Chronic Carbon Tetrachloride Applications Induced Hepatocyte Apoptosis in Lipocalin 2 Null Mice through Endoplasmic Reticulum Stress and Unfolded Protein Response



Abstract
1. Introduction
2. Results
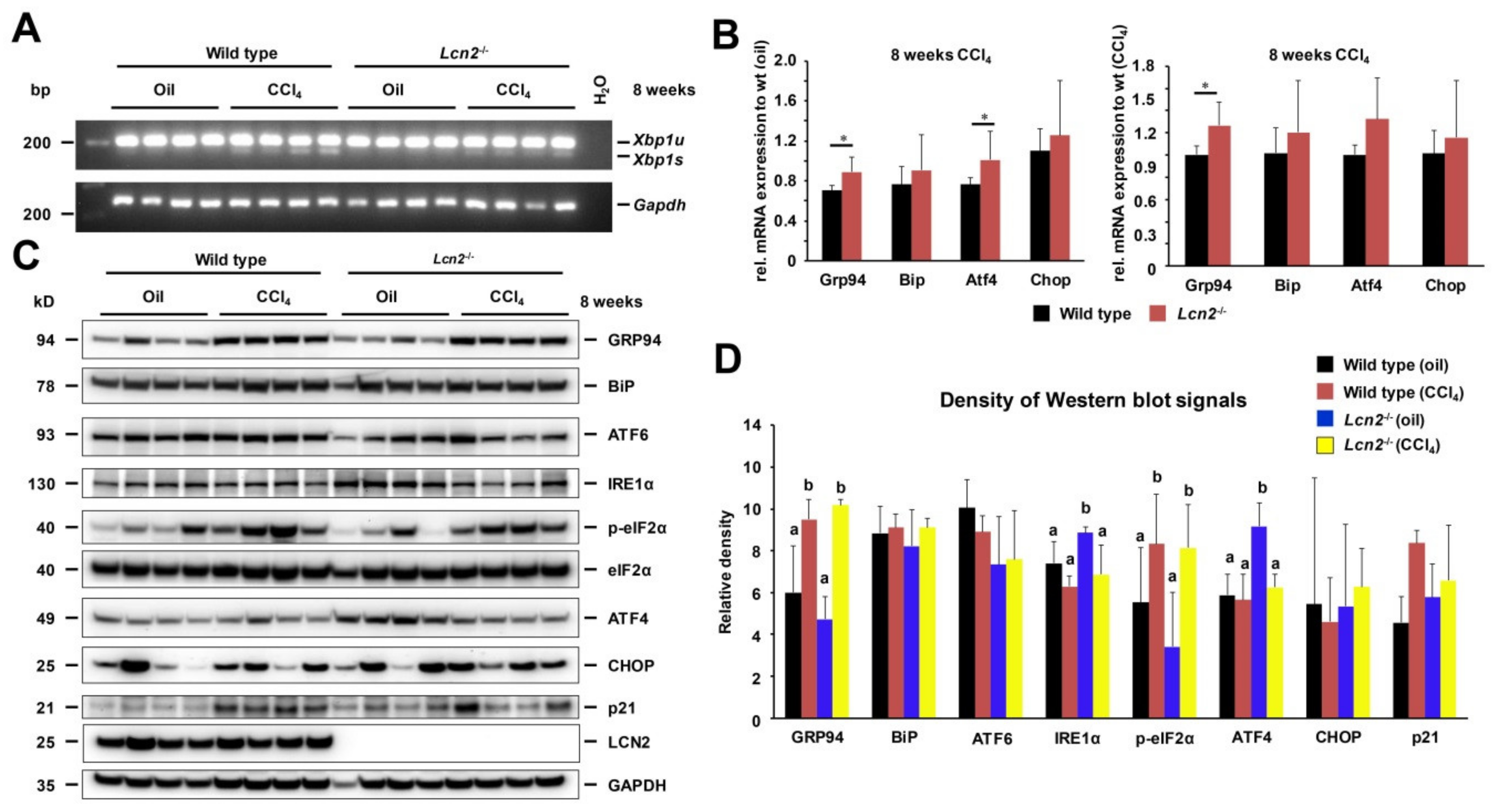
2.1. Repeated CCl4 Intoxication Induced ER Stress and UPR
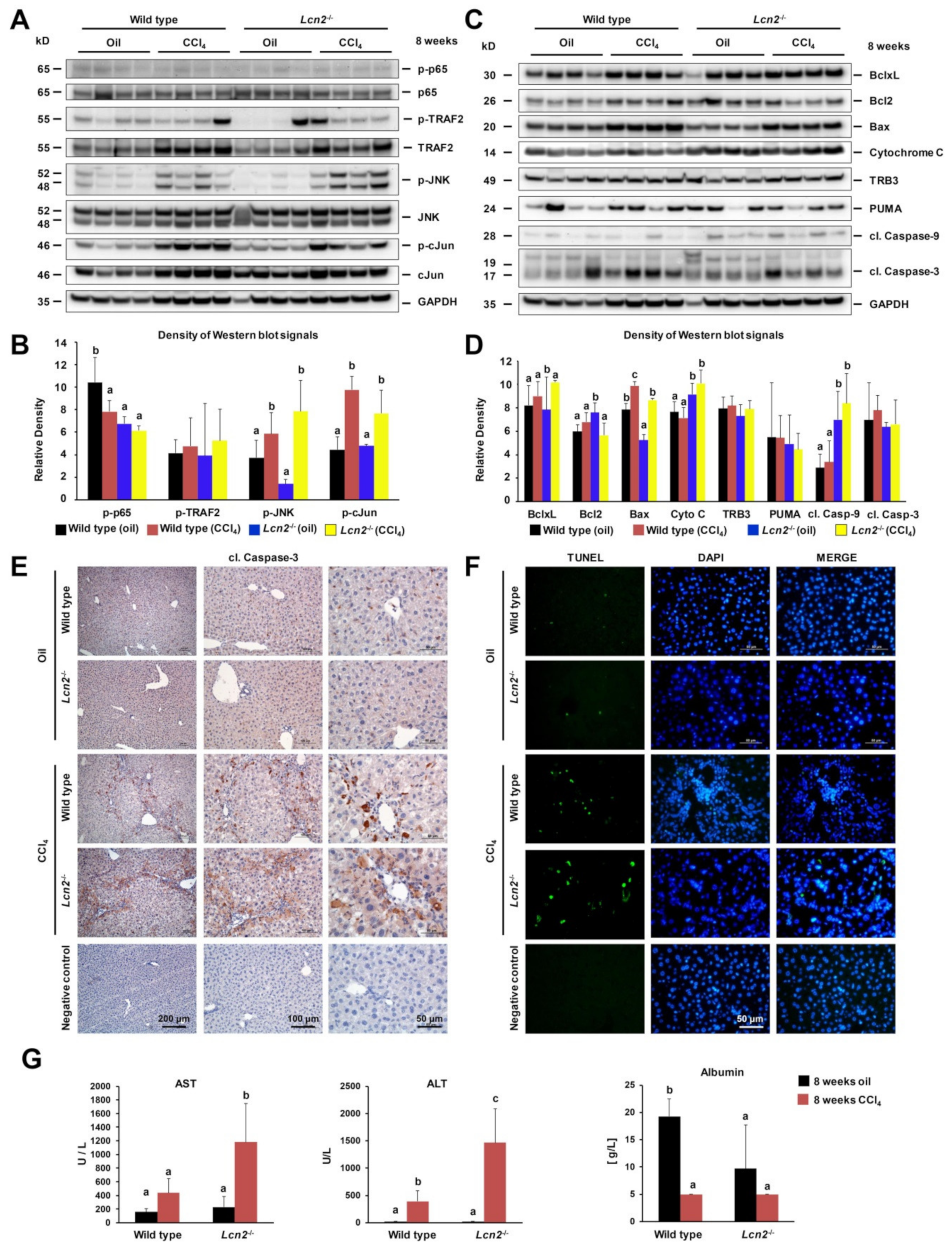
2.2. Chronic Repeated CCl4 Administration Activated c-Jun N-Terminal Kinase (JNK) and Intrinsic Hepatocyte Apoptosis
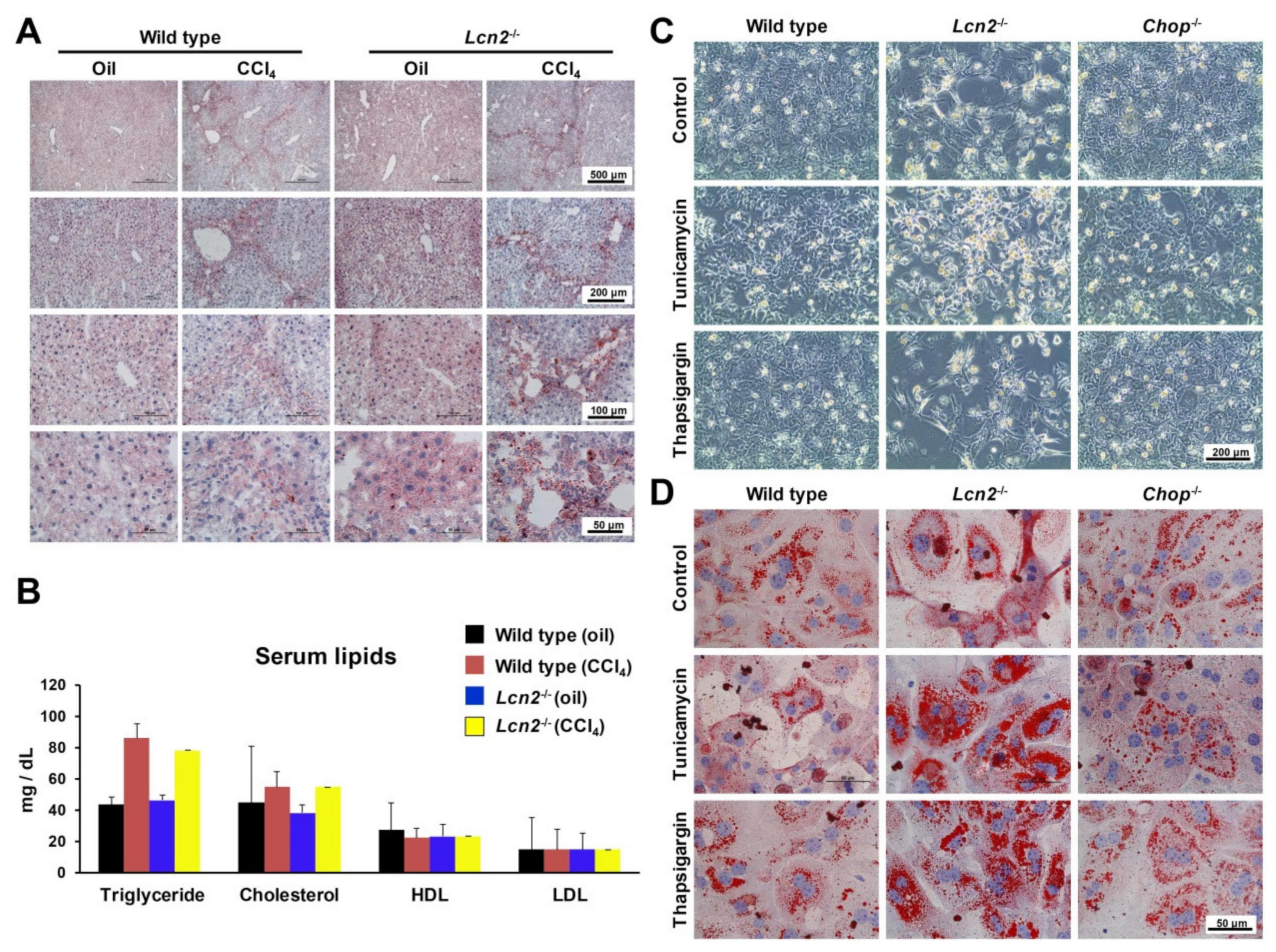
2.3. Lcn2 Null Mice Developed Slightly More Steatosis Than Wild Type Mice
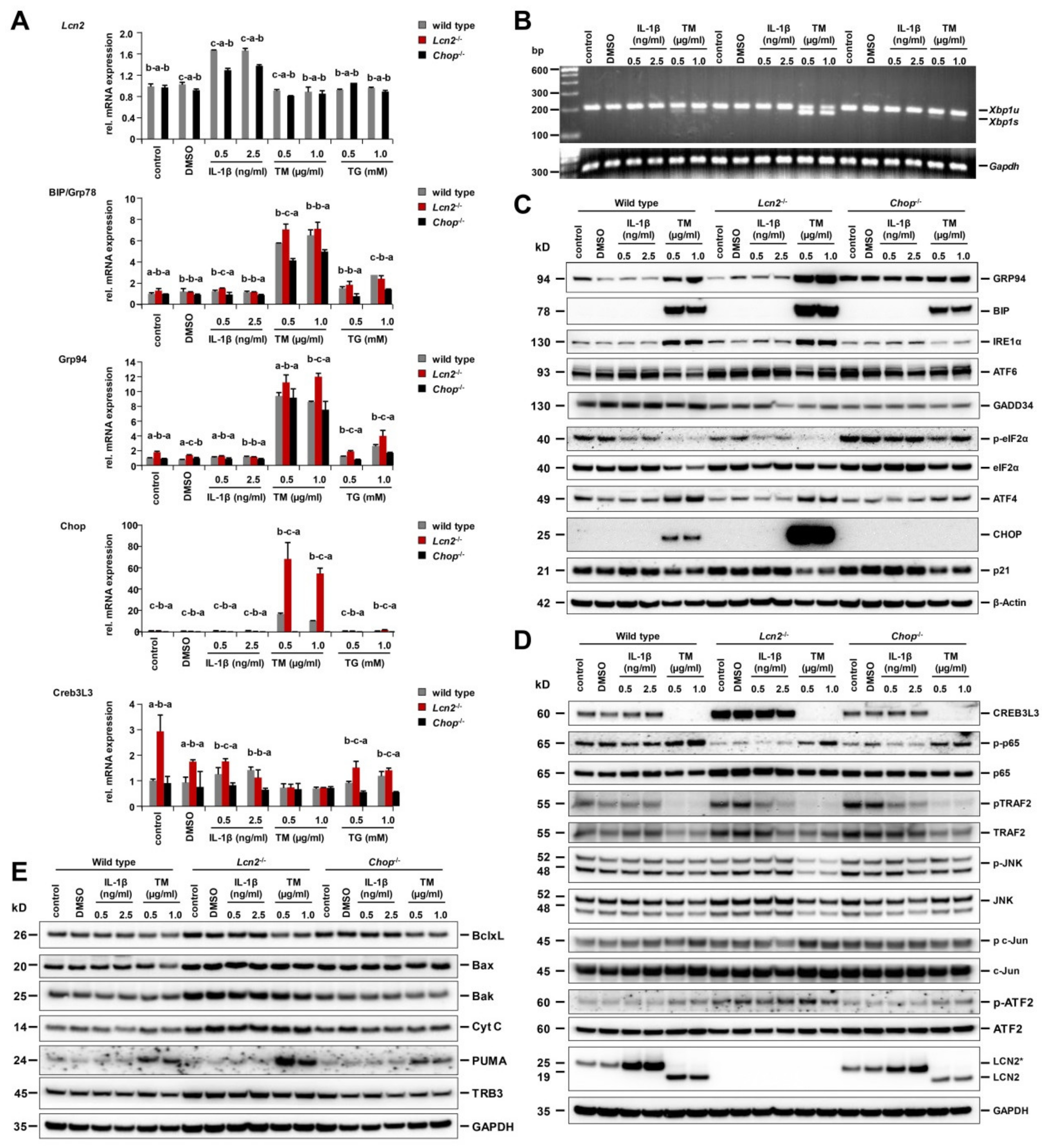
2.4. Strongest ER Stress Responses Were Observed in Lcn2−/− Hepatocytes That Also Expressed High Levels of Cyclic AMP-Responsive Element-Binding Protein 3-Like 3 (CREB3L3)
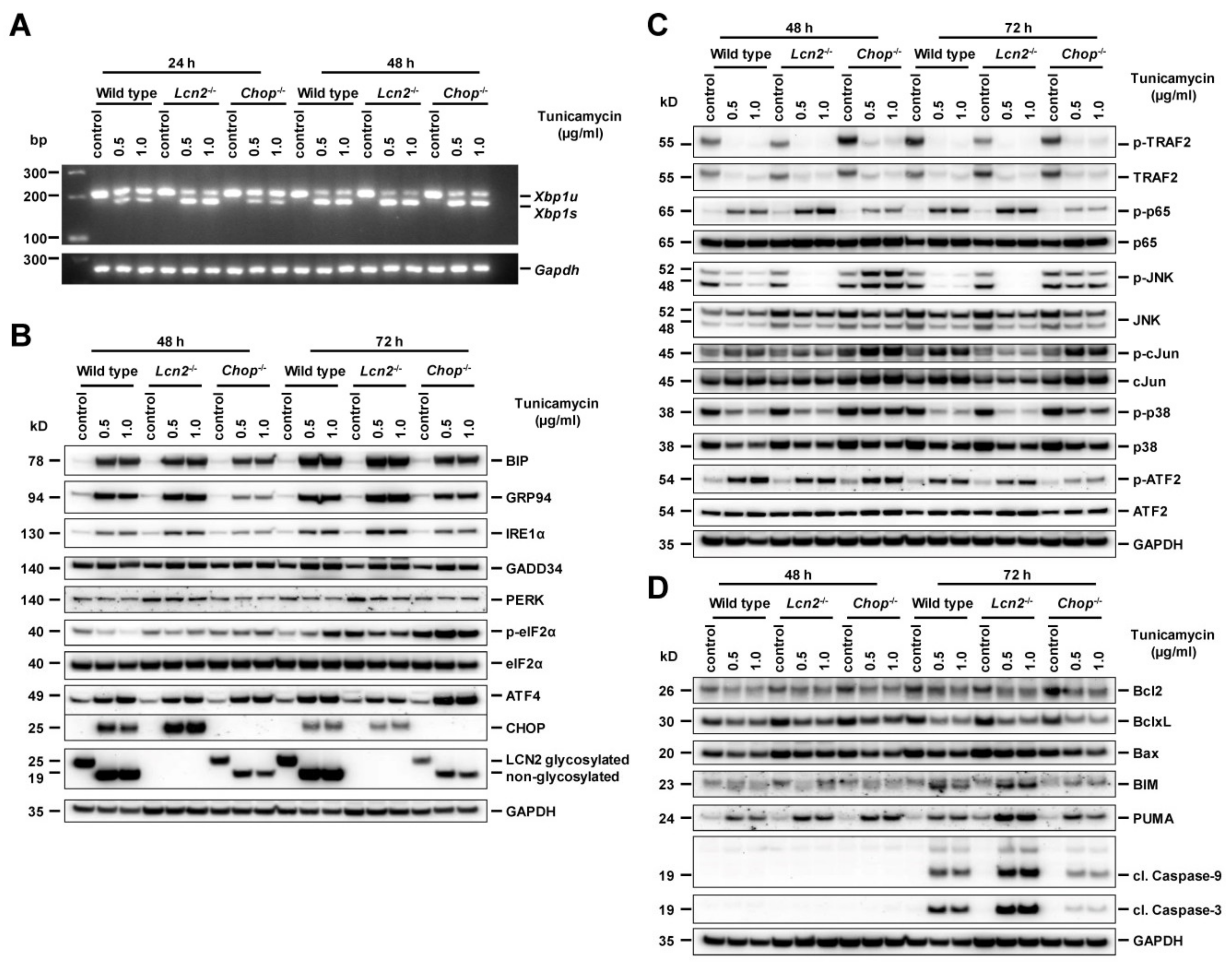
2.5. Pharmacological ER Stress Inducer Tunicamycin Activates TRAF2, c-Jun N-Terminal Kinase, NF-κB, and Mitochondrial Apoptotic Proteins in Hepatocytes
2.6. Chop−/− Alternates ER Stress Responses, Protects Against Tunicamycin-Activated ER Stress, and Induces Hepatocyte Apoptosis Through Delayed JNK Activation
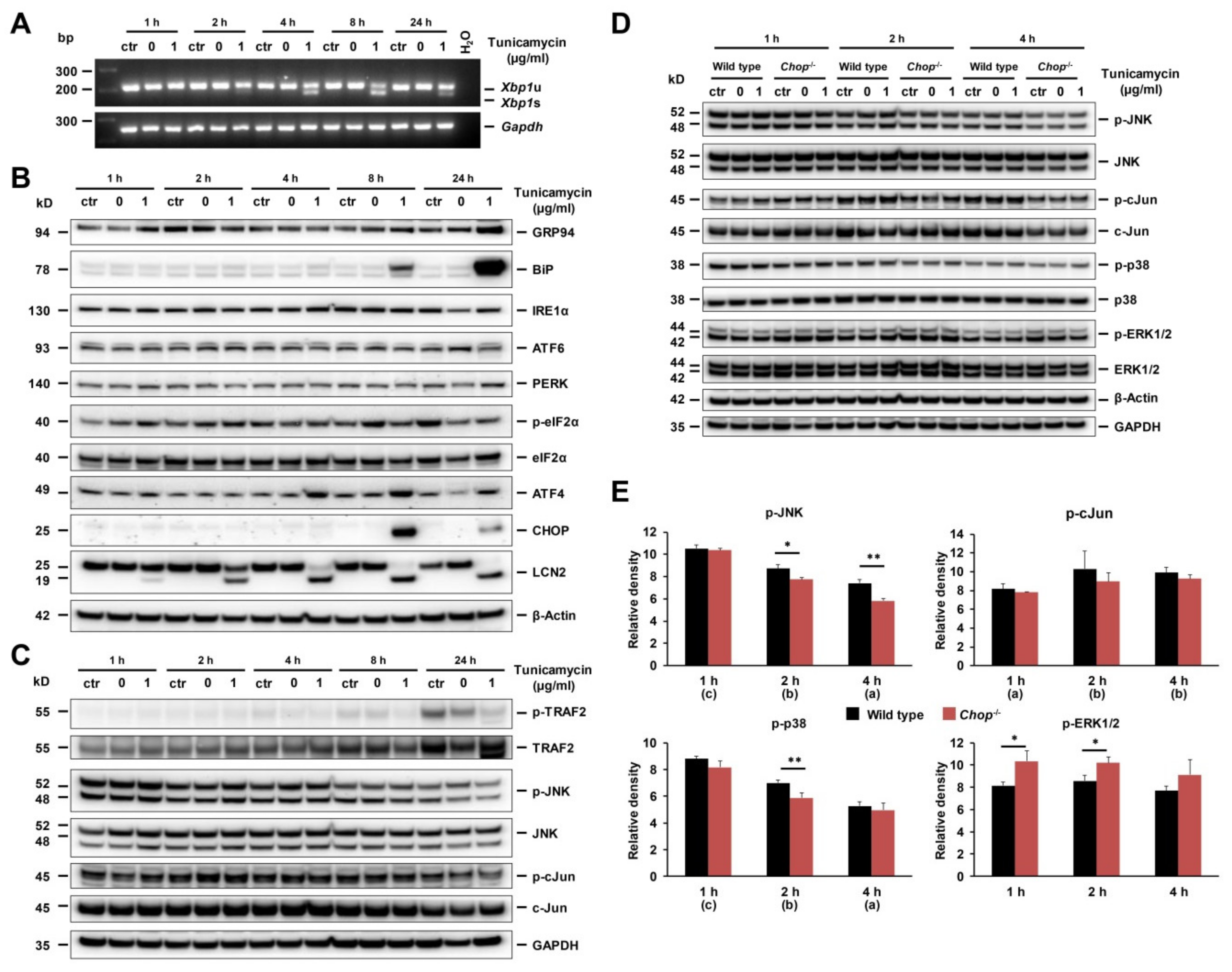
2.7. ER Stress and UPR in Early Stage Of Tunicamycin-Stimulated Primary Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Animal Experiments and Specimen Collections
4.2. RNA Isolation, cDNA Synthesis, and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
4.3. SDS-PAGE and Western Blot Analysis
4.4. Immunohistochemistry
4.5. Terminal Transferase dUTP Nick End-Labeling Assay (TUNEL)
4.6. Oil Red O Staining
4.7. Primary Liver Cell Isolation and Culturing
4.8. Tunicamycin and Thapsigargin Treatment
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | alanine transaminase |
| ASH | alcoholic steatohepatitis |
