Targeting Cannabinoid Receptors: Current Status and Prospects of Natural Products



Abstract
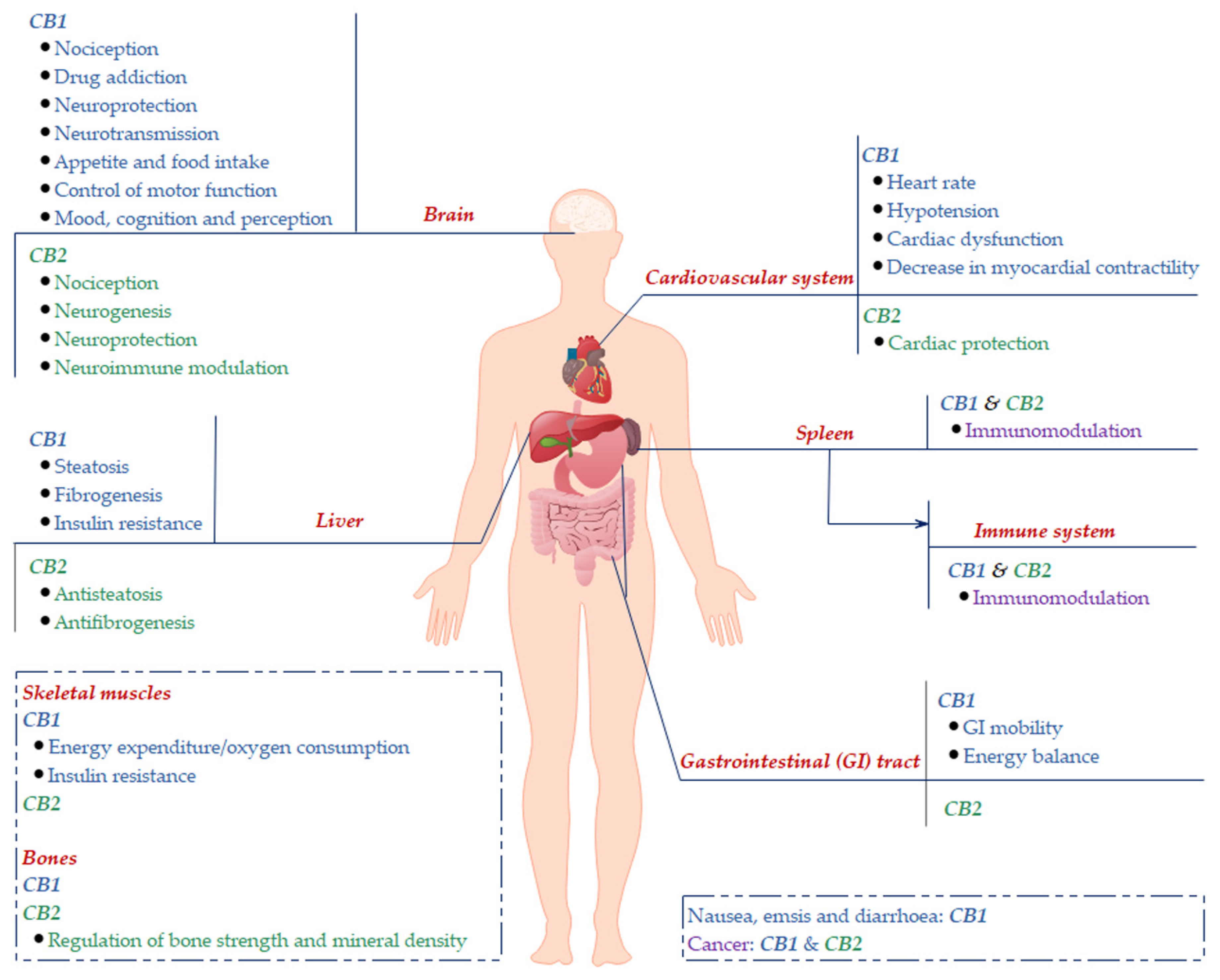
1. Introduction
2. Cannabinoid Receptors
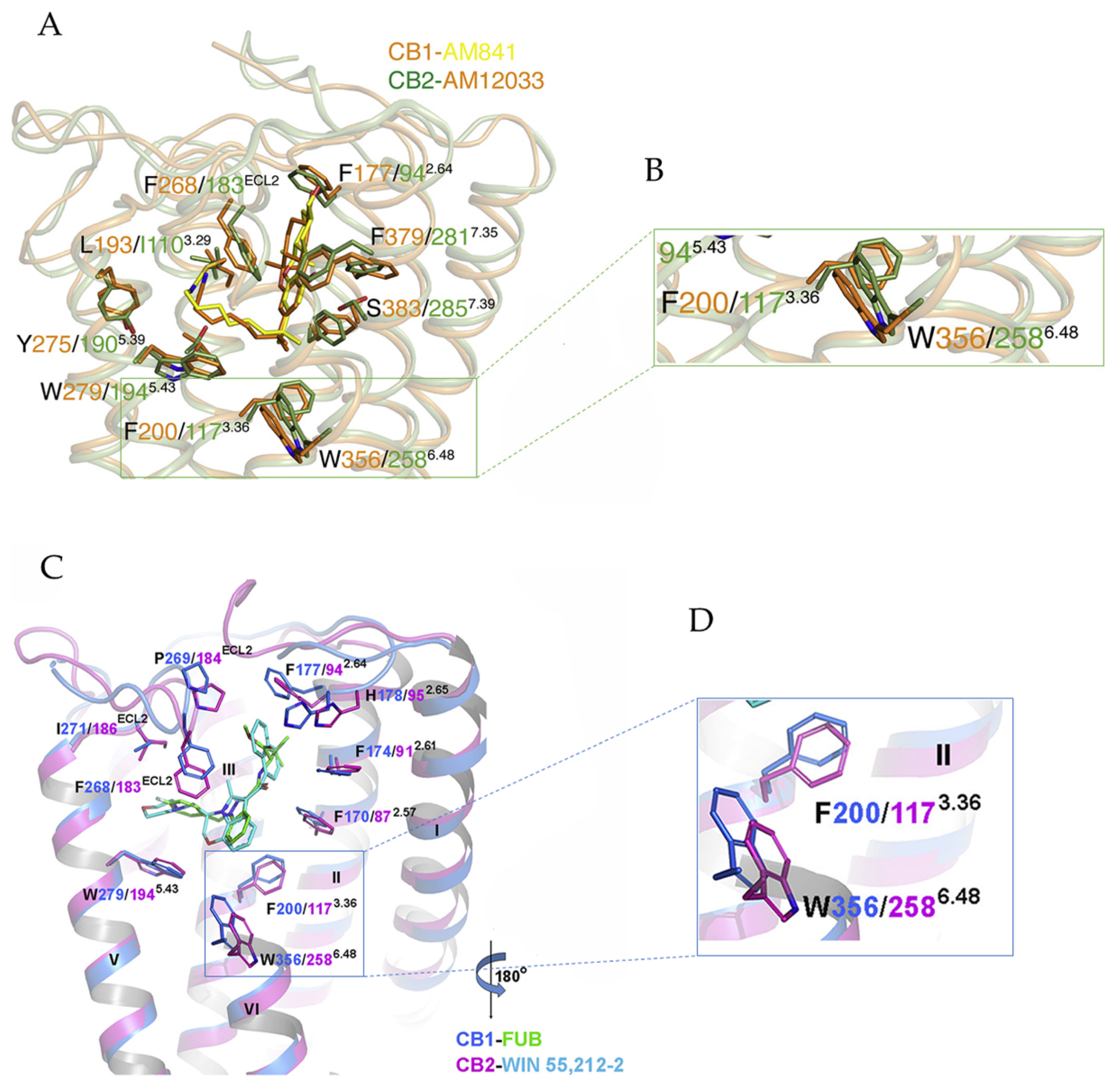
3. Ligand-Bound CB1/CB2-Gi Complex
3.1. Activation Mechanism of CB1 and CB2
3.2. Implications for CB1 and CB2 Ligand Selectivity
4. Phytocannabinoids and Synthetic Cannabinoids
4.1. Phytocannabinoids
4.1.1. Δ9-Tetrahydrocannabinol (Δ9-THC)
4.1.2. Cannabidiol (CBD)
4.1.3. Δ9-Tetrahydrocannabiphorol (Δ9-THCP) and Cannabidiphorol (CBDP)
4.2. Synthetic Cannabinoids
4.2.1. Mixed CB1/CB2 Agonists
4.2.2. CB1-Selective Agonists
4.2.3. CB2-Selective Agonists
4.2.4. CB1-Selective Antagonists/Inverse Agonists
4.2.5. CB2-Selective Antagonists/Inverse Agonists
4.2.6. Allosteric Modulators
5. Novel Cannabinoids from Animal Sources
5.1. Peptides Targeting CB1/CB2
5.1.1. Hemopressin and its Derivates VD-Hpα and RVD-Hpα
5.1.2. Pep19
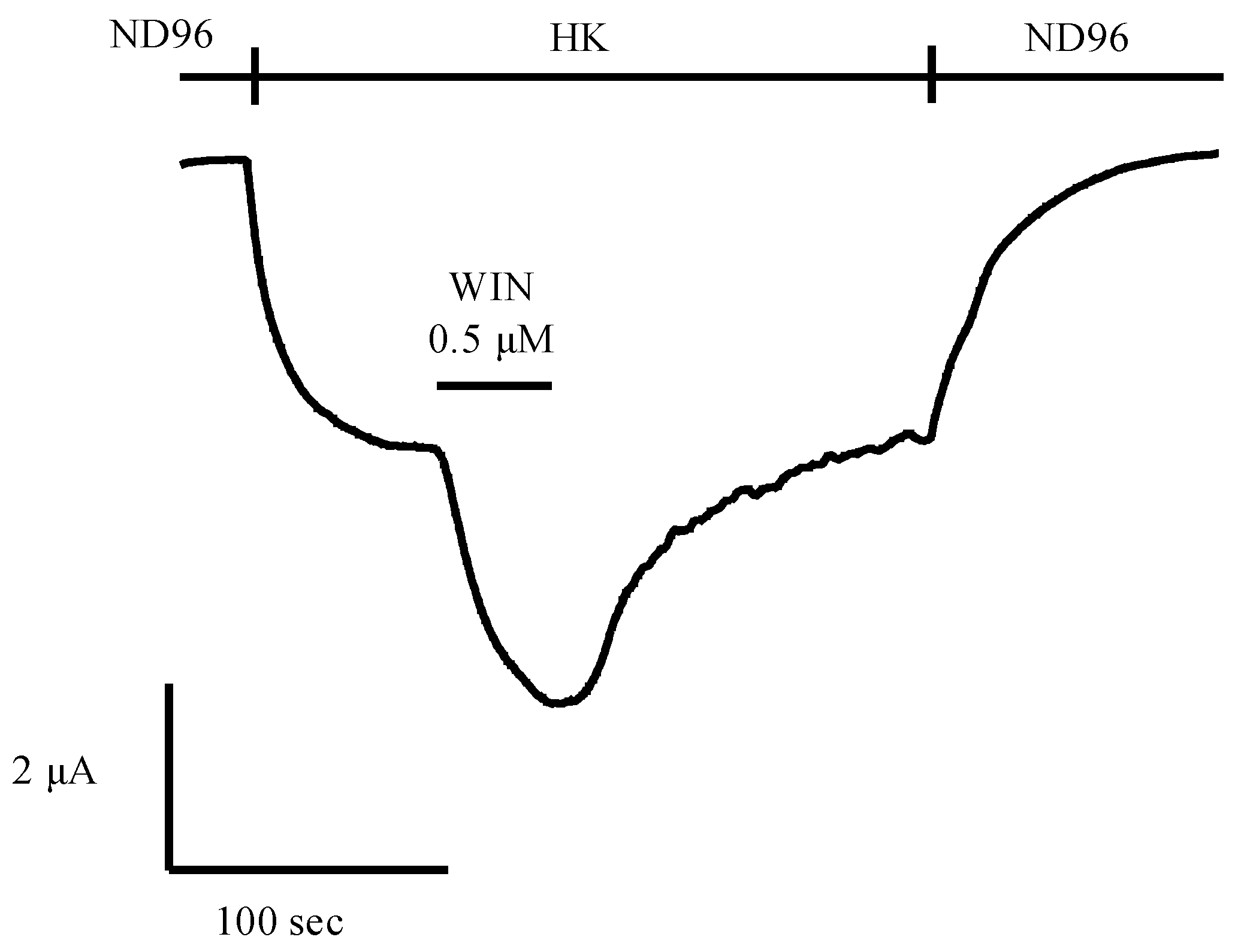
5.2. Potential of Animal Venoms as a Source for Novel Cannabinoids
6. Expression Systems of Cannabinoid Receptors Used for Screening Natural Products In Vitro
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Δ9-THC | Δ9-tetrahydrocannabinol |
| Δ9-THCP | Δ9-tetrahydrocannabiphorol |
| ACEA | Arachidonyl-2′-chloroethylamide |
| ACPA | Arachidonylcyclopropylamide |
| AEA | Anandamide or arachidonoylethanolamide |
| AM-251 | 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-piperidin-1-ylpyrazole-3-carboxamide |
| AM-281 | 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-morpholin-4-ylpyrazole-3-carboxamide |
| AM-630 | [6-iodo-2-methyl-1-(2-morpholin-4-ylethyl)indol-3-yl]-(4-methoxyphenyl)methanone or 6-iodopravadoline |
| CBD | Cannabidiol |
| CBDP | Cannabidiphorol |
| Compound C2 | N-[5-bromo-1,2-dihydro-1-(4′-fluorobenzyl)-4-methyl-2-ox-opyridin-3yl]cycloheptanecarboxamide |
| CP-55,940 | 2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol |
| GAT211 | 3-(2-nitro-1-phenylethyl)-2-phenyl-1H-indole |
| GAT229 | S-(-)-3-(2-nitro-1-phenylethyl)-2-phenyl-1H-indole |
| GW-405833 | (2,3-dichlorophenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-ylethyl)indol-1-yl]methanone |
| HK | High potassium solution (96mM KCl, 2 mM NaCl, 1 mMMgCl2, 1.8mM CaCl2, 5mM HEPES with a final pH of 7.5) |
| HU-210 | (6aR,10aR)-9-(hydroxymethyl)-6,6-dimethyl-3-(2-methyloctan-2-yl)-6a,7,10,10a tetrahydrobenzo[c]chromen-1-ol or 11-hydroxy-Δ8-THC-dimethylheptyl |
| HU-308 | [(1S,4S,5S)-4-[2,6-dimethoxy-4-(2-methyloctan-2-yl)phenyl]-6,6-dimethyl-2-bicyclo [3.1.1]hept-2-enyl]methanol |
| JWH-015 | (2-methyl-1-propylindol-3-yl)-naphthalen-1-ylmethanone |
| JWH-133 | (6aR,10aR)-6,6,9-trimethyl-3-(2-methylpentan-2-yl)-6a,7,10,10a-tetrahydrobenzo[c]chromene |
| L-759633 | (6aR,10aR)-1-methoxy-6,6,9-trimethyl-3-(2-methyloctan-2-yl)-6a,7,10,10a-tetrahydrobenzo[c]chromene |
| L-759656 | (6aR,10aR)-1-methoxy-6,6-dimethyl-9-methylidene-3-(2-methyloctan-2-yl)-7,8,10,10a-tetrahydro-6aH-benzo[c]chromene |
| ND96 | Physiological salt buffer solution (96 mM NaCl, 2 mM MgCl2, 2 mM KCl, 5 mM HEPES, and 1.8 mM CaCl2, with a final pH of 7.5) |
| Org27569 | 5-chloro-3-ethyl-N-[2-(4-piperidin-1-ylphenyl)ethyl]-1H-indole-2-carboxamide |
| Org27759 | N-[2-[4-(dimethylamino)phenyl]ethyl]-3-ethyl-5-fluoro-1H-indole-2-carboxamide |
| Org29647 | N-[(3R)-1-benzylpyrrolidin-3-yl]-5-chloro-3-ethyl-1H-indole-2-carboxamide;(E)-but-2-enedioic acid |
| PM-226 | 9-methoxy-4,4-dimethyl-7-(2-methyloctan-2-yl)chromeno[3,4-d][1,2]oxazole |
| PSNCBAM-1 | 1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl)phenyl]urea |
| Rimonabant or SR141716 | 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-piperidin-1-ylpyrazole-3-carboxamide |
| RTI-371 | 3-(4-chlorophenyl)-5-[(1R,2S,3S,5S)-8-methyl-3-(4-methylphenyl)-8-azabicyclo[3.2.1]octan-2-yl]-1,2-oxazole |
| SR144528 | 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethyl-2-bicyclo[2.2.1]heptanyl]pyrazole-3-carboxamide |
| Surinabant or SR147778 | 5-(4-bromophenyl)-1-(2,4-dichlorophenyl)-4-ethyl-N-piperidin-1-ylpyrazole-3-carboxamide |
| Taranabant or MK-0364 | N-[(2S,3S)-4-(4-chlorophenyl)-3-(3-cyanophenyl)butan-2-yl]-2-methyl-2-[5-(trifluoromethyl)pyridin-2-yl]oxypropanamide |
| THC | Tetrahydrocannabinol |
| WIN-55,212-2 | [(11R)-2-methyl-11-(morpholin-4-ylmethyl)-9-oxa-1-azatricyclo[6.3.1.04,12]dodeca-2,4(12),5,7-tetraen-3-yl]-naphthalen-1-ylmethanone |
| ZCZ011 | 6-methyl-3-(2-nitro-1-thiophen-2-ylethyl)-2-phenyl-1H-indole |
