Biophysical Characterization of Cancer-Related Carbonic Anhydrase IX

 , , and
, , and
Abstract

1. Introduction
2. Results and Discussion
2.1. Production of Recombinant Protein, SDS-PAGE and Degradation
2.2. MALDI-TOF Mass Spectrometry
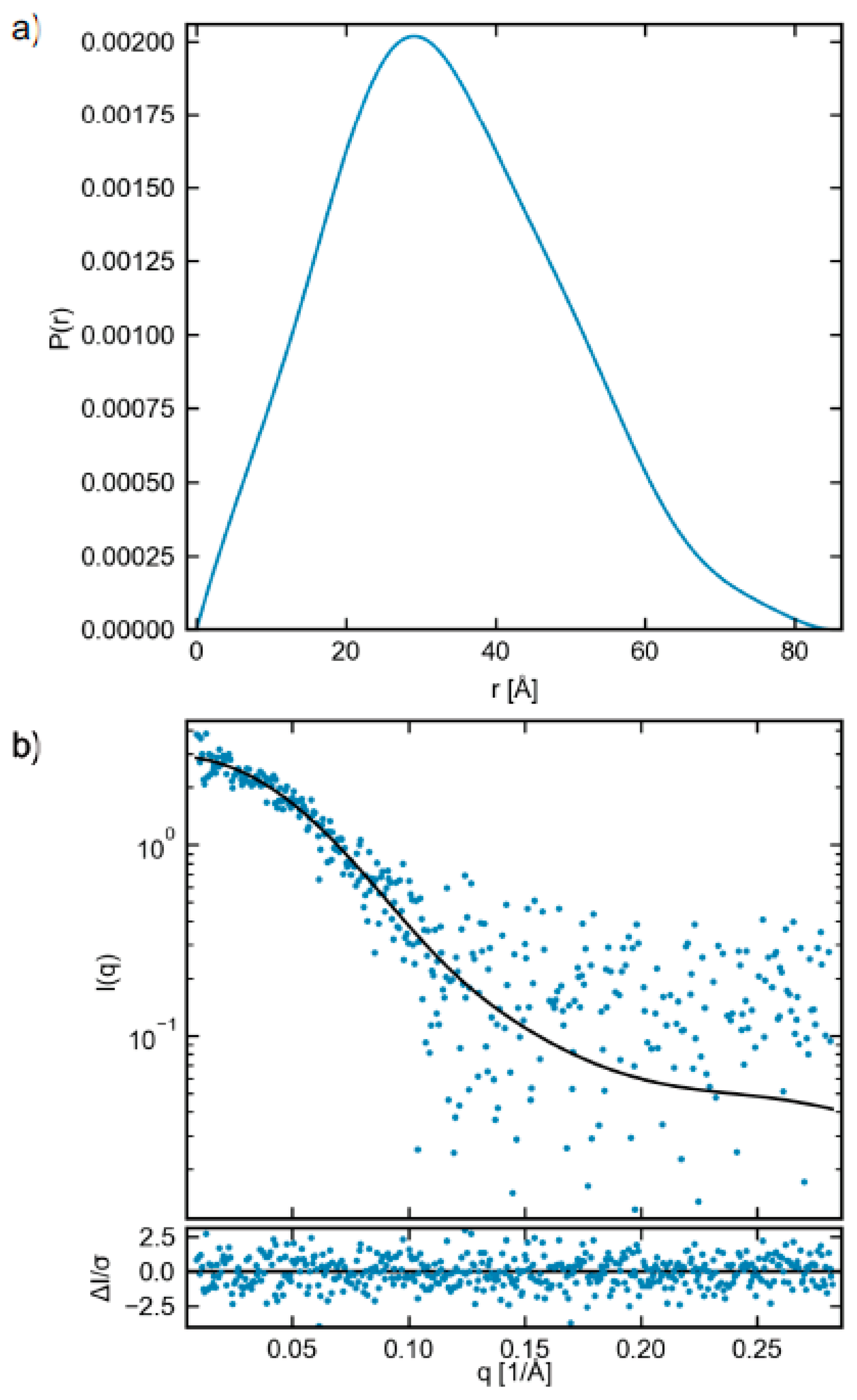
2.3. Small-Angle X-ray Scattering (SAXS)
2.4. Crystallography
3. Materials and Methods
3.1. Expression and Purification of ECD of CA IX
3.2. Construct 1 Protein Expression and Purification
3.3. Construct 2 Protein Expression and Purification
3.4. MALDI-TOF Mass Spectrometry of Construct 1
3.5. Crystallization of Construct 1
3.6. Crystallographic Data Collection and Structure Refinement
3.7. Small Angle X-ray Scattering (SAXS) of Construct 2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Silverman, D.N.; McKenna, R. Solvent-Mediated Proton Transfer in Catalysis by Carbonic Anhydrase. Acc. Chem. Res. 2007, 40, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.C. Physiological Functions of the Alpha Class of Carbonic Anhydrases. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Frost, S.C., McKenna, R., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 9–30. [Google Scholar]
- Mboge, M.Y.; Mahon, B.P.; McKenna, R.; Frost, S.C. Carbonic Anhydrases: Role in pH Control and Cancer. Metabolites 2018, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Pastorekova, S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: From biology to clinical use. Semin. Cancer Biol. 2015, 31, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.Y.; Chen, Z.; Wolff, A.; Mathias, J.V.; Tu, C.; Brown, K.D.; Bozdag, M.; Carta, F.; Supuran, C.T.; McKenna, R.; et al. Selective inhibition of carbonic anhydrase IX over carbonic anhydrase XII in breast cancer cells using benzene sulfonamides: Disconnect between activity and growth inhibition. PLoS ONE 2018, 13, e0207417. [Google Scholar] [CrossRef] [PubMed]
- Kats-Ugurlu, G.; Oosterwijk, E.; Muselaers, S.; Oosterwijk-Wakka, J.; Hulsbergen-van de Kaa, C.; de Weijert, M.; van Krieken, H.; Desar, I.; van Herpen, C.; Maass, C.; et al. Neoadjuvant sorafenib treatment of clear cell renal cell carcinoma and release of circulating tumor fragments. Neoplasia 2014, 16, 221–228. [Google Scholar] [CrossRef]
- Oosterwijk-Wakka, J.C.; de Weijert, M.C.; Franssen, G.M.; Leenders, W.P.; van der Laak, J.A.; Boerman, O.C.; Mulders, P.F.; Oosterwijk, E. Successful combination of sunitinib and girentuximab in two renal cell carcinoma animal models: A rationale for combination treatment of patients with advanced RCC. Neoplasia 2015, 17, 215–224. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor supp.ressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Svastova, E.; Hulikova, A.; Rafajova, M.; Zatovicova, M.; Gibadulinova, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Stock, C.; Schwab, A. Protons make tumor cells move like clockwork. Pflugers Arch. 2009, 458, 981–992. [Google Scholar] [CrossRef]