| AST | aspartate transaminase |
| ATF6 | activating transcription factor 6 |
| BIM | Bcl2-interacting protein/Bcl2-like protein 11 |
| BiP/GRP78 | binding immunoglobulin protein/glucose-regulated protein 78 |
| BSA | bovine serum albumin |
| CHOP/GADD153 | C/EBP homologous protein, growth arrest- and DNA damage-inducible gene |
| CREBH/CREB3L3 | cyclic AMP response element-binding protein H/cyclic AMP-responsive element-binding protein 3-like protein 3 |
| eIF2α | eukaryotic initiation factor 2α |
| ER | endoplasmic reticulum |
| GADD34 | growth arrest and DNA damage-inducible protein 34 |
| GRP94 | Glucose-regulated protein, 94kD |
| HCC | hepatocellular carcinoma |
| HDL | high-density lipoprotein |
| IRE1 | inositol-requiring enzyme 1 |
| LCN2 | Lipocalin 2 |
| LDL | low-density lipoprotein |
| PC | hepatocyte(s) |
| PBS | phosphate-buffered saline |
| PCR | polymerase chain reaction |
| PERK | protein kinase R (PKR)-like endoplasmic reticulum kinase |
| PUMA | p53-upregulated modulator of apoptosis |
| RT-qPCR | real-time quantitative polymerase chain reaction |
| TG | thapsigargin |
| TM | tunicamycin |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| UPR | unfolded protein response |
| Xbp1s | spliced X-box-binding protein 1 |
| Xbp1u | unspliced X-box-binding protein 1 |
References
- Kjeldsen, L.; Johnsen, A.H.; Sengelov, H.; Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 1993, 268, 10425–10432. [Google Scholar] [PubMed]
- Liu, Q.; Nilsen-Hamilton, M. Identification of a new acute phase protein. J. Biol. Chem. 1995, 270, 22565–22570. [Google Scholar] [CrossRef] [PubMed]
