T.; Chen, A.M. Association between ADAM12 Single-Nucleotide Polymorphisms and Knee Osteoarthritis: A Meta-Analysis. Biomed. Res. Int. 2017, 2017, 5398181. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Doherty, M.; Spector, T.D. The additive effect of individual genes in predicting risk of knee osteoarthritis. Ann. Rheum. Dis. 2008, 67, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhou, K.; Chen, Z.; Yang, F.; Zhang, C.; Zhou, Z.; Pei, F. The association between DVWA polymorphisms and osteoarthritis susceptibility: A genetic meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 12566–12574. [Google Scholar] [PubMed]
- Wang, T.; Liang, Y.; Li, H.; Li, H.; He, Q.; Xue, Y.; Shen, C.; Zhang, C.; Xiang, J.; Ding, J.; et al. Single Nucleotide Polymorphisms and Osteoarthritis: An Overview and a Meta-Analysis. Medicine (Baltim. ) 2016, 95, e2811. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Mabuchi, A.; Shi, D.; Kubo, T.; Takatori, Y.; Saito, S.; Fujioka, M.; Sudo, A.; Uchida, A.; Yamamoto, S.; et al. A functional polymorphism in the 5’ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat. Genet. 2007, 39, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.; Takahashi, A.; Meulenbelt, I.; Watson, C.; Rodriguez-Lopez, J.; Egli, R.; Tsezou, A.; Malizos, K.N.; Kloppenburg, M.; Shi, D.; et al. A meta-analysis of European and Asian cohorts reveals a global role of a functional SNP in the 5’ UTR of GDF5 with osteoarthritis susceptibility. Hum. Mol. Genet. 2008, 17, 1497–1504. [Google Scholar] [CrossRef]
- Williams, F.M.; Popham, M.; Hart, D.J.; de Schepper, E.; Bierma-Zeinstra, S.; Hofman, A.; Uitterlinden, A.G.; Arden, N.K.; Cooper, C.; Spector, T.D.; et al. GDF5 single-nucleotide polymorphism rs143383 is associated with lumbar disc degeneration in Northern European women. Arthritis Rheum. 2011, 63, 708–712. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Wang, Y.; Wang, X.; Zhang, J.; Tian, J. Association between GDF5 single nucleotide polymorphism rs143383 and lumbar disc degeneration. Exp. Ther. Med. 2018, 16, 1900–1904. [Google Scholar] [CrossRef]
- Southam, L.; Rodriguez-Lopez, J.; Wilkins, J.M.; Pombo-Suarez, M.; Snelling, S.; Gomez-Reino, J.J.; Chapman, K.; Gonzalez, A.; Loughlin, J. An SNP in the 5’-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum. Mol. Genet 2007, 16, 2226–2232. [Google Scholar] [CrossRef]
- Rickert, M.; Wang, H.; Wieloch, P.; Lorenz, H.; Steck, E.; Sabo, D.; Richter, W. Adenovirus-mediated gene transfer of growth and differentiation factor-5 into tenocytes and the healing rat Achilles tendon. Connect Tissue Res. 2005, 46, 175–183. [Google Scholar] [CrossRef]