- Li, Y.; Tu, C.; Wang, H.; Silverman, D.N.; Frost, S.C. Catalysis and pH control by membrane-associated carbonic anhydrase IX in MDA-MB-231 breast cancer cells. J. Biol. Chem. 2011, 286, 15789–15796. [Google Scholar] [CrossRef]
- Mahon, B.P.; Bhatt, A.; Socorro, L.; Driscoll, J.M.; Okoh, C.; Lomelino, C.L.; Mboge, M.Y.; Kurian, J.J.; Tu, C.; Agbandje-McKenna, M.; et al. The Structure of Carbonic Anhydrase IX Is Adapted for Low-pH Catalysis. Biochemistry 2016, 55, 4642–4653. [Google Scholar] [CrossRef]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef]
- Watson, P.H.; Chia, S.K.; Wykoff, C.C.; Han, C.; Leek, R.D.; Sly, W.S.; Gatter, K.C.; Ratcliffe, P.; Harris, A.L. Carbonic anhydrase XII is a marker of good prognosis in invasive breast carcinoma. Br. J. Cancer 2003, 88, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, J.; Hilvo, M.; Nordfors, K.; Haapasalo, H.; Parkkila, S.; Hyrskyluoto, A.; Rantala, I.; Waheed, A.; Sly, W.S.; Pastorekova, S.; et al. Identification of an alternatively spliced isoform of carbonic anhydrase XII in diffusely infiltrating astrocytic gliomas. Neuro. Oncol. 2008, 10, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Mujumdar, P.; Kopecka, J.; Bua, S.; Supuran, C.T.; Riganti, C.; Poulsen, S.-A. Carbonic Anhydrase XII Inhibitors Overcome Temozolomide Resistance in Glioblastoma. J. Med. Chem. 2019, 62, 4174–4192. [Google Scholar] [CrossRef] [PubMed]
- Pinard, M.A.; Mahon, B.; McKenna, R. Probing the surface of human carbonic anhydrase for clues towards the design of isoform specific inhibitors. Biomed. Res. Int. 2015, 2015, 453543. [Google Scholar] [CrossRef]
- Hilvo, M.; Baranauskiene, L.; Salzano, A.M.; Scaloni, A.; Matulis, D.; Innocenti, A.; Scozzafava, A.; Monti, S.M.; Di Fiore, A.; De Simone, G.; et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J. Biol. Chem. 2008, 283, 27799–27809. [Google Scholar] [CrossRef]
- Christianson, H.C.; Menard, J.A.; Chandran, V.I.; Bourseau-Guilmain, E.; Shevela, D.; Lidfeldt, J.; Mansson, A.S.; Pastorekova, S.; Messinger, J.; Belting, M. Tumor antigen glycosaminoglycan modification regulates antibody-drug conjugate delivery and cytotoxicity. Oncotarget 2017, 8, 66960–66974. [Google Scholar] [CrossRef]
- Dorai, T.; Sawczuk, I.S.; Pastorek, J.; Wiernik, P.H.; Dutcher, J.P. The role of carbonic anhydrase IX overexpression in kidney cancer. Eur. J. Cancer 2005, 41, 2935–2947. [Google Scholar] [CrossRef]
- Ditte, P.; Dequiedt, F.; Svastova, E.; Hulikova, A.; Ohradanova-Repic, A.; Zatovicova, M.; Csaderova, L.; Kopacek, J.; Supuran, C.T.; Pastorekova, S.; et al. Phosphorylation of Carbonic Anhydrase IX Controls Its Ability to Mediate Extracellular Acidification in Hypoxic Tumors. Cancer Res. 2011, 71, 7558–7567. [Google Scholar] [CrossRef]
- Opavský, R.; Pastoreková, S.; Zelnı́k, V.; Gibadulinová, A.; Stanbridge, E.J.; Závada, J.; Kettmann, R.; Pastorek, J. HumanMN/CA9Gene, a Novel Member of the Carbonic Anhydrase Family: Structure and Exon to Protein Domain Relationships. Genomics 1996, 33, 480–487. [Google Scholar] [CrossRef]