- Sultan, S.; Pascucci, M.; Ahmad, S.; Malik, I.A.; Bianchi, A.; Ramadori, P.; Ahmad, G.; Ramadori, G. Lipocalin-2 is a major acute-phase protein in a rat and mouse model of sterile abscess. Shock 2012, 37, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Van de Leur, E.; Haas, U.; Weiskirchen, R. Liver parenchymal cells lacking Lipocalin 2 (LCN2) are prone to endoplasmic reticulum stress and unfolded protein response. Cell Signal 2019, 55, 90–99. [Google Scholar] [CrossRef]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Berger, T.; Togawa, A.; Duncan, G.S.; Elia, A.J.; You-Ten, A.; Wakeham, A.; Fong, H.E.; Cheung, C.C.; Mak, T.W. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 1834–1839. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; van de Leur, E.; Zimmermann, H.W.; Karlmark, K.R.; Tihaa, L.; Haas, U.; Tacke, F.; Berger, T.; Mak, T.W.; Weiskirchen, R. Protective effects of lipocalin-2 (LCN2) in acute liver injury suggest a novel function in liver homeostasis. Biochim. Biophys. Acta 2013, 1832, 660–673. [Google Scholar] [CrossRef]
- Xu, M.J.; Feng, D.; Wu, H.; Wang, H.; Chan, Y.; Kolls, J.; Borregaard, N.; Porse, B.; Berger, T.; Mak, T.W.; et al. Liver is the major source of elevated serum lipocalin-2 levels after bacterial infection or partial hepatectomy: A critical role for IL-6/STAT3. Hepatology 2015, 61, 692–702. [Google Scholar] [CrossRef]
- Wieser, V.; Tymoszuk, P.; Adolph, T.E.; Grander, C.; Grabherr, F.; Enrich, B.; Pfister, A.; Lichtmanegger, L.; Gerner, R.; Drach, M.; et al. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J. Hepatol. 2016, 64, 872–880. [Google Scholar] [CrossRef]
- Cai, Y.; Jogasuria, A.; Yin, H.; Xu, M.J.; Hu, X.; Wang, J.; Kim, C.; Wu, J.; Lee, K.; Gao, B.; et al. The detrimental role played by lipocalin-2 in alcoholic fatty liver in mice. Am. J. Pathol. 2016, 186, 2417–2428. [Google Scholar] [CrossRef]