- Liang, H.; Ma, S.Y.; Feng, G.; Shen, F.H.; Joshua Li, X. Therapeutic effects of adenovirus-mediated growth and differentiation factor-5 in a mice disc degeneration model induced by annulus needle puncture. Spine J. 2010, 10, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Chujo, T.; An, H.S.; Akeda, K.; Miyamoto, K.; Muehleman, C.; Attawia, M.; Andersson, G.; Masuda, K. Effects of growth differentiation factor-5 on the intervertebral disc--in vitro bovine study and in vivo rabbit disc degeneration model study. Spine (Phila Pa 1976) 2006, 31, 2909–2917. [Google Scholar] [CrossRef] [PubMed]
- Byers, B.A.; Parrish, W.R.; Geesin, J.; Herzberg, U.; Wadsworth, S.; Bendele, A.M.; Story, B.J. Intra-articular supplementation with recombinant human GDF5 arrests disease progression and stimulates cartilage regeneration in the rat medial meniscus transection (MMT) model of osteoarthritis. Osteoarthr. Cartil. 2014, 22, S471–S472. [Google Scholar] [CrossRef][Green Version]
- Parrish, W.R.; Byers, B.A.; Su, D.; Geesin, J.; Herzberg, U.; Wadsworth, S.; Bendele, A.; Story, B. Intra-articular therapy with recombinant human GDF5 arrests disease progression and stimulates cartilage repair in the rat medial meniscus transection (MMT) model of osteoarthritis. Osteoarthr. Cartil. 2017, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- den Hollander, W.; Ramos, Y.F.; Bomer, N.; Elzinga, S.; van der Breggen, R.; Lakenberg, N.; de Dijcker, W.J.; Suchiman, H.E.; Duijnisveld, B.J.; Houwing-Duistermaat, J.J.; et al. Transcriptional associations of osteoarthritis-mediated loss of epigenetic control in articular cartilage. Arthritis Rheumatol. 2015, 67, 2108–2116. [Google Scholar] [CrossRef]
- Im, G.I.; Choi, Y.J. Epigenetics in osteoarthritis and its implication for future therapeutics. Expert Opin. Biol. 2013, 13, 713–721. [Google Scholar] [CrossRef]
- Kim, H.; Kang, D.; Cho, Y.; Kim, J.H. Epigenetic Regulation of Chondrocyte Catabolism and Anabolism in Osteoarthritis. Mol. Cells 2015, 38, 677–684. [Google Scholar] [CrossRef]
- Loughlin, J.; Reynard, L.N. Osteoarthritis: Epigenetics of articular cartilage in knee and hip OA. Nat. Rev. Rheumatol. 2015, 11, 6–7. [Google Scholar] [CrossRef]
- Raman, S.; FitzGerald, U.; Murphy, J.M. Interplay of Inflammatory Mediators with Epigenetics and Cartilage Modifications in Osteoarthritis. Front Bioeng. Biotechnol. 2018, 6, 22. [Google Scholar] [CrossRef]
- Reynard, L.N.; Loughlin, J. Genetics and epigenetics of osteoarthritis. Maturitas 2012, 71, 200–204. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, J. Epigenetics and Osteoarthritis. Genes Dis. 2015, 2, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Szczepanek, J. Role of microRNA dysregulation in childhood acute leukemias: Diagnostics, monitoring and therapeutics: A comprehensive review. World J. Clin. Oncol. 2020, 11, 348–369. [Google Scholar] [CrossRef]
- Endisha, H.; Rockel, J.; Jurisica, I.; Kapoor, M. The complex landscape of microRNAs in articular cartilage: Biology, pathology, and therapeutic targets. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.; Linsley, P.S. The therapeutic potential of microRNA modulation. Discov. Med. 2010, 9, 311–318. [Google Scholar] [PubMed]