- Langella, E.; Buonanno, M.; Vullo, D.; Dathan, N.; Leone, M.; Supuran, C.T.; De Simone, G.; Monti, S.M. Biochemical, biophysical and molecular dynamics studies on the proteoglycan-like domain of carbonic anhydrase IX. Cell Mol. Life Sci. 2018, 75, 3283–3296. [Google Scholar] [CrossRef]
- Innocenti, A.; Pastorekova, S.; Pastorek, J.; Scozzafava, A.; Simone, G.D.; Supuran, C.T. The proteoglycan region of the tumor-associated carbonic anhydrase isoform IX acts as anintrinsic buffer optimizing CO2 hydration at acidic pH values characteristic of solid tumors. Bioorg. Med. Chem. Lett. 2009, 19, 5825–5828. [Google Scholar] [CrossRef] [PubMed]
- Ames, S.; Pastorekova, S.; Becker, H.M. The proteoglycan-like domain of carbonic anhydrase IX mediates non-catalytic facilitation of lactate transport in cancer cells. Oncotarget 2018, 9, 27940–27957. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.L.; Miles, A.J.; Whitmore, L.; Wallace, B.A. Distinct circular dichroism spectroscopic signatures of polyproline II and unordered secondary structures: Applications in secondary structure analyses. Protein Sci. 2014, 23, 1765–1772. [Google Scholar] [CrossRef]
- Adzhubei, A.A.; Sternberg, M.J.; Makarov, A.A. Polyproline-II helix in proteins: Structure and function. J. Mol. Biol. 2013, 425, 2100–2132. [Google Scholar] [CrossRef] [PubMed]
- Zavada, J.; Zavadova, Z.; Pastorek, J.; Biesova, Z.; Jezek, J.; Velek, J. Human tumour-associated cell adhesion protein MN/CA IX: Identification of M75 epitope and of the region mediating cell adhesion. Br. J. Cancer 2000, 82, 1808–1813. [Google Scholar] [CrossRef] [PubMed]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient Expression and Crystallization System of Cancer-Associated Carbonic Anhydrase Isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef]
- Pinard, M.A.; Aggarwal, M.; Mahon, B.P.; Tu, C.; McKenna, R. A sucrose-binding site provides a lead towards an isoform-specific inhibitor of the cancer-associated enzyme carbonic anhydrase IX. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 1352–1358. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- Rambo, R.P.; Tainer, J.A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 2013, 496, 477–481. [Google Scholar] [CrossRef]
- Durand, D.; Vivès, C.; Cannella, D.; Pérez, J.; Pebay-Peyroula, E.; Vachette, P.; Fieschi, F. NADPH oxidase activator p67phox behaves in solution as a multidomain protein with semi-flexible linkers. J. Struct. Biol. 2010, 169, 45–53. [Google Scholar] [CrossRef]
- Bernado, P. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur. Biophys. J. 2010, 39, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Receveur-Brechot, V.; Durand, D. How random are intrinsically disordered proteins? A small angle scattering perspective. Curr. Protein Pept. Sci. 2012, 13, 55–75. [Google Scholar] [CrossRef] [PubMed]
- Putnam, C.D.; Hammel, M.; Hura, G.L.; Tainer, J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007, 40, 191–285. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.E. Mechanism of Action of Carbonic Anhydrase: Substrate, Sulfonamide, and Anion Binding. J. Biol. Chem. 1967, 242, 5212–5219. [Google Scholar]