- Ye, D.; Yang, K.; Zang, S.; Lin, Z.; Chau, H.T.; Wang, Y.; Zhang, J.; Shi, J.; Xu, A.; Lin, S.; et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J. Hepatol. 2016, 65, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Drews, F.; Weiskirchen, R. Induction of lipocalin-2 expression in acute and chronic experimental liver injury moderated by pro-inflammatory cytokines interleukin-1β through nuclear factor-κB activation. Liver Int. 2011, 31, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xue, J.; Yang, Y.; Zhou, X.; Qin, C.; Zheng, M.; Zhu, H.; Liu, Y.; Liu, W.; Lou, G.; et al. Lipocalin 2 upregulation protects hepatocytes from IL1-β-induced stress. Cell Physiol. Biochem. 2015, 36, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Cramer, E.P.; Dahl, S.L.; Rozell, B.; Knudsen, K.J.; Thomsen, K.; Moser, C.; Cowland, J.B.; Borregaard, N. Lipocalin-2 from both myeloid cells and the epithelium combats Klebsiella pneumoniae lung infection in mice. Blood 2017, 129, 2813–2817. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef]
- Gorman, A.M.; Healy, S.J.; Jäger, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012, 134, 306–316. [Google Scholar] [CrossRef]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab Anim. 2015, 49 (Suppl. 1), 4–11. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.W.; Boll, M.; Stampfl, A. Hepatotoxicity and mechanism of action of haloalkanes: Carbon tetrachloride as a toxicological model. Crit. Rev. Toxicol. 2003, 33, 105–136. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Mendez, R.; Heng, H.H.; Yang, Z.Q.; Zhang, K. Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am. J. Transl. Res. 2012, 4, 102–113. [Google Scholar] [PubMed]
- Zhang, C.; Wang, G.; Zheng, Z.; Maddipati, K.R.; Zhang, X.; Dyson, G.; Williams, P.; Duncan, S.A.; Kaufman, R.J.; Zhang, K. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 2012, 55, 1070–1082. [Google Scholar] [CrossRef]