- Jackson, A.L.; Levin, A.A. Developing microRNA therapeutics: Approaching the unique complexities. Nucleic Acid 2012, 22, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Sondag, G.R.; Haqqi, T.M. The Role of MicroRNAs and Their Targets in Osteoarthritis. Curr. Rheumatol. Rep. 2016, 18. [Google Scholar] [CrossRef]
- Genemaras, A.; Charles Huang, C.Y.D.; Kaplan, L. MicroRNA Therapies for Osteoarthritis and its Symptoms. Curr. Tissue Eng. 2015, 4, 86–94. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, L.; Fan, Y.; Liao, L.; Ma, P.X.; Xiao, G.; Chen, D. The microRNAs miR-204 and miR-211 maintain joint homeostasis and protect against osteoarthritis progression. Nat. Commun. 2019, 10, 2876. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Sun, Q.; Wang, Y.; Yang, J.; Yang, J.; Zhang, T.; Luo, S.; Wang, L.; Jiang, Y.; et al. Intra-articular Delivery of Antago-miR-483-5p Inhibits Osteoarthritis by Modulating Matrilin 3 and Tissue Inhibitor of Metalloproteinase 2. Mol. Ther. 2017, 25, 715–727. [Google Scholar] [CrossRef]
- Nagata, Y.; Nakasa, T.; Mochizuki, Y.; Ishikawa, M.; Miyaki, S.; Shibuya, H.; Yamasaki, K.; Adachi, N.; Asahara, H.; Ochi, M. Induction of apoptosis in the synovium of mice with autoantibody-mediated arthritis by the intraarticular injection of double-stranded MicroRNA-15a. Arthritis Rheum. 2009, 60, 2677–2683. [Google Scholar] [CrossRef]
- Ko, J.Y.; Lee, M.S.; Lian, W.S.; Weng, W.T.; Sun, Y.C.; Chen, Y.S.; Wang, F.S. MicroRNA-29a Counteracts Synovitis in Knee Osteoarthritis Pathogenesis by Targeting VEGF. Sci. Rep. 2017, 7, 3584. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Oreffo, R.O.; Gibson, M.B.; Goldring, M.B.; Roach, H.I. DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis Rheum. 2009, 60, 3303–3313. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Nakasa, T.; Miyaki, S.; Ishikawa, M.; Deie, M.; Adachi, N.; Yasunaga, Y.; Asahara, H.; Ochi, M. Expression of MicroRNA-146a in osteoarthritis cartilage. Arthritis Rheum. 2009, 60, 1035–1041. [Google Scholar] [CrossRef]
- Huh, Y.H.; Ryu, J.H.; Chun, J.S. Regulation of type II collagen expression by histone deacetylase in articular chondrocytes. J. Biol. Chem. 2007, 282, 17123–17131. [Google Scholar] [CrossRef] [PubMed]
- Martinek, V.; Ueblacker, P.; Imhoff, A.B. Current Concepts of Gene Therapy and Cartilage Repair. J. Bone Jt Surg. Br. Vol. 2003, 85-B, 782–788. [Google Scholar] [CrossRef]
- Venkatesan, J.K.; Rey-Rico, A.; Cucchiarini, M. Current Trends in Viral Gene Therapy for Human Orthopaedic Regenerative Medicine. Tissue Eng. Regen Med. 2019, 16, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.H.; Huard, J. Gene therapy approaches to regenerating the musculoskeletal system. Nat. Rev. Rheumatol. 2015, 11, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Pascher, A.; Palmer, G.D.; Steinert, A.; Oligino, T.; Gouze, E.; Gouze, J.N.; Betz, O.; Spector, M.; Robbins, P.D.; Evans, C.H.; et al. Gene delivery to cartilage defects using coagulated bone marrow aspirate. Gene 2004, 11, 133–141. [Google Scholar] [CrossRef]
