Pro-Fibrotic Phenotype in a Patient with Segmental Stiff Skin Syndrome via TGF-β Signaling Overactivation

 , ,
, ,  , , , and
, , , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
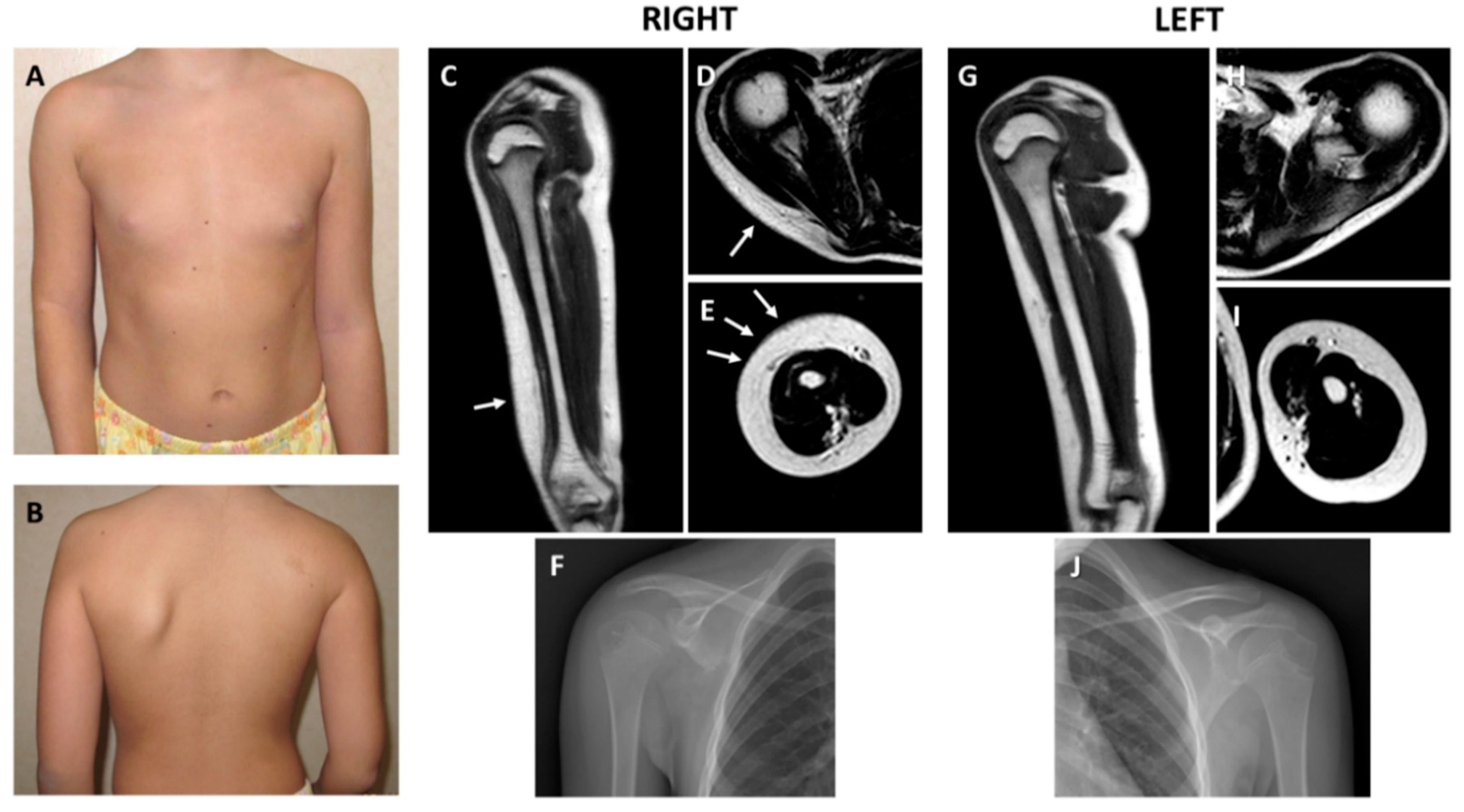
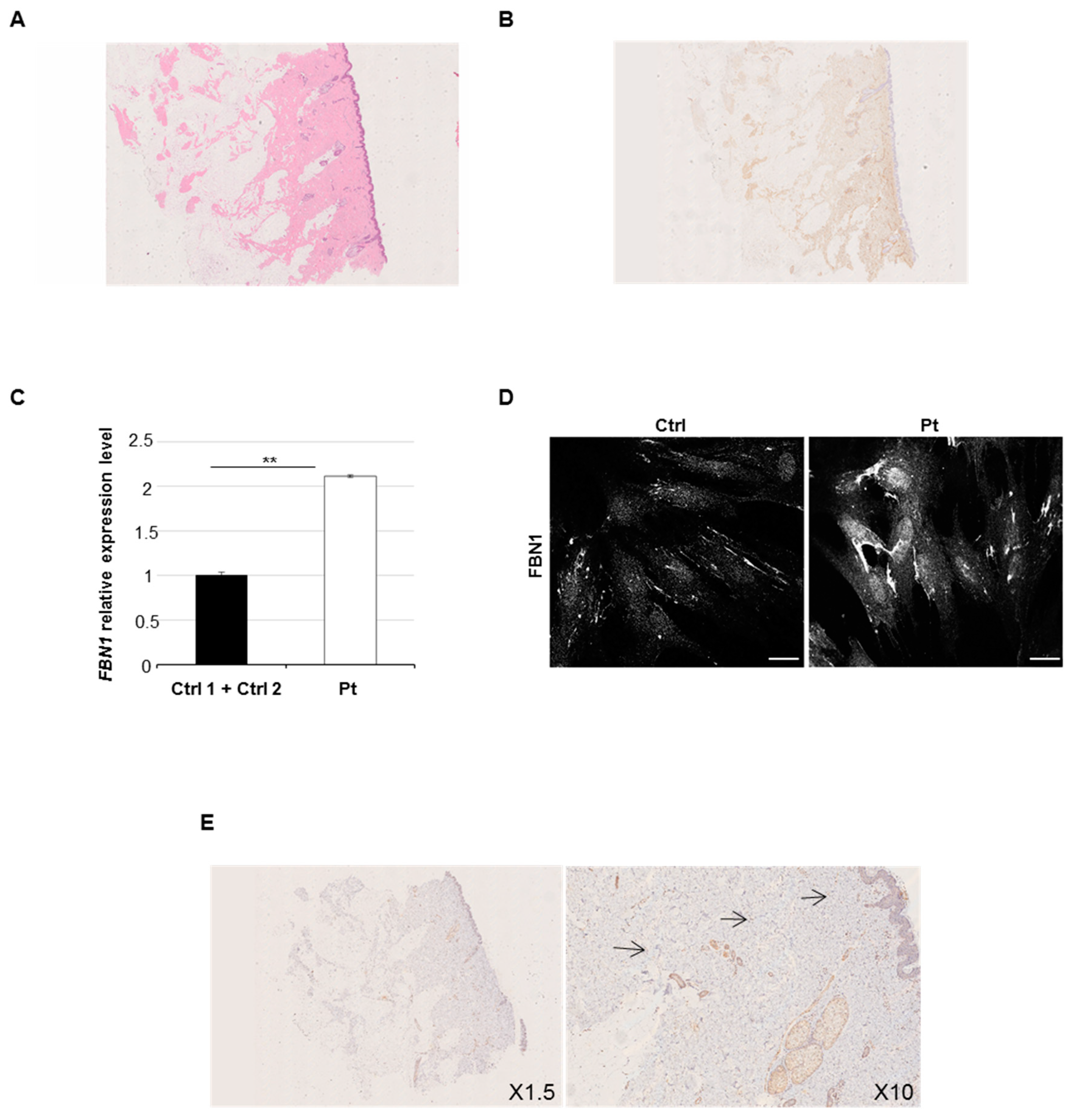
2.1. Clinical and Histological Findings
2.2. Germline and Somatic FBN1 Variants Analysis
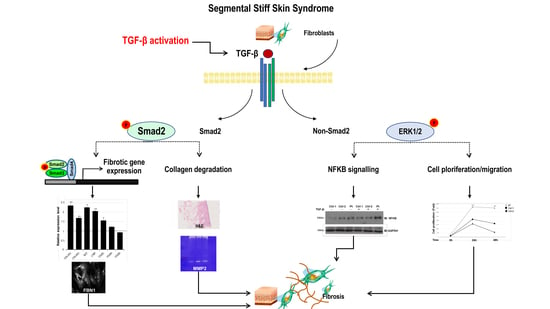
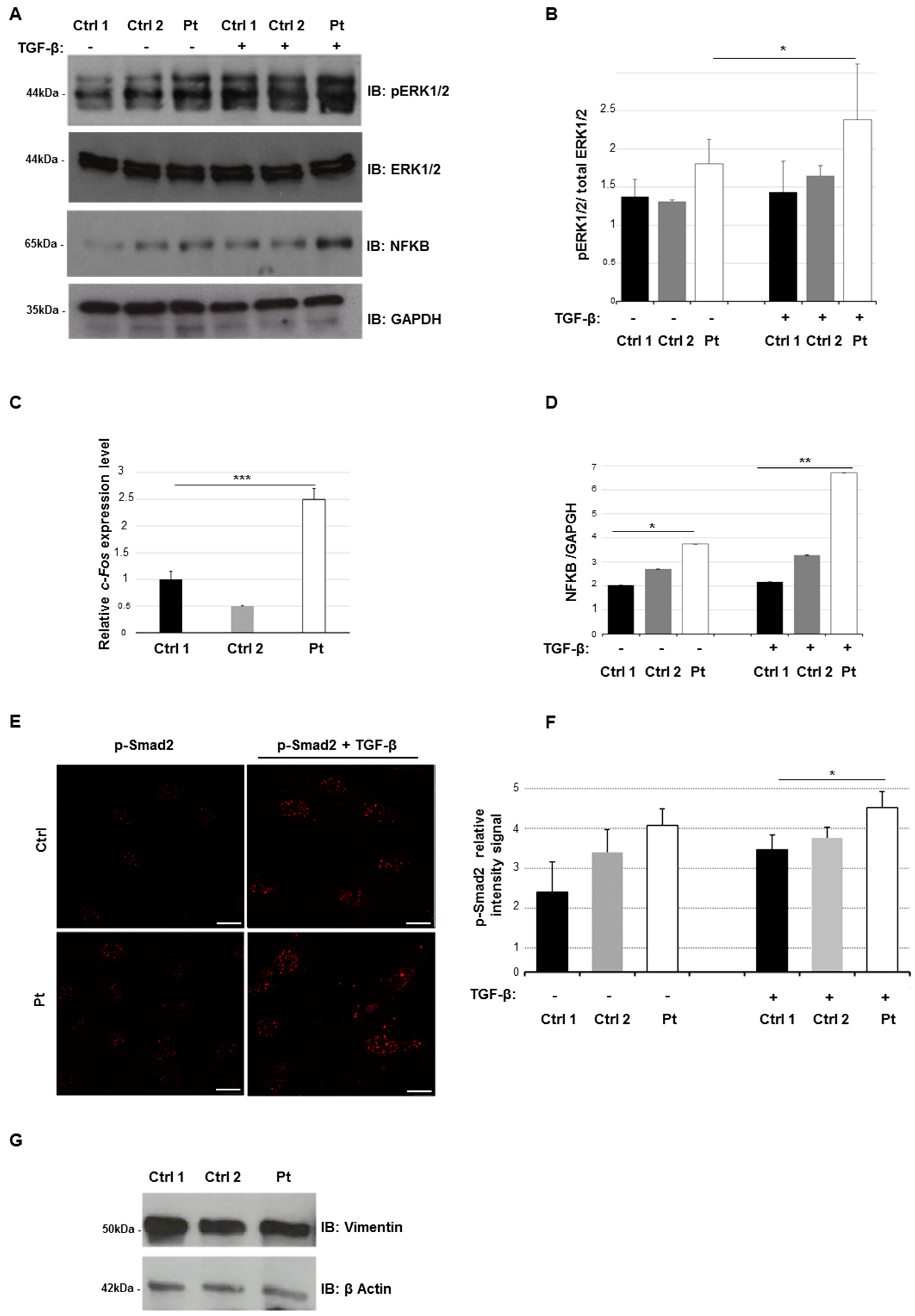
2.3. Altered Smad- and Non-Smad-Dependent TGF-β Signaling Pathways
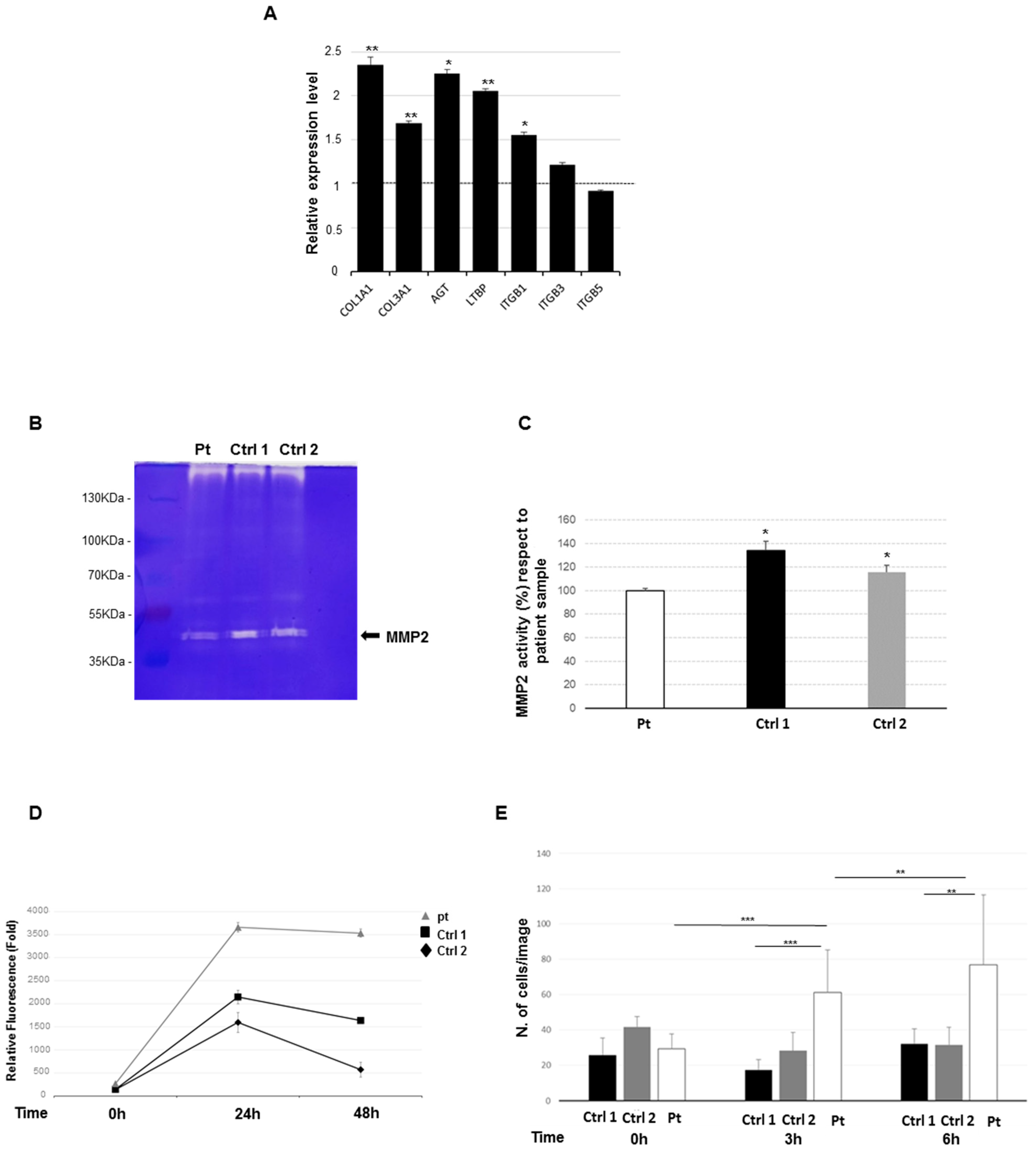
2.4. Increased Expression Profile of Key Pro-Fibrotic Molecules
2.5. Reduced Metalloproteinase 2 Activity
2.6. Increased Cell Proliferation and Migration
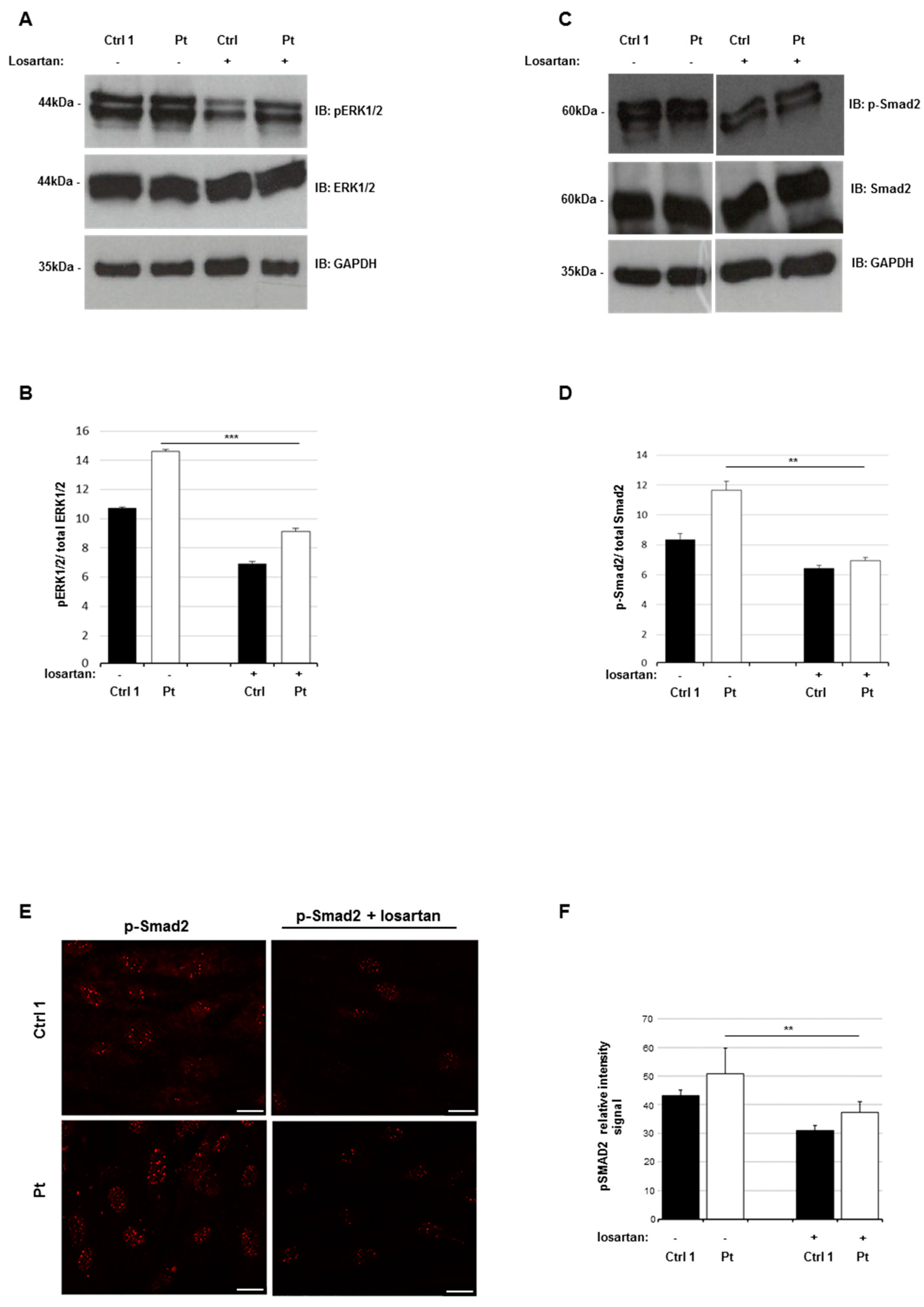
2.7. Losartan Reduces the Activation Status of TGF-β Signaling Pathways in SSS Cells
3. Discussion
4. Materials and Methods
4.1. Patient and Samples
4.2. Cell Cultures
4.3. DNA Extraction and Next-Generation Sequencing
4.4. CytoScan XON
4.5. Immunohistochemistry Analysis
4.6. RNA Extraction and Reverse Transcription
4.7. Quantitative Real-Time PCR
4.8. Western Blotting and Quantification of Phosphorylated Proteins
4.9. Confocal Study
4.10. Zymography
4.11. PrestoBlue Assay
4.12. Transwell Migration Assay
4.13. Losartan Treatment
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CNVs | copy number variations |
| COLL | collagens |
| Ctrl | control |
| D-MEM | Dulbecco’s Modified Eagle Medium |
| ECM | extracellular matrix |
| ERK | extracellular signal-regulated kinase |
| FBN1 | fibrillin-1 |
| LTBP | Latent transforming growth factor beta binding protein |
| MMP2 | Matrix metalloproteinase 2 |
| MRI | Magnetic Resonance Imaging |
| NGS | next generation sequencing |
| NF-κB | Nuclear factor κ-B |
| PBS | Phosphate-buffered saline |
| Pt | patient |
| qPCR | quantitative polymerase chain reaction |
| ROI | region of interest |
| SDS-PAGE | sulfate: polyacrylamide gel electrophoresis |
| SSS | stiff skin syndrome |
| TGF-β | Transforming growth factor β |
| Tsk | Tight skin mice |
References