- Koruza, K.; Lafumat, B.; Nyblom, M.; Mahon, B.P.; Knecht, W.; McKenna, R.; Fisher, S.Z. Structural comparison of protiated, H/D-exchanged and deuterated human carbonic anhydrase IX. Acta Crystallogr. D Biol. Crystallogr. 2019, 75, 895–903. [Google Scholar] [CrossRef]
- Yang, J.; Koruza, K.; Fisher, Z.; Knecht, W.; Baltzer, L. Improved molecular recognition of Carbonic Anhydrase IX by polypeptide conjugation to acetazolamide. Biorg. Med. Chem. 2017, 25, 5838–5848. [Google Scholar] [CrossRef]
- Gill, S.C.; von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Echols, N.; Headd, J.J.; Hung, L.W.; Jain, S.; Kapral, G.J.; Grosse Kunstleve, R.W.; et al. The Phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Schrödinger, L. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger LLC: New York, NY, USA, 2002; p. 10. [Google Scholar]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Acerbo, A.S.; Cook, M.J.; Gillilan, R.E. Upgrade of MacCHESS facility for X-ray scattering of biological macromolecules in solution. J. Synchrotron Radiat. 2015, 22, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Skou, S.; Gillilan, R.E.; Ando, N. Synchrotron-based small-angle X-ray scattering of proteins in solution. Nat. Protoc. 2014, 9, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.S.; Toft, K.N.; Snakenborg, D.; Jeppesen, M.G.; Jacobsen, J.K.; Vestergaard, B.; Kutter, J.P.; Arleth, L. BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. J. Appl. Crystallogr. 2009, 42, 959–964. [Google Scholar] [CrossRef]
- Hopkins, J.B.; Gillilan, R.E.; Skou, S. BioXTAS RAW: Improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 2017, 50, 1545–1553. [Google Scholar] [CrossRef]
- Piiadov, V.; Ares de Araújo, E.; Oliveira Neto, M.; Craievich, A.F.; Polikarpov, I. SAXSMoW 2.0: Online calculator of the molecular weight of proteins in dilute solution from experimental SAXS data measured on a relative scale. Protein Sci. 2019, 28, 454–463. [Google Scholar] [CrossRef]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Petoukhov, M.V.; Svergun, D.I. Ambiguity assessment of small-angle scattering curves from monodisperse systems. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1051–1058. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012, 45, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef]
- Tria, G.; Mertens, H.D.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef]
- Trewhella, J.; Duff, A.P.; Durand, D.; Gabel, F.; Guss, J.M.; Hendrickson, W.A.; Hura, G.L.; Jacques, D.A.; Kirby, N.M.; Kwan, A.H.; et al. 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: An update. Acta Crystallogr. D Biol. Crystallogr. 2017, 73, 710–728. [Google Scholar] [CrossRef] [PubMed]
- Wind, T.C.; Messenger, M.P.; Thompson, D.; Selby, P.J.; Banks, R.E. Measuring carbonic anhydrase IX as a hypoxia biomarker: Differences in concentrations in serum and plasma using a commercial enzyme-linked immunosorbent assay due to influences of metal ions. Ann. Clin. Biochem. 2011, 48, 112–120. [Google Scholar] [CrossRef]