References
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2006, 147. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Ye, L.; Cao, Z.; Wang, W.; Zhou, N. New insights in cannabinoid receptor structure and signaling. Curr. Mol. Pharmacol. 2019, 12, 239–248. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanuš, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Mazzola, C.; Micale, V.; Drago, F. Amnesia induced by β-amyloid fragments is counteracted by cannabinoid CB1 receptor blockade. Eur. J. Pharmacol. 2003, 477, 219–225. [Google Scholar] [CrossRef]
- Cerri, S.; Levandis, G.; Ambrosi, G.; Montepeloso, E.; Antoninetti, G.F.; Franco, R.; Lanciego, J.L.; Baqi, Y.; Müller, C.E.; Pinna, A.; et al. Neuroprotective potential of adenosine A2A and cannabinoid CB1 receptor antagonists in an animal model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2014, 73, 414–424. [Google Scholar] [CrossRef]
- Lunn, C.A.; Reich, E.P.; Fine, J.S.; Lavey, B.; Kozlowski, J.A.; Hipkin, R.W.; Lundell, D.J.; Bober, L. Biology and therapeutic potential of cannabinoid CB 2 receptor inverse agonists. Br. J. Pharmacol. 2008, 153, 226–239. [Google Scholar] [CrossRef]
- Dopart, R.; Lu, D.; Lichtman, A.H.; Kendall, D.A. Allosteric modulators of cannabinoid receptor 1: Developing compounds for improved specificity. Drug Metab. Rev. 2018, 50, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Cabral, G.A.; Pryce, G.; Fernández-Ruiz, J.; Rubino, T.; Katona, I.; Hillard, C.J.; Battista, N.; Gatta-Cherifi, B.; O’Sullivan, S.E.; et al. Endocannabinoids. In Handbook of Experimental Pharmacology; Rosenthal, W., Barrett, J.E., Flockerzi, V., Frohman, M.A., Geppetti, P., Hofmann, F.B., Michel, M.C., Page, C.P., Thorburn, A.M., Wang, K., Eds.; Springer International Publishing: Cham, Switzerland, 2015; Volume 231, pp. 1–473. [Google Scholar] [CrossRef]
- Turner, S.E.; Williams, C.M.; Iversen, L.; Whalley, B.J. Molecular Pharmacology of Phytocannabinoids. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer Nature: Cham, Switzerland, 2017; Volume 103, pp. 61–101. [Google Scholar] [CrossRef]
- Hervás, E.S. Synthetic cannabinoids: Characteristics, use and clinical implications. Arch. Psychiatry Psychother. 2017, 19, 42–48. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Aronne, L.J.; Tonstad, S.; Moreno, M.; Gantz, I.; Erondu, N.; Suryawanshi, S.; Molony, C.; Sieberts, S.; Nayee, J.; Meehan, A.G.; et al. A clinical trial assessing the safety and efficacy of taranabant, a CB1R inverse agonist, in obese and overweight patients: A high-dose study. Int. J. Obes. 2010, 34, 919–935. [Google Scholar] [CrossRef]
- Proietto, J.; Rissanen, A.; Harp, J.B.; Erondu, N.; Yu, Q.; Suryawanshi, S.; Jones, M.E.; Johnson-Levonas, A.O.; Heymsfield, S.B.; Kaufman, K.D.; et al. A clinical trial assessing the safety and efficacy of the CB1R inverse agonist taranabant in obese and overweight patients: Low-dose study. Int. J. Obes. 2010, 34, 1243–1254. [Google Scholar] [CrossRef][Green Version]
- Moreira, F.A.; Crippa, J.A.S. The psychiatric side-effects of rimonabant. Braz. J. Psychiatry 2009, 31, 145–153. [Google Scholar] [CrossRef]
- Jordan, C.J.; Zheng, X.X. Progress in brain cannabinoid CB2 receptor research: From genes to behavior. Neurosci. Biobehav. Rev. 2019, 98, 208–220. [Google Scholar] [CrossRef]
- Howlett, A.C.; Abood, M.E. CB(1) and CB(2) receptor pharmacology. Adv. Pharmacol. 2017, 80, 169–206. [Google Scholar] [CrossRef]
- Morales, P.; Hernandez-folgado, L.; Goya, P.; Jagerovic, N. Cannabinoid receptor 2 (CB 2) agonists and antagonists: A patent update. Expert Opin. Ther. Pat. 2016, 26, 843–856. [Google Scholar] [CrossRef]
- Hanuš, L.; Breuer, A.; Tchilibon, S.; Shiloah, S.; Goldenberg, D.; Horowitz, M.; Pertwee, R.G.; Ross, R.A.; Mechoulam, R.; Fride, E. HU-308: A specific agonist for CB2, a peripheral cannabinoid receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 14228–14233. [Google Scholar] [CrossRef]
- Valenzano, K.J.; Tafesse, L.; Lee, G.; Harrison, J.E.; Boulet, J.M.; Gottshall, S.L.; Mark, L.; Pearson, M.S.; Miller, W.; Shan, S.; et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology 2005, 48, 658–672. [Google Scholar] [CrossRef]
- Verty, A.N.A.; Stefanidis, A.; McAinch, A.J.; Hryciw, D.H.; Oldfield, B. Anti-Obesity effect of the CB2 receptor agonist JWH-015 in diet-induced obese mice. PLoS ONE 2015, 10, e0140592. [Google Scholar] [CrossRef]