- Esterly, N.B.; McKusick, V.A. Stiff skin syndrome. Pediatrics 1971, 47, 360–369. [Google Scholar] [PubMed]
- Amoric, J.C.; Stalder, J.F.; David, A.; Bureau, B.; Pierard, G.E.; Litoux, P. Dysmorphism in Stiff Skin syndrome. Annales de Dermatologie et de Vénéréologie 1991, 118, 802–804. [Google Scholar] [PubMed]
- Bodemer, C.; Habib, K.; Teillac, D.; Munich, A.; de Prost, Y. A new case of Stiff Skin syndrome. Annales de Dermatologie et de Vénéréologie 1991, 118, 805–806. [Google Scholar] [PubMed]
- Bundy, S.E.; Lie, K. Stiff skin syndrome. Birth Defects Orig. Artic. Ser. 1975, 11, 360–361. [Google Scholar]
- DiRocco, M. Clinical images: Stiff skin syndrome. Arthritis Rheum. 2000, 43, 1542. [Google Scholar]
- Ferrari, D.; Rossi, R.; Donzelli, O. Stiff-skin syndrome. La Chirurgia degli Organi di Movimento 2005, 90, 69–73. [Google Scholar]
- Gilaberte, Y.; Saenz-de-Santamaria, M.C.; Garcia-Latasa, F.J.; Gonzalez-Mediero, I.; Zambrano, A. Stiff skin syndrome: A case report and review of the literature. Dermatology 1995, 190, 148–151. [Google Scholar] [CrossRef]
- Helm, T.N.; Wirth, P.B.; Helm, K.F. Congenital fascial dystrophy: The stiff skin syndrome. Cutis 1997, 60, 153–154. [Google Scholar]
- Jablonska, S.; Blaszczyk, M. Stiff Skin Syndrome is Highly Heterogeneous, and Congenital Fascial Dystrophy is its Distinct Subset. Pediatr. Dermatol. 2004, 21, 508–510. [Google Scholar] [CrossRef]
- Jablonska, S.; Blaszczyk, M. Scleroderma-like indurations involving fascias: An abortive form of congenital fascial dystrophy (Stiff skin syndrome). Pediatr. Dermatol. 2000, 17, 105–110. [Google Scholar] [CrossRef]
- Lê, T.; Piérard, G.E. Stiff skin syndrome. Annales de Dermatologie et de Vénéréologie 1989, 116, 807–809. [Google Scholar] [PubMed]
- Loeys, B.; Gerber, E.E.; Riegert-Johnson, D.; Iqbal, S.; Whiteman, P.; McConnell, V.; Chillakuri, C.R.; Macaya, D.; Coucke, P.J.; De Paepe, A.; et al. Mutations in Fibrillin-1 Cause Congenital Scleroderma: Stiff Skin Syndrome. Sci. Transl. Med. 2010, 2, 23ra20. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; McCalmont, T.H.; Frieden, I.J.; Williams, M.L.; Connolly, M.K.; Gilliam, A.E. The stiff skin syndrome: Case series, differential diagnosis of the stiff skin phenotype, and review of the literature. Arch. Dermatol. 2008, 144, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Kurtzman, D.J.B.; Wright, N.A.; Patel, M.; Vleugels, R.A. Segmental stiff skin syndrome (SSS): Two additional cases with a positive response to mycophenolate mofetil and physical therapy. J. Am. Acad. Dermatol. 2016, 75, e237–e239. [Google Scholar] [CrossRef] [PubMed]
- Rangu, S.; Rubin, A.I.; Li, D.; Castelo-Soccio, L. Segmental stiff skin syndrome: A novel case with an interleukin-17C mutation successfully treated with secukinumab. Clin. Exp. Dermatol. 2020, 45, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Maillet-Lebel, N.; Kokta, V.; Coulombe, J.; Powell, J. A case of segmental stiff skin syndrome treated with systemic losartan. Pediatr. Dermatol. 2017, 35, e66–e67. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Yagi, H.; Hara, H.; Fujiwara, T.; Fujita, D.; Nawata, K.; Inuzuka, R.; Taniguchi, Y.; Harada, M.; Toko, H.; et al. Pathophysiology and Management of Cardiovascular Manifestations in Marfan and Loeys–Dietz Syndromes. Int. Hear. J. 2016, 57, 271–277. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Verrecchia, F.; Vindevoghel, L.; Lechleider, R.J.; Uitto, J.; Roberts, A.B.; Mauviel, A. Smad3/AP-1 interactions control transcriptional responses to TGF-β in a promoter-specific manner. Oncogene 2001, 20, 3332–3340. [Google Scholar] [CrossRef]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef]
- Dennler, S.; Itoh, S.; Vivien, D.; Dijke, P.T.; Huet, S.; Gauthier, J. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Nakerakanti, S.S.; Bujor, A.M.; Trojanowska, M. CCN2 Is Required for the TGF-β Induced Activation of Smad1 - Erk1/2 Signaling Network. PLoS ONE 2011, 6, e21911. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J. Immunol. 2005, 175, 7708–7718. [Google Scholar] [CrossRef]