- Xu, X.; Park, J.G.; So, J.S.; Lee, A.H. Transcriptional activation of Fsp27 by the liver-enriched transcription factor CREBH promotes lipid droplet growth and hepatic steatosis. Hepatology 2015, 61, 857–869. [Google Scholar] [CrossRef]
- Gupta, S.; Campbell, D.; Dérijard, B.; Davis, R.J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 1995, 267, 389–393. [Google Scholar] [CrossRef]
- Waas, W.F.; Lo, H.H.; Dalby, K.N. The kinetic mechanism of the dual phosphorylation of the ATF2 transcription factor by p38 mitogen-activated protein (MAP) kinase alpha. Implications for signal/response profiles of MAP kinase pathways. J. Biol. Chem. 2001, 276, 5676–5684. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Wu, J.; Back, S.H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 2008, 15, 829–840. [Google Scholar] [CrossRef]
- Kammoun, H.L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferré, P.; Foufelle, F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Investig. 2009, 119, 1201–1215. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Campos, G.; Schmidt-Heck, W.; Ghallab, A.; Rochlitz, K.; Pütter, L.; Medinas, D.B.; Hetz, C.; Widera, A.; Cadenas, C.; Begher-Tibbe, B.; et al. The transcription factor CHOP, a central component of the transcriptional regulatory network induced upon CCl4 intoxication in mouse liver, is not a critical mediator of hepatotoxicity. Arch. Toxicol. 2014, 88, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, S.C.; Elmi, N.A.; Akazawa, Y.; Bronk, S.F.; Mott, J.L.; Gores, G.J. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G236–G243. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, Y.; Jadhav, K.; Li, Y.; Sun, H.; Yin, L.; Kasumov, T.; Chen, X.; Zhang, Y. Lipocalin-2 protects against diet-induced nonalcoholic fatty liver disease by targeting hepatocytes. Hepatol. Commun. 