- Poleszak, E.; Wośko, S.; Sławińska, K.; Wyska, E.; Szopa, A.; Sobczyński, J.; Wróbel, A.; Doboszewska, U.; Wlaź, P.; Wlaź, A.; et al. Ligands of the CB2 cannabinoid receptors augment activity of the conventional antidepressant drugs in the behavioural tests in mice. Behav. Brain Res. 2019. [Google Scholar] [CrossRef]
- Çakır, M.; Tekin, S.; Doğanyiğit, Z.; Erden, Y.; Soytürk, M.; Çiğremiş, Y.; Sandal, S. Cannabinoid type 2 receptor agonist JWH-133, attenuates Okadaic acid induced spatial memory impairment and neurodegeneration in rats. Life Sci. 2019, 217, 25–33. [Google Scholar] [CrossRef]
- Xing, C.; Zhuang, Y.; Xu, T.H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell 2020, 180, 645–654.e13. [Google Scholar] [CrossRef]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Johnston, C.A.; Nikas, S.P.; Song, F.; et al. Activation and signaling mechanism revealed by Cannabinoid Receptor-Gi complex structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR structures on drug discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467.e13. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB 1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef]
- Lynch, D.L.; Hurst, D.P.; Reggio, P.H. The Nucleotide-Free state of the cannabinoid CB2/Gi complex. Cell 2020, 180, 603–604. [Google Scholar] [CrossRef]
- Reckziegel, P.; Festuccia, W.T.; Britto, L.R.G.; Jang, K.L.L.; Romão, C.M.; Heimann, J.C.; Fogaça, M.V.; Rodrigues, N.S.; Silva, N.R.; Guimarães, F.S.; et al. A novel peptide that improves metabolic parameters without adverse central nervous system effects. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Wei, F.; Zhao, L.; Jing, Y. Signaling molecules targeting cannabinoid receptors: Hemopressin and related peptides. Neuropeptides 2020, 79, 101998. [Google Scholar] [CrossRef] [PubMed]
- Muratspahić, E.; Freissmuth, M.; Gruber, C.W. Nature-Derived peptides: A growing niche for GPCR ligand discovery. Trends Pharmacol. Sci. 2019, 40, 309–326. [Google Scholar] [CrossRef]
- Holford, M.; Daly, M.; King, G.F.; Norton, R.S. Venoms to the rescue. Science 2018, 361, 842–844. [Google Scholar] [CrossRef]
- Citti, C.; Linciano, P.; Russo, F.; Luongo, L.; Iannotta, M.; Maione, S.; Laganà, A.; Capriotti, A.L.; Forni, F.; Vandelli, M.A.; et al. A novel phytocannabinoid isolated from Cannabis sativa L. with an in vivo cannabimimetic activity higher than Δ9-tetrahydrocannabinol: Δ9-Tetrahydrocannabiphorol. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Amin, M.R.; Ali, D.W. Pharmacology of Medical Cannabis. In Advances in Experimental Medicine and Biology; Cohen, I.R., Lajtha, A., Lambris, J.D., Paoletti, R., Rezaei, N., Eds.; Springer Nature: Cham, Switzerland, 2019; Volume 1162, pp. 151–165. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The diverse CB 1 and CB 2 receptor pharmacology of three plant cannabinoids: Δ 9-tetrahydrocannabinol, cannabidiol and Δ 9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef]
- Boggs, D.L.; Nguyen, J.D.; Morgenson, D.; Taffe, M.A.; Ranganathan, M. Clinical and preclinical evidence for functional interactions of cannabidiol and Δ9-Tetrahydrocannabinol. Neuropsychopharmacology 2018, 43, 142–154. [Google Scholar] [CrossRef]
- Novotna, A.; Mares, J.; Ratcliffe, S.; Novakova, I.; Vachova, M.; Zapletalova, O.; Gasperini, C.; Pozzilli, C.; Cefaro, L.; Comi, G.; et al. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex®), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur. J. Neurol. 2011, 18, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Grof, C.P.L. Cannabis, from plant to pill. Br. J. Clin. Pharmacol. 2018, 84, 2463–2467. [Google Scholar] [CrossRef] [PubMed]
- Celius, E.G.; Vila, C. The influence of THC:CBD oromucosal spray on driving ability in patients with multiple sclerosis-related spasticity. Brain Behav. 2018, 8, e00962. [Google Scholar] [CrossRef] [PubMed]
- Hložek, T.; Uttl, L.; Kadeřábek, L.; Balíková, M.; Lhotková, E.; Horsley, R.R.; Nováková, P.; Šíchová, K.; Štefková, K.; Tylš, F.; et al. Pharmacokinetic and behavioural profile of THC, CBD, and THC+CBD combination after pulmonary, oral, and subcutaneous administration in rats and confirmation of conversion in vivo of CBD to THC. Eur. Neuropsychopharmacol. 2017, 27, 1223–1237. [Google Scholar] [CrossRef]
- Javadi-Paydar, M.; Nguyen, J.D.; Kerr, T.M.; Grant, Y.; Vandewater, S.A.; Cole, M.; Taffe, M.A. Effects of Δ9-THC and cannabidiol vapor inhalation in male and female rats. Psychopharmacology 2018, 235, 2541–2557. [Google Scholar] [CrossRef]