- Pietzsch, S.; Ricke-Hoch, M.; Stapel, B.; Hilfiker-Kleiner, D. Modulation of cardiac AKT and STAT3 signalling in preclinical cancer models and their impact on the heart. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1867, 118519. [Google Scholar] [CrossRef]
- Weekes, D.; Kashima, T.G.; Zandueta, C.; Perurena, N.; Thomas, D.P.; Sunters, A.; Vuillier, C.; Bozec, A.; El-Emir, E.; Miletich, I.; et al. Regulation of osteosarcoma cell lung metastasis by the c-Fos/AP-1 target FGFR1. Oncogene 2015, 35, 2852–2861. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. NF-κB signaling, liver disease and hepatoprotective agents. Oncogene 2008, 27, 6228–6244. [Google Scholar] [CrossRef]
- Van Caam, A.; Vonk, M.; Hoogen, F.V.D.; Van Lent, P.; Van Der Kraan, P. Unraveling SSc Pathophysiology; The Myofibroblast. Front. Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef]
- Tidin, O.; Friman, E.; Naef, F.; Suter, D.M. Quantitative relationships between SMAD dynamics and target gene activation kinetics in single live cells. Sci. Rep. 2019, 9, 5372. [Google Scholar] [CrossRef]
- Qian, W.; Cai, X.; Qian, Q.; Peng, W.; Yu, J.; Zhang, X.; Tian, L.; Wang, C. lncRNA ZEB1-AS1 promotes pulmonary fibrosis through ZEB1-mediated epithelial–mesenchymal transition by competitively binding miR-141-3p. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Onozuka, I.; Kakinuma, S.; Kamiya, A.; Miyoshi, M.; Sakamoto, N.; Kiyohashi, K.; Watanabe, T.; Funaoka, Y.; Ueyama, M.; Nakagawa, M.; et al. Cholestatic liver fibrosis and toxin-induced fibrosis are exacerbated in matrix metalloproteinase-2 deficient mice. Biochem. Biophys. Res. Commun. 2011, 406, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Model. Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Droppelmann, C.A.; Gutiérrez, J.; Vial, C.; Brandan, E. Matrix Metalloproteinase-2-deficient Fibroblasts Exhibit an Alteration in the Fibrotic Response to Connective Tissue Growth Factor/CCN2 because of an Increase in the Levels of Endogenous Fibronectin. J. Boil. Chem. 2009, 284, 13551–13561. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Peng, Z.; Zu, C.; Ma, J.; Lu, S.; Zhong, J.; Zhang, S. Losartan Attenuates Myocardial Endothelial-To-Mesenchymal Transition in Spontaneous Hypertensive Rats via Inhibiting TGF-beta/Smad Signaling. PLoS ONE 2016, 11, e0155730. [Google Scholar] [CrossRef]