2019, 3, 763–775. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Borkham-Kamphorst, E.; Henning, M.; Yagmur, E.; Gassler, N.; Liedtke, C.; Berger, T.; Mak, T.W.; Weiskirchen, R. Lipocalin-2 (LCN2) regulates PLIN5 expression and intracellular lipid droplet formation in the liver. Biochim. Biophys. Acta 2014, 1842, 1513–1524. [Google Scholar] [CrossRef]
- Lee, E.K.; Kim, H.J.; Lee, K.J.; Lee, H.J.; Lee, J.S.; Kim, D.G.; Hong, S.W.; Yoon, Y.; Kim, J.S. Inhibition of the proliferation and invasion of hepatocellular carcinoma cells by lipocalin 2 through blockade of JNK and PI3K/Akt signaling. Int. J. Oncol. 2011, 38, 325–333. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; Huss, S.; Van de Leur, E.; Haas, U.; Weiskirchen, R. Adenoviral CCN3/NOV gene transfer fails to mitigate liver fibrosis in an experimental bile duct ligation model because of hepatocyte apoptosis. Liver Int. 2012, 32, 1342–1353. [Google Scholar] [CrossRef]
- Mueller, K.; Sunami, Y.; Stuetzle, M.; Guldiken, N.; Kucukoglu, O.; Mueller, S.; Kulaksiz, H.; Schwarz, P.; Strnad, P. CHOP-mediated hepcidin suppression modulates hepatic iron load. J. Pathol. 2013, 231, 532–542. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Murine Gene | Acc. No. | Primer |
|---|---|---|
| Xbp-1 | NM_001271730 | for: 5′-GAACCAGGAGTTAAGAACACG-3′ |
| rev: 5′-AGGCAACAGTGTCAGAGTCC-3′ | ||
| Xbp1u = 205 bp, Xbp1s = 179 bp | ||
| Gapdh | NM_008084 | for: 5′-TCGTGGATCTGACGTGCCGCCTG-3′ |
| rev: 5′-CACCACCCTGTTGCTGTAGCCGTAT-3′ | ||
| semi-qPCR = 251 bp | ||
| Bip | NM_001163434 | for: 5′-CTGAGGCGTATTTGGGAAAG-3′ |
| rev: 5′-TCATGACATTCAGTCCAGCAA-3′ | ||
| Grp94 | NM_011631 | for: 5′-AGGGTCCTGTGGGTGTTG-3′ |
| rev: 5′-CATCATCAGCTCTGACGAACC-3′ | ||