- Nguyen, J.D.; Grant, Y.; Kerr, T.M.; Gutierrez, A.; Cole, M.; Taffe, M.A. Tolerance to hypothermic and antinoceptive effects of ∆9-tetrahydrocannabinol (THC) vapor inhalation in rats. Pharmacol. Biochem. Behav. 2018, 172, 33–38. [Google Scholar] [CrossRef]
- Atwal, N.; Casey, S.L.; Mitchell, V.A.; Vaughan, C.W. THC and gabapentin interactions in a mouse neuropathic pain model. Neuropharmacology 2019, 144, 115–121. [Google Scholar] [CrossRef]
- Li, H.; Liu, Y.; Tian, D.; Tian, L.; Ju, X.; Qi, L.; Wang, Y.; Liang, C. Overview of cannabidiol (CBD) and its analogues: Structures, biological activities, and neuroprotective mechanisms in epilepsy and Alzheimer’s disease. Eur. J. Med. Chem. 2020, 192, 112163. [Google Scholar] [CrossRef]
- McAllister, S.D.; Soroceanu, L.; Desprez, P.-Y. The antitumor activity of plant-derived Non-Psychoactive cannabinoids. J. Neuroimmune Pharmacol. 2015, 10, 255–267. [Google Scholar] [CrossRef]
- Badal, S.; Smith, K.N.; Rajnarayanan, R. Analysis of natural product regulation of cannabinoid receptors in the treatment of human disease. Pharmacol. Ther. 2017, 180, 24–48. [Google Scholar] [CrossRef]
- Tham, M.; Yilmaz, O.; Alaverdashvili, M.; Kelly, M.E.M.; Denovan-Wright, E.M.; Laprairie, R.B. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br. J. Pharmacol. 2019, 176, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Fierro, A.; Pessoa-Mahana, C.D. Cannabidiol binding and negative allosteric modulation at the cannabinoid type 1 receptor in the presence of delta-9-tetrahydrocannabinol: An in silico study. PLoS ONE 2019, 14, e0220025. [Google Scholar] [CrossRef] [PubMed]
- Maione, S.; Piscitelli, F.; Gatta, L.; Vita, D.; De Petrocellis, L.; Palazzo, E.; de Novellis, V.; Di Marzo, V. Non-psychoactive cannabinoids modulate the descending pathway of antinociception in anaesthetized rats through several mechanisms of action. Br. J. Pharmacol. 2011, 162, 584–596. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid ligands targeting TRP channels. Front. Mol. Neurosci. 2019, 11, 487. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, B.; Laurino, C.; Vadalà, M. A therapeutic effect of cbd-enriched ointment in inflammatory skin diseases and cutaneous scars. La Clin. Ter. 2019, 170, e93–e99. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Fernández-Carballido, A.; Simancas-Herbada, R.; Martin-Sabroso, C.; Torres-Suárez, A.I. CBD loaded microparticles as a potential formulation to improve paclitaxel and doxorubicin-based chemotherapy in breast cancer. Int. J. Pharm. 2020, 574, 118916. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef]
- Bow, E.W.; Rimoldi, J.M. The Structure–Function relationships of classical cannabinoids: CB1/CB2 modulation. Perspect. Med. Chem. 2016, 8, PMC.S32171. [Google Scholar] [CrossRef]
- FDA and Cannabis: Research and Drug Approval Process. Available online: https://www.fda.gov/news-events/public-health-focus/fda-and-cannabis-research-and-drug-approval-process (accessed on 29 June 2020).
- Le Boisselier, R.; Alexandre, J.; Lelong-Boulouard, V.; Debruyne, D. Focus on cannabinoids and synthetic cannabinoids. Clin. Pharmacol. Ther. 2017, 101, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Pharmacological Actions of Cannabinoids. In Handbook of Experimental Pharmacology; Starke, K., Born, G.V.R., Eichelbaum, M., Ganten, D., Hofmann, F., Kobilka, B., Rosenthal, W., Rubanyi, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 168, pp. 1–51. [Google Scholar] [CrossRef]
- Wiley, J.L.; Barrett, R.L.; Lowe, J.; Balster, R.L.; Martin, B.R. Discriminative stimulus effects of CP 55,940 and structurally dissimilar cannabinoids in rats. Neuropharmacology 1995, 34, 669–676. [Google Scholar] [CrossRef]
- Griffin, G.; Atkinson, P.J.; Showalter, V.M.; Martin, B.R.; Abood, M.E. Evaluation of cannabinoid receptor agonists and antagonists using the guanosine-5′-O-(3-[35S]thio)-triphosphate binding assay in rat cerebellar membranes. J. Pharmacol. Exp. Ther. 1998, 285, 553–560. [Google Scholar] [PubMed]
- Rinaldi-Carmona, M.; Pialot, F.; Congy, C.; Redon, E.; Barth, F.; Bachy, A.; Brelière, J.-C.; Soubrié, P.; Le Fur, G. Characterization and distribution of binding sites for [3H]-SR 141716A, a selective brain (CB1) cannabinoid receptor antagonist, in rodent brain. Life Sci. 1996, 58, 1239–1247. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 29 June 2020).