- Sieker, J.T.; Kunz, M.; Weissenberger, M.; Gilbert, F.; Frey, S.; Rudert, M.; Steinert, A.F. Direct bone morphogenetic protein 2 and Indian hedgehog gene transfer for articular cartilage repair using bone marrow coagulates. Osteoarthr. Cartil. 2015, 23, 433–442. [Google Scholar] [CrossRef]
- Ivkovic, A.; Pascher, A.; Hudetz, D.; Maticic, D.; Jelic, M.; Dickinson, S.; Loparic, M.; Haspl, M.; Windhager, R.; Pecina, M. Articular cartilage repair by genetically modified bone marrow aspirate in sheep. Gene 2010, 17, 779–789. [Google Scholar] [CrossRef]
- Cucchiarini, M.; Madry, H. Overexpression of human IGF-I via direct rAAV-mediated gene transfer improves the early repair of articular cartilage defects in vivo. Gene 2014, 21, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Cucchiarini, M.; Orth, P.; Madry, H. Direct rAAV SOX9 administration for durable articular cartilage repair with delayed terminal differentiation and hypertrophy in vivo. J. Mol. Med. (Berl.) 2013, 91, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Cucchiarini, M.; Madry, H.; Ma, C.; Thurn, T.; Zurakowski, D.; Menger, M.D.; Kohn, D.; Trippel, S.B.; Terwilliger, E.F. Improved tissue repair in articular cartilage defects in vivo by rAAV-mediated overexpression of human fibroblast growth factor 2. Mol. Ther. 2005, 12, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, A.; Kon, E.; Filardo, G.; Delcogliano, M.; Montaperto, C.; Boldrini, L.; Bathan, L.; Marcacci, M., 2nd. Gen ACI Vs Microfracture in Knee Chondral Defect Treatment: Comparative study at 5 years (SS-61). Arthrosc. J. Arthrosc. Relat. Surg. 2009, 25, e33–e34. [Google Scholar] [CrossRef]
- Brittberg, M.; Lindahl, A.; Nilsson, A.; Ohlsson, C.; Isaksson, O.; Peterson, L. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N. Engl. J. Med. 1994, 331, 889–895. [Google Scholar] [CrossRef]
- Peterson, L.; Brittberg, M.; Kiviranta, I.; Akerlund, E.L.; Lindahl, A. Autologous chondrocyte transplantation. Biomechanics and long-term durability. Am. J. Sports Med. 2002, 30, 2–12. [Google Scholar] [CrossRef]
- Kang, R.; Marui, T.; Ghivizzani, S.C.; Nita, I.M.; Georgescu, H.I.; Suh, J.K.; Robbins, P.D.; Evans, C.H. Ex vivo gene transfer to chondrocytes in full-thickness articular cartilage defects: A feasibility study. Osteoarthr. Cartil. 1997, 5, 139–143. [Google Scholar] [CrossRef]
- Orth, P.; Kaul, G.; Cucchiarini, M.; Zurakowski, D.; Menger, M.D.; Kohn, D.; Madry, H. Transplanted articular chondrocytes co-overexpressing IGF-I and FGF-2 stimulate cartilage repair in vivo. Knee Surg. Sports Traumatol. Arthrosc. 2011, 19, 2119–2130. [Google Scholar] [CrossRef]
- Ortved, K.F.; Begum, L.; Mohammed, H.O.; Nixon, A.J. Implantation of rAAV5-IGF-I transduced autologous chondrocytes improves cartilage repair in full-thickness defects in the equine model. Mol. Ther. 2015, 23, 363–373. [Google Scholar] [CrossRef]