- Rodriguez-Vita, J.; Sanchez-Lopez, E.; Esteban, V.; Rupérez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II Activates the Smad Pathway in Vascular Smooth Muscle Cells by a Transforming Growth Factor-β–Independent Mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef]
- Habashi, J.P.; Doyle, J.J.; Holm, T.M.; Aziz, H.; Schoenhoff, F.; Bedja, D.; Chen, Y.; Modiri, A.N.; Judge, D.P.; Dietz, H.C. Angiotensin II Type 2 Receptor Signaling Attenuates Aortic Aneurysm in Mice Through ERK Antagonism. Science 2011, 332, 361–365. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Huang, W.; Zhang, P.; Guo, Y.; Körner, H.; Wu, H.; Wei, W. Losartan suppresses the inflammatory response in collagen-induced arthritis by inhibiting the MAPK and NF-kappaB pathways in B and T cells. Inflammopharmacology 2018, 27, 487–502. [Google Scholar] [CrossRef]
- Richard, M.-A.; Grob, J.-J.; Philip, N.; Rey, J.; Chamson, A.; Mege, J.; Andrac, L.; Faure, F.; Basseres, N.; Bonerandi, J. Physiopathogenic investigations in a case of familial stiff-skin syndrome. Dermatology 1998, 197, 127–131. [Google Scholar] [CrossRef]
- Kikuchi, I.; Inoue, S.; Hamada, K.; Ando, H. Stiff Skin Syndrome. Pediatr. Dermatol. 1985, 3, 48–53. [Google Scholar] [CrossRef]
- Jablonska, S.; Schubert, H.; Kikuchi, I. Congenital fascial dystrophy: Stiff skin syndrome—A human counterpart of the tight-skin mouse. J. Am. Acad. Dermatol. 1989, 21, 943–950. [Google Scholar] [CrossRef]
- Jablonska, S.; Groniowski, J.; Krieg, T.; Nerlich, A.G.; Peltonen, L.; Oikarinen, A.; Debrowski, J.; Pietrow, D. Congenital Fascial Dystrophy—A Noninflammatory Disease of Fascia: The Stiff Skin Syndrome. Pediatr. Dermatol. 1984, 2, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Fidzianska, A.; Jablonska, S. Congenital fascial dystrophy: Abnormal composition of the fascia. J. Am. Acad. Dermatol. 