- Kapur, A.; Hurst, D.P.; Fleischer, D.; Whitnell, R.; Thakur, G.A.; Makriyannis, A.; Reggio, P.H.; Abood, M.E. Mutation studies of Ser7.39 and Ser2.60 in the human CB1 cannabinoid receptor: Evidence for a serine-induced bend in CB1 transmembrane helix 7. Mol. Pharmacol. 2007, 71, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Lax, P.; Esquiva, G.; Altavilla, C.; Cuenca, N. Neuroprotective effects of the cannabinoid agonist HU210 on retinal degeneration. Exp. Eye Res. 2014, 120, 175–185. [Google Scholar] [CrossRef]
- Lax, P.; Kutsyr, O.; Esquiva, G.; Altavilla, C.; Maneu, V.; Cuenca, N. Cannabinoid-mediated retinal rescue correlates with improved circadian parameters in retinal dystrophic rats. Exp. Eye Res. 2019, 180, 192–199. [Google Scholar] [CrossRef]
- Hillard, C.J.; Manna, S.; Greenberg, M.J.; DiCamelli, R.; Ross, R.A.; Stevenson, L.A.; Murphy, V.; Pertwee, R.G.; Campbell, W.B. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J. Pharmacol. Exp. Ther. 1999, 289, 1427–1433. [Google Scholar]
- Bajo, M.; Roberto, M.; Schweitzer, P. Differential alteration of hippocampal excitatory synaptic transmission by cannabinoid ligands. J. Neurosci. Res. 2009, 87, 766–775. [Google Scholar] [CrossRef][Green Version]
- Vaughan, C.W.; Connor, M.; Bagley, E.E.; Christie, M.J. Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Mol. Pharmacol. 2000, 57, 288–295. [Google Scholar]
- Schweitzer, P. Cannabinoids decrease the K+ M-current in hippocampal CA1 neurons. J. Neurosci. 2000, 20, 51–58. [Google Scholar] [CrossRef]
- Morales, P.; Gómez-Cañas, M.; Navarro, G.; Hurst, D.P.; Carrillo-Salinas, F.J.; Lagartera, L.; Pazos, R.; Goya, P.; Reggio, P.H.; Guaza, C.; et al. Chromenopyrazole, a versatile cannabinoid scaffold with in vivo activity in a model of multiple sclerosis. J. Med. Chem. 2016, 59, 6753–6771. [Google Scholar] [CrossRef]
- Huizenga, M.N.; Forcelli, P.A. Neuroprotective Action of the CB1/2 Receptor Agonist, WIN 55,212-2, against DMSO but not phenobarbital-induced neurotoxicity in immature rats. Neurotox. Res. 2019, 35, 173–182. [Google Scholar] [CrossRef]
- Peball, M.; Werkmann, M.; Ellmerer, P.; Stolz, R.; Valent, D.; Knaus, H.G.; Ulmer, H.; Djamshidian, A.; Poewe, W.; Seppi, K. Nabilone for non-motor symptoms of Parkinson’s disease: A randomized placebo-controlled, double-blind, parallel-group, enriched enrolment randomized withdrawal study (The NMS-Nab Study). J. Neural Transm. 2019, 126, 1061–1072. [Google Scholar] [CrossRef]
- Ruthirakuhan, M.T.; Herrmann, N.; Gallagher, D.; Andreazza, A.C.; Kiss, A.; Verhoeff, N.P.L.G.; Black, S.E.; Lanctôt, K.L. Investigating the safety and efficacy of nabilone for the treatment of agitation in patients with moderate-to-severe Alzheimer’s disease: Study protocol for a cross-over randomized controlled trial. Contemp. Clin. Trials Commun. 2019, 15. [Google Scholar] [CrossRef]
- Guindon, J.; Hohmann, A.G. The endocannabinoid system and cancer: Therapeutic implication. Br. J. Pharmacol. 2011, 163, 1447–1463. [Google Scholar] [CrossRef]
- Ebrahimi-Ghiri, M.; Nasehi, M.; Zarrindast, M.-R. Anxiolytic and antidepressant effects of ACPA and harmaline co-treatment. Behav. Brain Res. 2019, 364, 296–302. [Google Scholar] [CrossRef]
- Rutkowska, M.; Fereniec-Gołtbiewska, L. ACEA (arachidonyl-2-chloroethylamide), the selective cannabinoid CB1 receptor agonist, protects against aspirin-induced gastric ulceration. Die Pharm. 2006, 61, 341–342. [Google Scholar]
- Jafari, M.R.; Ghiasvand, F.; Golmohammadi, S.; Zarrindast, M.R.; Djahanguiri, B. Influence of central nicotinic receptors on arachidonylcyclopropylamide (ACPA)-induced antinociception in mice. Int. J. Neurosci. 2008, 118, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Luszczki, J.J.; Czuczwar, P.; Cioczek-Czuczwar, A.; Czuczwar, S.J. Arachidonyl-2′-chloroethylamide, a highly selective cannabinoid CB1 receptor agonist, enhances the anticonvulsant action of valproate in the mouse maximal electroshock-induced seizure model. Eur. J. Pharmacol. 2006, 547, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Luszczki, J.J.; Patrzylas, P.; Zagaja, M.; Andres-Mach, M.; Zaluska, K.; Kondrat-Wrobel, M.W.; Szpringer, M.; Chmielewski, J.; Florek-Luszczki, M. Effects of arachidonyl-2′-chloroethylamide (ACEA) on the protective action of various antiepileptic drugs in the 6-Hz corneal stimulation model in mice. PLoS ONE 2017, 12, e0183873. [Google Scholar] [CrossRef]
- Florek-Luszczki, M.; Zagaja, M.; Luszczki, J.J. Influence of arachidonyl-2′-chloroethylamide, a selective cannabinoid CB 1 receptor agonist, on the anticonvulsant and acute side-effect potentials of clobazam, lacosamide, and pregabalin in the maximal electroshock-induced seizure model and chimney test. Fundam. Clin. Pharmacol. 2015, 29, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Andres-Mach, M.; Haratym-Maj, A.; Zagaja, M.; Rola, R.; Maj, M.; Chrościńska-Krawczyk, M.; Luszczki, J.J. ACEA (a highly selective cannabinoid CB1 receptor agonist) stimulates hippocampal neurogenesis in mice treated with antiepileptic drugs. Brain Res. 2015, 1624, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Zagaja, M.; Haratym-Maj, A.; Szewczyk, A.; Rola, R.; Maj, M.; Łuszczki, J.J.; Andres-Mach, M. Levetiracetam combined with ACEA, highly selective cannabinoid CB1 receptor agonist changes neurogenesis in mouse brain. Neurosci. Lett. 2019, 696, 79–86. [Google Scholar] [CrossRef]
- Aso, E.; Palomer, E.; Juvés, S.; Maldonado, R.; Muñoz, F.J.; Ferrer, I. CB1 agonist ACEA protects neurons and reduces the cognitive impairment of AβPP/PS1 mice. J. Alzheimer’s Dis. 2012, 30, 439–459. [Google Scholar] [CrossRef]
- Ma, L.; Jia, J.; Niu, W.; Jiang, T.; Zhai, Q.; Yang, L.; Bai, F.; Wang, Q.; Xiong, L. Mitochondrial CB1 receptor is involved in ACEA-induced protective effects on neurons and mitochondrial functions. Sci. Rep. 2015, 5, 12440. [Google Scholar] [CrossRef]
- Wiley, J.L.; Smith, F.L.; Razdan, R.K.; Dewey, W.L. Task specificity of cross-tolerance between Delta9-tetrahydrocannabinol and anandamide analogs in mice. Eur. J. Pharmacol. 2005, 510, 59–68. [Google Scholar] [CrossRef]
- Wallace, M.J.; Martin, B.R.; DeLorenzo, R.J. Evidence for a physiological role of endocannabinoids in the modulation of seizure threshold and severity. Eur. J. Pharmacol. 2002, 452, 295–301. [Google Scholar] [CrossRef]
- Gómez-Cañas, M.; Morales, P.; García-Toscano, L.; Navarrete, C.; Muñoz, E.; Jagerovic, N.; Fernández-Ruiz, J.; García-Arencibia, M.; Pazos, M.R. Biological characterization of PM226, a chromenoisoxazole, as a selective CB2 receptor agonist with neuroprotective profile. Pharmacol. Res. 2016, 110, 205–215. [Google Scholar] [CrossRef]