- Ha, C.W.; Noh, M.J.; Choi, K.B.; Lee, K.H. Initial phase I safety of retrovirally transduced human chondrocytes expressing transforming growth factor-beta-1 in degenerative arthritis patients. Cytotherapy 2012, 14, 247–256. [Google Scholar] [CrossRef]
- Evans, C.H.; Liu, F.J.; Glatt, V.; Hoyland, J.A.; Kirker-Head, C.; Walsh, A.; Betz, O.; Wells, J.W.; Betz, V.; Porter, R.M.; et al. Use of genetically modified muscle and fat grafts to repair defects in bone and cartilage. Eur. Cell Mater. 2009, 18, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Rowland, C.R.; Glass, K.A.; Ettyreddy, A.R.; Gloss, C.C.; Matthews, J.R.L.; Huynh, N.P.T.; Guilak, F. Regulation of decellularized tissue remodeling via scaffold-mediated lentiviral delivery in anatomically-shaped osteochondral constructs. Biomaterials 2018, 177, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Zaslav, K.; McAdams, T.; Scopp, J.; Theosadakis, J.; Mahajan, V.; Gobbi, A. New Frontiers for Cartilage Repair and Protection. Cartilage 2012, 3, 77S–86S. [Google Scholar] [CrossRef] [PubMed]
- Cucchiarini, M.; Madry, H. Biomaterial-guided delivery of gene vectors for targeted articular cartilage repair. Nat. Rev. Rheumatol. 2019, 15, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Moutos, F.T.; Glass, K.A.; Compton, S.A.; Ross, A.K.; Gersbach, C.A.; Guilak, F.; Estes, B.T. Anatomically shaped tissue-engineered cartilage with tunable and inducible anticytokine delivery for biological joint resurfacing. Proc. Natl. Acad. Sci. USA 2016, 113, E4513–E4522. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Lu, S.; Kasper, F.K.; Mikos, A.G. Strategies for controlled delivery of biologics for cartilage repair. Adv. Drug Deliv. Rev. 2015, 84, 123–134. [Google Scholar] [CrossRef]
- Rey-Rico, A.; Frisch, J.; Venkatesan, J.K.; Schmitt, G.; Rial-Hermida, I.; Taboada, P.; Concheiro, A.; Madry, H.; Alvarez-Lorenzo, C.; Cucchiarini, M. PEO-PPO-PEO Carriers for rAAV-Mediated Transduction of Human Articular Chondrocytes in Vitro and in a Human Osteochondral Defect Model. ACS Appl. Mater. Interfaces 2016, 8, 20600–20613. [Google Scholar] [CrossRef]
- Pannier, A.K.; Shea, L.D. Controlled release systems for DNA delivery. Mol. Ther. 2004, 10, 19–26. [Google Scholar] [CrossRef]
- Glass, K.A.; Link, J.M.; Brunger, J.M.; Moutos, F.T.; Gersbach, C.A.; Guilak, F. Tissue-engineered cartilage with inducible and tunable immunomodulatory properties. Biomaterials 2014, 35, 5921–5931. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Chen, Y.R.; Song, Y.F.; Yang, M.; Ye, J.; Zhou, G.; Yu, J.K. Scaffold-Based Gene Therapeutics for Osteochondral Tissue Engineering. Front Pharm. 2019, 10, 1534. [Google Scholar] [CrossRef]