2000, 43, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Corson, G.M.; Chalberg, S.C.; Dietz, H.C.; Charbonneau, N.L.; Sakai, L.Y. Fibrillin Binds Calcium and Is Coded by cDNAs That Reveal a Multidomain Structure and Alternatively Spliced Exons at the 5′ End. Genomics 1993, 17, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Gerber, E.E.; Gallo, E.M.; Fontana, S.C.; Davis, E.C.; Wigley, F.M.; Huso, D.L.; Dietz, H.C. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature 2013, 503, 126–130. [Google Scholar] [CrossRef]
- Del Cid, J.S.; Reed, N.I.; Molnar, K.; Liu, S.; Dang, B.; Jensen, S.; DeGrado, W.; Handford, P.A.; Sheppard, D.; Sundaram, A.A. A disease-associated mutation in fibrillin-1 differentially regulates integrin-mediated cell adhesion. J. Boil. Chem. 2019, 294, 18232–18243. [Google Scholar] [CrossRef]
- Siracusa, L.D.; McGrath, R.; Ma, Q.; Moskow, J.J.; Manné, J.; Christner, P.J.; Buchberg, A.M.; A Jimenez, S. A tandem duplication within the fibrillin 1 gene is associated with the mouse tight skin mutation. Genome Res. 1996, 6, 300–313. [Google Scholar] [CrossRef][Green Version]
- Barisic-Dujmovic, T.; Boban, I.; Adams, U.J.; Clark, S.H. Marfan-Like Skeletal Phenotype in the Tight Skin (Tsk) Mouse. Calcif. Tissue Int. 2007, 81, 305–315. [Google Scholar] [CrossRef]
- Saito, S.; Nishimura, H.; Brumeanu, T.-D.; Casares, S.; Stan, A.C.; Honjo, T.; Bona, C.A. Characterization of mutated protein encoded by partially duplicated fibrillin-1 gene in tight skin (TSK) mice. Mol. Immunol. 1999, 36, 169–176. [Google Scholar] [CrossRef]
- Danen, E.H.; Sonnenberg, A. Integrins in regulation of tissue development and function. J. Pathol. 2003, 201, 632–641. [Google Scholar] [CrossRef]
- Varga, J.; Pasche, B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat. Rev. Rheumatol. 2009, 5, 200–206. [Google Scholar] [CrossRef]
- Zeng, Z.-S.; Cohen, A.M.; Guillem, J.G. Loss of basement membrane type IV collagen is associated with increased expression of metalloproteinases 2 and 9 (MMP-2 and MMP-9) during human colorectal tumorigenesis. Carcinogenesis 1999, 20, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Mahalanobish, S.; Saha, S.; Dutta, S.; Sil, P.C. Matrix metalloproteinase: An upcoming therapeutic approach for idiopathic pulmonary fibrosis. Pharmacol. Res. 2020, 152, 104591. [Google Scholar] [CrossRef] [PubMed]