- Thapa, D.; Cairns, E.A.; Szczesniak, A.-M.; Toguri, J.T.; Caldwell, M.D.; Kelly, M.E.M. The cannabinoids Δ(8)THC, CBD, and HU-308 Act via distinct receptors to reduce corneal pain and inflammation. Cannabis Cannabinoid Res. 2018, 3, 11–20. [Google Scholar] [CrossRef]
- Ossola, C.A.; Surkin, P.N.; Mohn, C.E.; Elverdin, J.C.; Fernández-Solari, J. Anti-inflammatory and osteoprotective effects of Cannabinoid-2 receptor agonist HU-308 in a rat model of lipopolysaccharide-induced periodontitis. J. Periodontol. 2016, 87, 725–734. [Google Scholar] [CrossRef]
- Rentsch, P.; Stayte, S.; Egan, T.; Clark, I.; Vissel, B. Targeting the cannabinoid receptor CB2 in a mouse model of l-dopa induced dyskinesia. Neurobiol. Dis. 2020, 134, 104646. [Google Scholar] [CrossRef]
- Li, A.-L.; Carey, L.M.; Mackie, K.; Hohmann, A.G. Cannabinoid CB(2) Agonist GW405833 suppresses inflammatory and neuropathic pain through a CB(1) mechanism that is independent of CB(2) receptors in mice. J. Pharmacol. Exp. Ther. 2017, 362, 296–305. [Google Scholar] [CrossRef]
- Craft, R.M.; Greene, N.Z.; Wakley, A.A. Antinociceptive effects of JWH015 in female and male rats. Behav. Pharmacol. 2018, 29, 280–289. [Google Scholar] [CrossRef]
- Ghonim, A.E.; Ligresti, A.; Rabbito, A.; Mahmoud, A.M.; Di Marzo, V.; Osman, N.A.; Abadi, A.H. Structure-activity relationships of thiazole and benzothiazole derivatives as selective cannabinoid CB2 agonists with in vivo anti-inflammatory properties. Eur. J. Med. Chem. 2019, 180, 154–170. [Google Scholar] [CrossRef]
- Mugnaini, C.; Rabbito, A.; Brizzi, A.; Palombi, N.; Petrosino, S.; Verde, R.; Di Marzo, V.; Ligresti, A.; Corelli, F. Synthesis of novel 2-(1-adamantanylcarboxamido)thiophene derivatives. Selective cannabinoid type 2 (CB2) receptor agonists as potential agents for the treatment of skin inflammatory disease. Eur. J. Med. Chem. 2019, 161, 239–251. [Google Scholar] [CrossRef]
- Gado, F.; Meini, S.; Bertini, S.; Digiacomo, M.; Macchia, M.; Manera, C. Allosteric modulators targeting cannabinoid cb1 and cb2 receptors: Implications for drug discovery. Future Med. Chem. 2019, 11, 2019–2037. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G. International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB 1 and CB 2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef]
- Yamamoto, W.; Mikami, T.; Iwamura, H. Involvement of central cannabinoid CB2 receptor in reducing mechanical allodynia in a mouse model of neuropathic pain. Eur. J. Pharmacol. 2008, 583, 56–61. [Google Scholar] [CrossRef]
- Bisogno, T.; Oddi, S.; Piccoli, A.; Fazio, D.; Maccarrone, M. Type-2 cannabinoid receptors in neurodegeneration. Pharmacol. Res. 2016, 111, 721–730. [Google Scholar] [CrossRef]
- Çakır, M.; Tekin, S.; Okan, A.; Çakan, P.; Doğanyiğit, Z. The ameliorating effect of cannabinoid type 2 receptor activation on brain, lung, liver and heart damage in cecal ligation and puncture-induced sepsis model in rats. Int. Immunopharmacol. 2020, 78, 105978. [Google Scholar] [CrossRef]
- Yuill, M.B.; Hale, D.E.; Guindon, J.; Morgan, D.J. Anti-nociceptive interactions between opioids and a cannabinoid receptor 2 agonist in inflammatory pain. Mol. Pain 2017, 13, 1744806917728227. [Google Scholar] [CrossRef] [PubMed]
- Çakır, M.; Tekin, S.; Doğanyiğit, Z.; Çakan, P.; Kaymak, E. The protective effect of cannabinoid type 2 receptor activation on renal ischemia-reperfusion injury. Mol. Cell. Biochem. 2019, 462, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dong, X.; Tu, C.; Li, X.; Peng, Z.; Zhou, Y.; Zhang, D.; Jiang, J.; Burke, A.; Zhao, Z.; et al. Opposite effects of cannabinoid CB(1) and CB(2) receptors on antipsychotic clozapine-induced cardiotoxicity. Br. J. Pharmacol. 2019, 176, 890–905. [Google Scholar] [CrossRef]
- Castany, S.; Carcolé, M.; Leánez, S.; Pol, O. The role of carbon monoxide on the anti-nociceptive effects and expression of cannabinoid 2 receptors during painful diabetic neuropathy in mice. Psychopharmacology 2016, 233, 2209–2219. [Google Scholar] [CrossRef]
- Hanlon, K.E.; Lozano-Ondoua, A.N.; Umaretiya, P.J.; Symons-Liguori, A.M.; Chandramouli, A.; Moy, J.K.; Kwass, W.K.; Mantyh, P.W.; Nelson, M.A.; Vanderah, T.W. Modulation of breast cancer cell viability by a cannabinoid receptor 2 agonist, JWH-015, is calcium dependent. Breast Cancer 2016, 8, 59–71. [Google Scholar] [CrossRef]
- Liu, A.P.; Yuan, Q.H.; Zhang, B.; Yang, L.; He, Q.W.; Chen, K.; Liu, Q.S.; Li, Z.; Zhan, J. Cannabinoid receptor 2 activation alleviates septic lung injury by promoting autophagy via inhibition of inflammatory mediator release. Cell. Signal. 2020, 69, 109556. [Google Scholar] [CrossRef]
- Huang, Z.-B.; Zheng, Y.-X.; Li, N.; Cai, S.-L.; Huang, Y.; Wang, J.; Hu, X.-W.; Wang, Y.; Wu, J.; Fan, X.-G. Protective effects of specific cannabinoid receptor 2 agonist GW405833 on concanavalin A-induced acute liver injury in mice. Acta Pharmacol. Sin. 2019, 40, 1404–1411. [Google Scholar] [CrossRef]
- Ross, R.A.; Brockie, H.C.; Stevenson, L.A.; Murphy, V.L.; Templeton, F.; Makriyannis, A.; Pertwee, R.G. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656 and AM630. Br. J. Pharmacol. 1999, 126, 665–672. [Google Scholar] [CrossRef]
- Rinaldi-Carmona, M.; Barth, F.; Héaulme, M.; Shire, D.; Calandra, B.; Congy, C.; Martinez, S.; Maruani, J.; Néliat, G.; Caput, D.; et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994, 350, 240–244. [Google Scholar] [CrossRef]
- Pertwee, R.G. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005, 76, 1307–1324. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Tsuruma, K.; Inoue, Y.; Otsuka, T.; Ohno, Y.; Ogami, S.; Yamane, S.; Shimazawa, M.; Hara, H. Rimonabant, a selective cannabinoid(1) receptor antagonist, protects against light-induced retinal degeneration in vitro and in vivo. Eur. J. Pharmacol. 2017, 803, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Luo, X.; Liu, S.; Shen, Y. Neuroprotective effect of cannabinoid receptor 1 antagonist in the MNU-induced retinal degeneration model. Exp. Eye Res. 2018, 167, 145–151. [Google Scholar] [CrossRef]
- Janero, D.R.; Makriyannis, A. Cannabinoid receptor antagonists: Pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin. Emerg. Drugs 2009, 14, 43–65. [Google Scholar] [CrossRef]
- Kipnes, M.S.; Hollander, P.; Fujioka, K.; Gantz, I.; Seck, T.; Erondu, N.; Shentu, Y.; Lu, K.; Suryawanshi, S.; Chou, M.; et al. A one-year study to assess the safety and efficacy of the CB1R inverse agonist taranabant in overweight and obese patients with type 2 diabetes. Diabetes Obes. Metab. 2010, 12, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Dossou, K.S.S.; Devkota, K.P.; Kavanagh, P.V.; Beutler, J.A.; Egan, J.M.; Moaddel, R. Development and preliminary validation of a plate-based CB1/CB2 receptor functional assay. Anal. Biochem. 2013, 437, 138–143. [Google Scholar] [CrossRef]