- Rey-Rico, A.; Venkatesan, J.K.; Schmitt, G.; Speicher-Mentges, S.; Madry, H.; Cucchiarini, M. Effective Remodelling of Human Osteoarthritic Cartilage by sox9 Gene Transfer and Overexpression upon Delivery of rAAV Vectors in Polymeric Micelles. Mol. Pharm. 2018, 15, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Rodriguez, P.; Rey-Rico, A.; Madry, H.; Landin, M.; Cucchiarini, M. Effective genetic modification and differentiation of hMSCs upon controlled release of rAAV vectors using alginate/poloxamer composite systems. Int. J. Pharm. 2015, 496, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Rey-Rico, A.; Babicz, H.; Madry, H.; Concheiro, A.; Alvarez-Lorenzo, C.; Cucchiarini, M. Supramolecular polypseudorotaxane gels for controlled delivery of rAAV vectors in human mesenchymal stem cells for regenerative medicine. Int. J. Pharm. 2017, 531, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Rey-Rico, A.; Venkatesan, J.K.; Schmitt, G.; Concheiro, A.; Madry, H.; Alvarez-Lorenzo, C.; Cucchiarini, M. rAAV-mediated overexpression of TGF-beta via vector delivery in polymeric micelles stimulates the biological and reparative activities of human articular chondrocytes in vitro and in a human osteochondral defect model. Int. J. Nanomed. 2017, 12, 6985–6996. [Google Scholar] [CrossRef]
- Brunger, J.M.; Huynh, N.P.; Guenther, C.M.; Perez-Pinera, P.; Moutos, F.T.; Sanchez-Adams, J.; Gersbach, C.A.; Guilak, F. Scaffold-mediated lentiviral transduction for functional tissue engineering of cartilage. Proc. Natl. Acad. Sci. USA 2014, 111, E798–E806. [Google Scholar] [CrossRef]
- Zhao, R.; Peng, X.; Li, Q.; Song, W. Effects of phosphorylatable short peptide-conjugated chitosan-mediated IL-1Ra and igf-1 gene transfer on articular cartilage defects in rabbits. PLoS ONE 2014, 9, e112284. [Google Scholar] [CrossRef]
- Rey-Rico, A.; Venkatesan, J.K.; Frisch, J.; Schmitt, G.; Monge-Marcet, A.; Lopez-Chicon, P.; Mata, A.; Semino, C.; Madry, H.; Cucchiarini, M. Effective and durable genetic modification of human mesenchymal stem cells via controlled release of rAAV vectors from self-assembling peptide hydrogels with a maintained differentiation potency. Acta Biomater. 2015, 18, 118–127. [Google Scholar] [CrossRef]
- Rey-Rico, A.; Venkatesan, J.K.; Frisch, J.; Rial-Hermida, I.; Schmitt, G.; Concheiro, A.; Madry, H.; Alvarez-Lorenzo, C.; Cucchiarini, M. PEO-PPO-PEO micelles as effective rAAV-mediated gene delivery systems to target human mesenchymal stem cells without altering their differentiation potency. Acta Biomater. 2015, 27, 42–52. [Google Scholar] [CrossRef]
- Meng, W.; Rey-Rico, A.; Claudel, M.; Schmitt, G.; Speicher-Mentges, S.; Pons, F.; Lebeau, L.; Venkatesan, J.K.; Cucchiarini, M. rAAV-Mediated Overexpression of SOX9 and TGF-beta via Carbon Dot-Guided Vector Delivery Enhances the Biological Activities in Human Bone Marrow-Derived Mesenchymal Stromal Cells. Nanomaterials 2020, 10. [Google Scholar] [CrossRef]
- Leng, P.; Ding, C.R.; Zhang, H.N.; Wang, Y.Z. Reconstruct large osteochondral defects of the knee with hIGF-1 gene enhanced Mosaicplasty. Knee 2012, 19, 804–811. [Google Scholar] [CrossRef]