- Rongioletti, F.; Ferreli, C.; Atzori, L.; Bottoni, U.; Soda, G. Scleroderma with an update about clinico-pathological correlation. G. Ital. Dermatol. Venereol. 2018, 153, 208–215. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Amorim, A.G.; Aide, M.K.; Duraes, S.M.; Rochael, M.C. Stiff skin syndrome—Case report. An. Bras. Dermatol. 2011, 86, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Filocamo, M.; Baldo, C.; Goldwurm, S.; Renieri, A.; Angelini, C.; Moggio, M.; Mora, M.; Merla, G.; Politano, L.; Garavaglia, B.; et al. Telethon Network of Genetic Biobanks: A key service for diagnosis and research on rare diseases. Orphanet J. Rare Dis. 2013, 8, 129. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.M.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for General Users and for Biologist Programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Fusco, C.; Mandriani, B.; Di Rienzo, M.; Micale, L.; Malerba, N.; Cocciadiferro, D.; Sjottem, E.; Augello, B.; Squeo, G.M.; Pellico, M.T.; et al. TRIM50 regulates Beclin 1 proautophagic activity. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Altamura, G.; Martano, M.; Licenziato, L.; Maiolino, P.; Borzacchiello, G. Telomerase Reverse Transcriptase (TERT) Expression, Telomerase Activity, and Expression of Matrix Metalloproteinases (MMP)-1/-2/-9 in Feline Oral Squamous Cell Carcinoma Cell Lines Associated With Felis catus Papillomavirus Type-2 Infection. Front. Vet. Sci. 2020, 7, 148. [Google Scholar] [CrossRef] [PubMed]
- Deepe, G.S.; Buesing, W.R. Deciphering the pathways of death of Histoplasma capsulatum-infected macrophages: Implications for the immunopathogenesis of early infection. J. Immunol. 2011, 188, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, P.; Mithbaokar, P.; Sabatino, V.; Tolmie, J.; Melis, D.; Schiaffino, M.C.; Filocamo, M.; Andria, G.; Brunetti-Pierri, N. SMAD4 mutations causing Myhre syndrome result in disorganization of extracellular matrix improved by losartan. Eur. J. Hum. Genet. 2014, 22, 988–994. [Google Scholar] [CrossRef]