- Poleszak, E.; Wośko, S.; Sławińska, K.; Wyska, E.; Szopa, A.; Świąder, K.; Wróbel, A.; Doboszewska, U.; Wlaź, P.; Wlaź, A.; et al. Influence of the CB(1) and CB(2) cannabinoid receptor ligands on the activity of atypical antidepressant drugs in the behavioural tests in mice. Pharmacol. Biochem. Behav. 2020, 188, 172833. [Google Scholar] [CrossRef]
- Jenkin, K.A.; O’Keefe, L.; Simcocks, A.C.; Grinfeld, E.; Mathai, M.L.; McAinch, A.J.; Hryciw, D.H. Chronic administration of AM251 improves albuminuria and renal tubular structure in obese rats. J. Endocrinol. 2015, 225, 113–124. [Google Scholar] [CrossRef]
- Bialuk, I.; Winnicka, M.M. AM251, cannabinoids receptors ligand, improves recognition memory in rats. Pharmacol. Rep. 2011, 63, 670–679. [Google Scholar] [CrossRef]
- Bialuk, I.; Winnicka, M.M. Facilitatory effect of AM281 on recognition memory in rats. Pharmacol. Rep. 2016, 68, 301–309. [Google Scholar] [CrossRef]
- Xu, X.; Jiang, S.; Xu, E.; Wu, X.; Zhao, R. Inhibition of CB1 receptor ameliorates spatial learning and memory impairment in mice with traumatic brain injury. Neurosci. Lett. 2019, 696, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Hamill, T.G.; Lin, L.S.; Hagmann, W.; Liu, P.; Jewell, J.; Sanabria, S.; Eng, W.S.; Ryan, C.; Fong, T.M.; Connolly, B.; et al. PET imaging studies in rhesus monkey with the cannabinoid-1 (CB1) receptor ligand [11C]CB-119. Mol. Imaging Biol. 2009, 11, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J.; Paquot, N. Use of cannabinoid CB1 receptor antagonists for the treatment of metabolic disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Fremming, B.A.; Boyd, S.T. Taranabant, a novel cannabinoid type 1 receptor inverse agonist. Curr. Opin. Investig. Drugs 2008, 9, 1116–1129. [Google Scholar] [PubMed]
- Fichna, J.; Sibaev, A.; Sałaga, M.; Sobczak, M.; Storr, M. The cannabinoid-1 receptor inverse agonist taranabant reduces abdominal pain and increases intestinal transit in mice. Neurogastroenterol. Motil. 2013, 25, e550–e559. [Google Scholar] [CrossRef] [PubMed]
- Hadcock, J.R.; Griffith, D.A.; Iredale, P.A.; Carpino, P.A.; Dow, R.L.; Black, S.C.; O’Connor, R.; Gautreau, D.; Lizano, J.S.; Ward, K.; et al. In vitro and in vivo pharmacology of CP-945,598, a potent and selective cannabinoid CB1 receptor antagonist for the management of obesity. Biochem. Biophys. Res. Commun. 2010, 394, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Griffith, D.A.; Hadcock, J.R.; Black, S.C.; Iredale, P.A.; Carpino, P.A.; Dasilva-Jardine, P.; Day, R.; Dibrino, J.; Dow, R.L.; Landis, M.S.; et al. Discovery of 1-[9-(4-chlorophenyl)-8-(2-chlorophenyl)-9H-purin-6-yl]-4- ethylamino-piperidine-4-carboxylic Acid Amide Hydrochloride (CP-945,598), a novel, potent, and selective cannabinoid type 1 receptor antagonist. J. Med. Chem. 2009, 52, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi-Carmona, M.; Barth, F.; Congy, C.; Martinez, S.; Oustric, D.; Pério, A.; Poncelet, M.; Maruani, J.; Arnone, M.; Finance, O.; et al. SR147778 [5-(4-bromophenyl)-1-(2,4-dichlorophenyl)-4-ethyl-N-(1-piperidinyl)-1H-pyrazole-3-carboxamide], a new potent and selective antagonist of the CB1 cannabinoid receptor: Biochemical and pharmacological characterization. J. Pharmacol. Exp. Ther. 2004, 310, 905–914. [Google Scholar] [CrossRef]
- Rinaldi-Carmona, M.; Barth, F.; Millan, J.; Derocq, J.M.; Casellas, P.; Congy, C.; Oustric, D.; Sarran, M.; Bouaboula, M.; Calandra, B.; et al. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J. Pharmacol. Exp. Ther. 1998, 284, 644–650. [Google Scholar]
- Geng, D.C.; Xu, Y.Z.; Yang, H.L.; Zhu, X.S.; Zhu, G.M.; Wang, X.B. Inhibition of titanium particle-induced inflammatory osteolysis through inactivation of cannabinoid receptor 2 by AM630. J. Biomed. Mater. Res. Part A 2010, 95, 321–326. [Google Scholar] [CrossRef]
- Kruk-Slomka, M.; Boguszewska-Czubara, A.; Slomka, T.; Budzynska, B.; Biala, G. Correlations between the memory-related behavior and the level of oxidative stress biomarkers in the mice brain, provoked by an acute administration of CB receptor ligands. Neural Plast. 2016, 2016, 9815092. [Google Scholar] [CrossRef] [PubMed]
- Price, M.R.; Baillie, G.L.; Thomas, A.A.; Stevenson, L.A.; Easson, M.; Goodwin, R.; McLean, A.A.; McIntosh, L.; Goodwin, G.; Walker, G.; et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol. Pharmacol. 2005, 68, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Goya, P.; Jagerovic, N.; Hernandez-Folgado, L. Allosteric modulators of the CB(1) cannabinoid receptor: A structural update review. Cannabis Cannabinoid Res. 2016, 1, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Yan, W.; Chapman, K.; Ramesh, K.; Ferrell, A.J.; Yin, J.; Wang, X.; Xu, Q.; Rosenbaum, D.M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. [Google Scholar] [CrossRef]
- Ignatowska-Jankowska, B.M.; Baillie, G.L.; Kinsey, S.; Crowe, M.; Ghosh, S.; Owens, R.A.; Damaj, I.M.; Poklis, J.; Wiley, J.L.; Zanda, M.; et al. A cannabinoid CB1 receptor-positive allosteric modulator reduces neuropathic pain in the mouse with No Psychoactive effects. Neuropsychopharmacology 2015, 40, 2948–2959. [Google Scholar] [CrossRef]
- Slivicki, R.A.; Xu, Z.; Kulkarni, P.M.; Pertwee, R.G.; Mackie, K.; Thakur, G.A.; Hohmann, A.G. Positive allosteric modulation of cannabinoid receptor type 1 suppresses pathological pain without producing tolerance or dependence. Biol. Psychiatry 2018, 84, 722–733. [Google Scholar] [CrossRef]
- Thapa, D.; Cairns, E.A.; Szczesniak, A.-M.M.; Kulkarni, P.M.; Straiker, A.J.; Thakur, G.A.; Kelly, M.E.M.M. Allosteric cannabinoid receptor 1 (CB1) ligands reduce ocular pain and inflammation. Molecules 2020, 25, 417. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Kulkarni, P.M.; Deschamps, J.R.; Kelly, M.E.M.; Janero, D.R.; Cascio, M.G.; Stevenson, L.A.; Pertwee, R.G.; Kenakin, T.P.; Denovan-Wright, E.M.; et al. Enantiospecific allosteric modulation of cannabinoid 1 receptor. ACS Chem. Neurosci. 2017, 8, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Gado, F.; Di Cesare Mannelli, L.; Lucarini, E.; Bertini, S.; Cappelli, E.; Digiacomo, M.; Stevenson, L.A.; Macchia, M.; Tuccinardi, T.; Ghelardini, C.; et al. Identification of the first synthetic allosteric modulator of the CB2 receptors and evidence of its efficacy for neuropathic pain relief. J. Med. Chem. 2019, 62, 276–287. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Rourke, J.L.; Zrein, A.; Cairns, E.A.; Kelly, M.E.M.; Sinal, C.J.; Kulkarni, P.M.; Thakur, G.A.; Denovan-Wright, E.M. Positive allosteric modulation of the type 1 cannabinoid receptor reduces the signs and symptoms of Huntington’s disease in the R6/2 mouse model. Neuropharmacology 2019, 151, 1–12. [Google Scholar] [CrossRef]