- Yang, S.; Qian, Z.; Liu, D.; Wen, N.; Xu, J.; Guo, X. Integration of C-type natriuretic peptide gene-modified bone marrow mesenchymal stem cells with chitosan/silk fibroin scaffolds as a promising strategy for articular cartilage regeneration. Cell Tissue Bank 2019, 20, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.K.; Gardner, O.; Rey-Rico, A.; Eglin, D.; Alini, M.; Stoddart, M.J.; Cucchiarini, M.; Madry, H. Improved Chondrogenic Differentiation of rAAV SOX9-Modified Human MSCs Seeded in Fibrin-Polyurethane Scaffolds in a Hydrodynamic Environment. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Pi, Y.; Zhang, X.; Shi, J.; Zhu, J.; Chen, W.; Zhang, C.; Gao, W.; Zhou, C.; Ao, Y. Targeted delivery of non-viral vectors to cartilage in vivo using a chondrocyte-homing peptide identified by phage display. Biomaterials 2011, 32, 6324–6332. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.H. The vicissitudes of gene therapy. Bone Jt. Re 2019, 8, 469–471. [Google Scholar] [CrossRef] [PubMed]



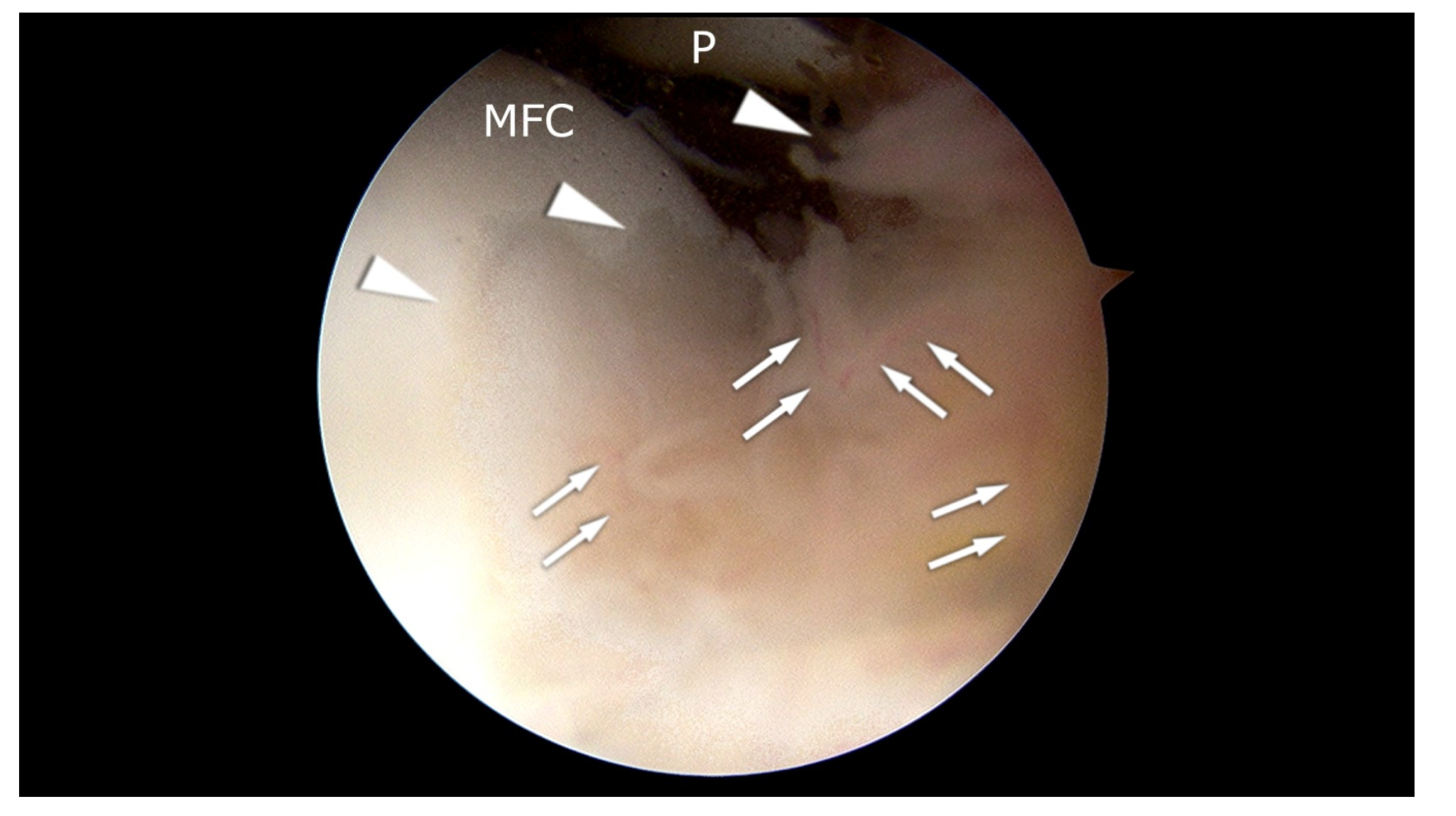{kind=link}
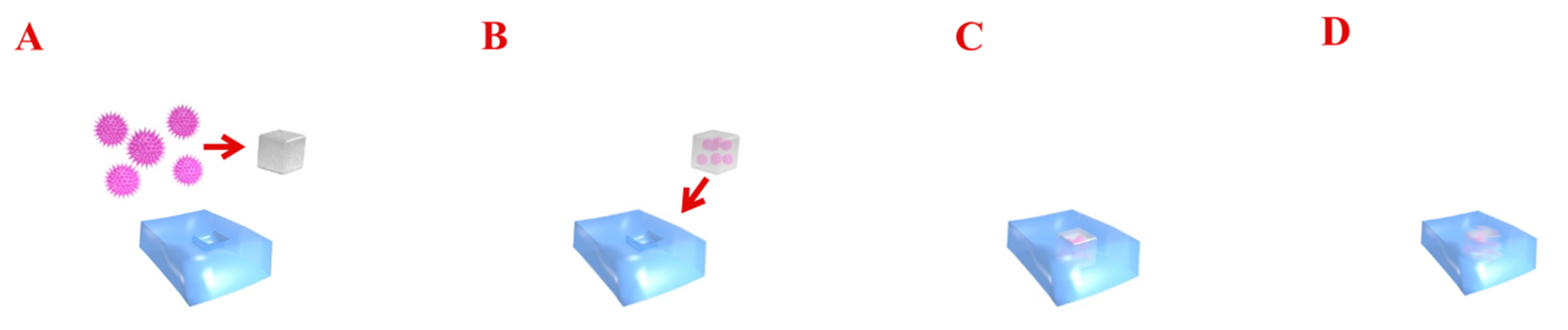{kind=link}
| Gene Category | Gene(s) | Therapeutic Potential | Ref. |
|---|---|---|---|
| extracellular matrix components | PIIANP, CTX-II, PIINP, COMP, type II collagen | inhibition of collagen and proteoglycan degradation, stimulation of cartilage matrix synthesis and/or cell proliferation | [13] |
| matrix-degrading enzymes proteolytic enzymes | metalloproteinases (MMPs such as MMP-3, MMP-9 and MMP-13); aggrecanases (e.g., ADAMTS-4 or ADAMTS-5) and their inhibitors (TIMP-1, TIMP -2) | inhibition of ECM degradation | [14,15,16,17,18,19,20,21,22,23,24] |
| circulating or locally occurring cytokines, inflammatory mediators and anabolic growth factors | TGF-β1,2,3; BMP-2,-4,-7; CDMP-1,-2,-3; IGF-1, PDGF, EGF, HGF | stimulation of chondrogenic differentiation, cartilage matrix synthesis, and/or cell proliferation | [25,26,27] |
| inflammatory factors | IL-1Ra, IL-1R, ICE inhibitor, TNF-R, anti-TNF-antibodies, TACE inhibitor, TIMP-1,-2, MMP inhibitors, IL-4,-10,-11,-13 | anti-inflammatory mechanism: IL-1 blockage, TNF-α and MMPs inhibition | [21,28,29,30,31,32,33,34,35] |
| transcription factors | Runx2, CEBPβ, HIF2α, Sox4, Sox9, and Sox11 | stimulation of chondrogenic differentiation | [36,37,38,39,40] |
| growth factors and signal transduction molecules | PTHrP, IHH, SHH, DHH, Smad 6, -7, mLAP-1 | inhibition of osteogenesis/hypertrophy, TGF-β/BMP action, and terminal differentiation | [41,42,43,44,45,46] |
| apoptosis regulators | Bcl-2, Bcl-XL, Anti-FasL, Akt, PI3-kinase, NF-κB | apoptosis inhibition: caspase inhibition, FAS-l blockage no-induced apoptosis, TNF-α and trail inhibition | [47,48,49,50] |
| Gene | SNPs | Affected Joint and Impact | Reference |
|---|---|---|---|
| MMP-3 | rs639752, rs520540, rs602128, rs650108, rs679620 |
| [73,114] |
| MMP-8 | rs1940475, rs3765620 |
| [115] |
| TIMP-3 | rs715572 (G/A), rs1962223 (G/C) |
| [114,116] |