- Zatovicova, M.; Sedlakova, O.; Svastova, E.; Ohradanova, A.; Ciampor, F.; Arribas, J.; Pastorek, J.; Pastorekova, S. Ectodomain shedding of the hypoxia-induced carbonic anhydrase IX is a metalloprotease-dependent process regulated by TACE/ADAM17. Br. J. Cancer 2005, 93, 1267–1276. [Google Scholar] [CrossRef]
- Fu, Z.; Thorpe, M.; Akula, S.; Chahal, G.; Hellman, L.T. Extended Cleavage Specificity of Human Neutrophil Elastase, Human Proteinase 3, and Their Distant Ortholog Clawed Frog PR3-Three Elastases With Similar Primary but Different Extended Specificities and Stability. Front. Immunol 2018, 9, 2387. [Google Scholar] [CrossRef]
- Takahashi, H.; Nukiwa, T.; Yoshimura, K.; Quick, C.D.; States, D.J.; Holmes, M.D.; Whang-Peng, J.; Knutsen, T.; Crystal, R.G. Structure of the human neutrophil elastase gene. J. Biol. Chem. 1988, 263, 14739–14747. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection and Processing | PDB ID 6y74 |
|---|---|
| Source | BioMAX (MAX IV Laboratory) |
| 1 Wavelength (Å) | 0.979 |
| Detector | DECTRIS, Eiger 16M Hybrid-pixel detector |
| 2 Rotation range per image (°) | 0.1 |
| Total number of images | 3600 |
| Space group, unit cell (Å, °) | C2, a = 113.9, b =78.3, c = 74.3; β = 128.2 |
| Resolution range (Å) | 37.13–1.53 (1.56–1.53) |
| Total No. of reflections | 519,006 (21,203) |
| No. of unique reflections | 75,610 (3288) |
| Multiplicity | 6.9 (6.4) |
| Completeness (%) | 85.0 (98.3) |
| I/σ(I) | 10.6 (2.2) |
| 3 R-merge (%) | 0.110 (0.815) |
| CC(1/2) | 0.76 (0.99) |
| Wilson B | 15.6 |
| 4 Rcryst/Rfree (%) | 17.3/19.8 |
| Rmsd bond lengths, angles (Å, °) | 0.016/1.723 |
| Ramachandran favored/outliers/rotamer outliers % | 97.1/0.0/1.34 |
| Model clash score | 6.31 |
| No. solvent molecules | 606 |
| Mean B factor protein/solvent/ | 20.2/29.0 |
| Mean B factor Zn/sugar/acetate | 13.3/44.1/24.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koruza, K.; Murray, A.B.; Mahon, B.P.; Hopkins, J.B.; Knecht, W.; McKenna, R.; Fisher, S.Z. Biophysical Characterization of Cancer-Related Carbonic Anhydrase IX. Int. J. Mol. Sci. 2020, 21, 5277. https://doi.org/10.3390/ijms21155277
Koruza K, Murray AB, Mahon BP, Hopkins JB, Knecht W, McKenna R, Fisher SZ. Biophysical Characterization of Cancer-Related Carbonic Anhydrase IX. International Journal of Molecular Sciences. 2020; 21(15):5277. https://doi.org/10.3390/ijms21155277
Chicago/Turabian StyleKoruza, Katarina, A. Briana Murray, Brian P. Mahon, Jesse B. Hopkins, Wolfgang Knecht, Robert McKenna, and S. Zoë Fisher. 2020. "Biophysical Characterization of Cancer-Related Carbonic Anhydrase IX" International Journal of Molecular Sciences 21, no. 15: 5277. https://doi.org/10.3390/ijms21155277
APA StyleKoruza, K., Murray, A. B., Mahon, B. P., Hopkins, J. B., Knecht, W., McKenna, R., & Fisher, S. Z. (2020). Biophysical Characterization of Cancer-Related Carbonic Anhydrase IX. International Journal of Molecular Sciences, 21(15), 5277. https://doi.org/10.3390/ijms21155277