| Chop | NM_007837 | for: 5′-GCGACAGAGCCAGAATAACA-3′ |
| rev: 5′-GATGCACTTCCTTCTGGAACA-3′ | ||
| Creb3L3 | NM_145365 | for: 5′-CTCCCGCTTCAACCTCACT-3′ |
| rev: 5′-GCCAAGGAATGCTGTTGC-3′ | ||
| Atf4 | NM_001287180 | for: 5′-GGCTGGTCGTCAACCTATAAA-3′ |
| rev: 5′-CAGGCACTGCTGCCTCTAAT-3′ |
| Antibody | Cat. No. | Supplier | Dilution |
|---|---|---|---|
| ATF2 | 242 | Santa Cruz, Santa Cruz Biotech, CA, USA | 1:1000 |
| p-ATF2 | 8398 | Santa Cruz | 1:1000 |
| ATF4 | sc-390063 | Santa Cruz | 1:1000 |
| ATF6 | sc-166659 | Santa Cruz | 1:1000 |
| β-actin | A5441 | Sigma, Taufkirchen, Germany | 1:10,000 |
| Bak | 12105 | Cell Signaling, Darmstadt, Germany | 1:1000 |
| Bax | 14796 | Cell Signaling | 1:1000 |
| Bcl-xL | 2764 | Cell Signaling | 1:1000 |
| Bcl2 | 3498 | Cell Signaling | 1:1000 |
| BIM | 374358 | Santa Cruz | 1:1000 |
| BiP | 3177 | Cell Signaling | 1:1000 |
| c-Jun | 9165 | Cell Signaling | 1:1000 |
| CHOP/GADD153 | sc-7351 | Santa Cruz | 1:1000 |
| cleaved Caspase-3 | 9664 | Cell Signaling | 1:1000 |
| cleaved Caspase-9 | 9507 | Cell Signaling | 1:1000 |
| CREB3L3/CREBH | sc-377331 | Santa Cruz | 1:1000 |
| Cytochrome C | 11940 | Cell Signaling | 1:1000 |
| eIF2α | 9722 | Cell Signaling | 1:1000 |
| p-eIF2α | 3597 | Cell Signaling | 1:1000 |
| ERK1/2 | 9102 | Cell Signaling | 1:1000 |
| p-ERK1/2 | 9101 | Cell Signaling | 1:1000 |
| GADD34 | 373815 | Santa Cruz | 1:1000 |
| GAPDH | sc-32233 | Santa Cruz | 1:1000 |
| GRP94 | sc-393402 | Santa Cruz | 1:1000 |
| IRE1α | 3294 | Cell Signaling | 1:1000 |
| LCN2 | AF1857 | R&D Systems, Wiesbaden, Germany | 1:1000 |
| p21 | 556430 | BD Bioscience, Heidelberg, Germany | 1:1000 |
| p38 | 612168 | BD Biosciences | 1:2500 |
| p-p38 | 612288 | BD Biosciences | 1:2500 |
| p65 | sc-8008 | Santa Cruz | 1:1000 |
| p-p65 | 3033 | Cell Signaling | 1:1000 |