- Piccolo, P.; Sabatino, V.; Mithbaokar, P.; Polishchuk, E.; Hicks, J.; Polishchuk, R.; Bacino, C.A.; Brunetti-Pierri, N. Skin fibroblasts of patients with geleophysic dysplasia due to FBN1 mutations have lysosomal inclusions and losartan improves their microfibril deposition defect. Mol. Genet. Genom. Med. 2019, 7, e844. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fusco, C.; Nardella, G.; Augello, B.; Boccafoschi, F.; Palumbo, O.; Fusaro, L.; Notarangelo, A.; Barbano, R.; Parrella, P.; Annicchiarico, G.; et al. Pro-Fibrotic Phenotype in a Patient with Segmental Stiff Skin Syndrome via TGF-β Signaling Overactivation. Int. J. Mol. Sci. 2020, 21, 5141. https://doi.org/10.3390/ijms21145141
Fusco C, Nardella G, Augello B, Boccafoschi F, Palumbo O, Fusaro L, Notarangelo A, Barbano R, Parrella P, Annicchiarico G, et al. Pro-Fibrotic Phenotype in a Patient with Segmental Stiff Skin Syndrome via TGF-β Signaling Overactivation. International Journal of Molecular Sciences. 2020; 21(14):5141. https://doi.org/10.3390/ijms21145141
Chicago/Turabian StyleFusco, Carmela, Grazia Nardella, Bartolomeo Augello, Francesca Boccafoschi, Orazio Palumbo, Luca Fusaro, Angelantonio Notarangelo, Raffaela Barbano, Paola Parrella, Giuseppina Annicchiarico, and et al. 2020. "Pro-Fibrotic Phenotype in a Patient with Segmental Stiff Skin Syndrome via TGF-β Signaling Overactivation" International Journal of Molecular Sciences 21, no. 14: 5141. https://doi.org/10.3390/ijms21145141
APA StyleFusco, C., Nardella, G., Augello, B., Boccafoschi, F., Palumbo, O., Fusaro, L., Notarangelo, A., Barbano, R., Parrella, P., Annicchiarico, G., De Meco, C., Micale, L., Graziano, P., & Castori, M. (2020). Pro-Fibrotic Phenotype in a Patient with Segmental Stiff Skin Syndrome via TGF-β Signaling Overactivation. International Journal of Molecular Sciences, 21(14), 5141. https://doi.org/10.3390/ijms21145141