| COL11A1 | rs1241164, rs4907986, rs2615977 |
| [117] |
| COL9A2 | rs7533552 |
| [118] |
| VEGF | rs833058 |
| [117] |
| DVWA | rs7639618, rs9864422, rs11718863 |
| [119,120,121] |
| FRZB | rs7775, rs288326 |
| [122,123] |
| GDF5 | rs143383 (risk allele T) |
| [123,124] |
| CALM1 | rs12885713 (-16C/T transition SNP) |
| [125,126,127,128] |
| TGF-1β | rs1982073, rs1800470 (TT genotype), rs180046 (TT genotype and T allele), rs1800469 (TT genotype and T allele) |
| [116,129] |
| IGF1 | rs2195239, rs11247361 (4488C>G) |
| [130,131,132,133] |
| IL1RN | rs9005, rs315952, rs419598, rs315943, rs315920 (C/t) |
| [134,135,136,137,138,139] |
| IL-6 | rs1800795 (−174G>C) |
| [132,140] |
| MCF2L | rs11842874 (risk allele G) |
| [141,142,143] |
| SMAD3 | rs12901499 (GA and GG genotypes), rs12102171 (CC and CT+TT and TT and TG+GG), rs2289263, rs6494629 (T/C), |
| [116,144,145] |
| BMP5 | rs921126 (GA and GG genotypes) |
| [145] |
| ADAM12 | rs1871054, rs3740199 (Gly48Arg) |
| [132,146] |
| ADAMTS14 | rs4747096 |
| [147,148,149,150] |
| Gene(s) | Vector(s) | Scaffold | Reference |
|---|---|---|---|
| Sox9 | rAAV | polymeric micelles | [211] |
| rAAV | poloxamers, poloxamines, micellar systems | [211] | |
| IL-1Ra | lentiviral | poly-e-caprolactone | [202,205,209] |
| lacZ | rAAV | poloxamers, poloxamines, micellar systems | [207] |
| rAAV | alginate, alginate/poloxamers | [212] | |
| rAAV | polypseudorotaxane gels | [213] | |
| TGF-β1 | rAAV | poloxamers, poloxamines, micellar systems | [214] |
| lentiviral | poly-e-caprolactone | [215] | |
| rAAV | polymeric micelles | [214] | |
| IL1RN and IGF1 | non-viral | chitosan | [216] |
| lacZ, RFP | rAAV | self-assembling peptide hydrogels | [212,217] |
| lacZ, RFP, Sox9 | rAAV | poloxamers, poloxamines, micellar systems | [213,218] |
| Sox9, TGF-β | rAAV | carbon dots | [219] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szwedowski, D.; Szczepanek, J.; Paczesny, Ł.; Pękała, P.; Zabrzyński, J.; Kruczyński, J. Genetics in Cartilage Lesions: Basic Science and Therapy Approaches. Int. J. Mol. Sci. 2020, 21, 5430. https://doi.org/10.3390/ijms21155430
Szwedowski D, Szczepanek J, Paczesny Ł, Pękała P, Zabrzyński J, Kruczyński J. Genetics in Cartilage Lesions: Basic Science and Therapy Approaches. International Journal of Molecular Sciences. 2020; 21(15):5430. https://doi.org/10.3390/ijms21155430
Chicago/Turabian StyleSzwedowski, Dawid, Joanna Szczepanek, Łukasz Paczesny, Przemysław Pękała, Jan Zabrzyński, and Jacek Kruczyński. 2020. "Genetics in Cartilage Lesions: Basic Science and Therapy Approaches" International Journal of Molecular Sciences 21, no. 15: 5430. https://doi.org/10.3390/ijms21155430
APA StyleSzwedowski, D., Szczepanek, J., Paczesny, Ł., Pękała, P., Zabrzyński, J., & Kruczyński, J. (2020). Genetics in Cartilage Lesions: Basic Science and Therapy Approaches. International Journal of Molecular Sciences, 21(15), 5430. https://doi.org/10.3390/ijms21155430