| p-c-Jun | 3270 | Cell Signaling | 1:1000 |
| PERK | 377400 | Santa Cruz | 1:1000 |
| PUMA | 374223 | Santa Cruz | 1:1000 |
| SAPK/JNK2 | 9258 | Cell Signaling | 1:1000 |
| p-SAPK/JNK | 4668 | Cell Signaling | 1:1000 |
| TRAF2 | 4712 | Cell Signaling | 1:1000 |
| p-TRAF2 | 13908 | Cell Signaling | 1:1000 |
| TRB3 | 390242 | Santa Cruz | 1:1000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borkham-Kamphorst, E.; Haas, U.; Van de Leur, E.; Trevanich, A.; Weiskirchen, R. Chronic Carbon Tetrachloride Applications Induced Hepatocyte Apoptosis in Lipocalin 2 Null Mice through Endoplasmic Reticulum Stress and Unfolded Protein Response. Int. J. Mol. Sci. 2020, 21, 5230. https://doi.org/10.3390/ijms21155230
Borkham-Kamphorst E, Haas U, Van de Leur E, Trevanich A, Weiskirchen R. Chronic Carbon Tetrachloride Applications Induced Hepatocyte Apoptosis in Lipocalin 2 Null Mice through Endoplasmic Reticulum Stress and Unfolded Protein Response. International Journal of Molecular Sciences. 2020; 21(15):5230. https://doi.org/10.3390/ijms21155230
Chicago/Turabian StyleBorkham-Kamphorst, Erawan, Ute Haas, Eddy Van de Leur, Anothai Trevanich, and Ralf Weiskirchen. 2020. "Chronic Carbon Tetrachloride Applications Induced Hepatocyte Apoptosis in Lipocalin 2 Null Mice through Endoplasmic Reticulum Stress and Unfolded Protein Response" International Journal of Molecular Sciences 21, no. 15: 5230. https://doi.org/10.3390/ijms21155230
APA StyleBorkham-Kamphorst, E., Haas, U., Van de Leur, E., Trevanich, A., & Weiskirchen, R. (2020). Chronic Carbon Tetrachloride Applications Induced Hepatocyte Apoptosis in Lipocalin 2 Null Mice through Endoplasmic Reticulum Stress and Unfolded Protein Response. International Journal of Molecular Sciences, 21(15), 5230. https://doi.org/10.3390/ijms21155230