- Hiranita, T.; Wilkinson, D.S.; Hong, W.C.; Zou, M.-F.; Kopajtic, T.A.; Soto, P.L.; Lupica, C.R.; Newman, A.H.; Katz, J.L. 2-isoxazol-3-phenyltropane derivatives of cocaine: Molecular and atypical system effects at the dopamine transporter. J. Pharmacol. Exp. Ther. 2014, 349, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Juknat, A.; Kozela, E.; Kaushansky, N.; Mechoulam, R.; Vogel, Z. Anti-inflammatory effects of the cannabidiol derivative dimethylheptyl-cannabidiol-studies in BV-2 microglia and encephalitogenic T cells. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, F.S.; de Aguiar, J.C.; Mechoulam, R.; Breuer, A. Anxiolytic effect of cannabidiol derivatives in the elevated plus-maze. Gen. Pharmacol. 1994, 25, 161–164. [Google Scholar] [CrossRef]
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 19, 389–413. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2016, 16, 19–34. [Google Scholar] [CrossRef]
- Gomes, I.; Grushko, J.S.; Golebiewska, U.; Hoogendoorn, S.; Gupta, A.; Heimann, A.S.; Ferro, E.S.; Scarlata, S.; Fricker, L.D.; Devi, L.A. Novel endogenous peptide agonists of cannabinoid receptors. FASEB J. 2009, 23, 3020–3029. [Google Scholar] [CrossRef]
- Rioli, V.; Gozzo, F.C.; Heimann, A.S.; Linardi, A.; Krieger, J.E.; Shida, C.S.; Almeida, P.C.; Hyslop, S.; Eberlin, M.N.; Ferro, E.S. Novel natural peptide substrates for endopeptidase 24.15, neurolysin, and angiotensin-converting enzyme. J. Biol. Chem. 2003, 278, 8547–8555. [Google Scholar] [CrossRef]
- Heimann, A.S.; Gomes, I.; Dale, C.S.; Pagano, R.L.; Gupta, A.; de Souza, L.L.; Luchessi, A.D.; Castro, L.M.; Giorgi, R.; Rioli, V.; et al. Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 20588–20593. [Google Scholar] [CrossRef]
- Dvorácskó, S.; Tömböly, C.; Berkecz, R.; Keresztes, A. Investigation of receptor binding and functional characteristics of hemopressin(1–7). Neuropeptides 2016, 58, 15–22. [Google Scholar] [CrossRef]
- Straiker, A.; Mitjavila, J.; Yin, D.; Gibson, A.; Mackie, K. Aiming for allosterism: Evaluation of allosteric modulators of CB1 in a neuronal model. Pharmacol. Res. 2015, 99, 370–376. [Google Scholar] [CrossRef]
- Bauer, M.; Chicca, A.; Tamborrini, M.; Eisen, D.; Lerner, R.; Lutz, B.; Poetz, O.; Pluschke, G.; Gertsch, J. Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB 1 receptors. J. Biol. Chem. 2012, 287, 36944–36967. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, V.; Chicca, A.; Glasmacher, S.; Paloczi, J.; Cao, Z.; Pacher, P.; Gertsch, J. Pepcan-12 (RVD-hemopressin) is a CB2 receptor positive allosteric modulator constitutively secreted by adrenals and in liver upon tissue damage. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.T.; Mancini, G.; Lutz, B.; Luckman, S.M. The peptide hemopressin acts through CB1 cannabinoid receptors to reduce food intake in rats and mice. J. Neurosci. 2010, 30, 7369–7376. [Google Scholar] [CrossRef]
- Mahmoud, M.F.; El Swefy, S.; Hasan, R.A.; Ibrahim, A. Role of cannabinoid receptors in hepatic fibrosis and apoptosis associated with bile duct ligation in rats. Eur. J. Pharmacol. 2014, 742, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Fang, Q.; Wang, Z.; Li, X.; Li, N.; Chang, X.; Pan, J.; Tang, H.; Wang, R. Antinociceptive effects of central administration of the endogenous cannabinoid receptor type 1 agonist VDPVNFKLLSH-OH [(m)VD-hemopressin (α)], an N-Terminally extended hemopressin peptide. J. Pharmacol. Exp. Ther. 2014, 348, 316–323. [Google Scholar] [CrossRef]
- Li, X.-H.; Lin, M.-L.; Wang, Z.-L.; Wang, P.; Tang, H.-H.; Lin, Y.-Y.; Li, N.; Fang, Q.; Wang, R. Central administrations of hemopressin and related peptides inhibit gastrointestinal motility in mice. Neurogastroenterol. Motil. 2016, 28, 891–899. [Google Scholar] [CrossRef]
- Zhang, R.; He, Z.; Jin, W.; Wang, R. Effects of the cannabinoid 1 receptor peptide ligands hemopressin, (m)RVD-hemopressin(α) and (m)VD-hemopressin(α) on memory in novel object and object location recognition tasks in normal young and Aβ1–42-treated mice. Neurobiol. Learn. Mem. 2016, 134, 264–274. [Google Scholar] [CrossRef]
- Leone, S.; Recinella, L.; Chiavaroli, A.; Martinotti, S.; Ferrante, C.; Mollica, A.; Macedonio, G.; Stefanucci, A.; Dvorácskó, S.; Tömböly, C.; et al. Emotional disorders induced by Hemopressin and RVD-hemopressin(α) administration in rats. Pharmacol. Rep. 2017, 69, 1247–1253. [Google Scholar] [CrossRef]
- McDermott, A. News Feature: Venom back in vogue as a wellspring for drug candidates. Proc. Natl. Acad. Sci. USA 2020, 10100–10104. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Ciolek, J.; Reinfrank, H.; Quinton, L.; Viengchareun, S.; Stura, E.A.; Vera, L.; Sigismeau, S.; Mouillac, B.; Orcel, H.; Peigneur, S.; et al. Green mamba peptide targets type-2 vasopressin receptor against polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2017, 114, 7154–7159. [Google Scholar] [CrossRef] [PubMed]
- Emerich, B.L.; Ferreira, R.C.M.; Cordeiro, M.N.; Borges, M.H.; Pimenta, A.M.C.; Figueiredo, S.G.; Duarte, I.D.G.; De Lima, M.E. Δ-Ctenitoxin-Pn1a, a peptide from phoneutria nigriventer spider venom, shows antinociceptive effect involving opioid and cannabinoid systems, in rats. Toxins 2016, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- MacHado, F.C.; Zambelli, V.O.; Fernandes, A.C.O.; Heimann, A.S.; Cury, Y.; Picolo, G. Peripheral interactions between cannabinoid and opioid systems contribute to the antinociceptive effect of crotalphine. Br. J. Pharmacol. 2014, 171, 961–972. [Google Scholar] [CrossRef]
- Fonseca Pacheco, D.; Freitas, A.C.N.; Pimenta, A.M.C.; Duarte, I.D.G.; Lima, M.E. A spider derived peptide, pnpp-19, induces central antinociception mediated by opioid and cannabinoid systems. J. Venom. Anim. Toxins Incl. Trop. Dis. 2016, 22. [Google Scholar] [CrossRef]
- Freitas, A.C.N.; Pacheco, D.F.; MacHado, M.F.M.; Carmona, A.K.; Duarte, I.D.G.; De Lima, M.E. PnPP-19, a spider toxin peptide, induces peripheral antinociception through opioid and cannabinoid receptors and inhibition of neutral endopeptidase. Br. J. Pharmacol. 2016, 173, 1491–1501. [Google Scholar] [CrossRef]
- Konno, K.; Picolo, G.; Gutierrez, V.P.; Brigatte, P.; Zambelli, V.O.; Camargo, A.C.M.; Cury, Y. Crotalphine, a novel potent analgesic peptide from the venom of the South American rattlesnake Crotalus durissus terrificus. Peptides 2008, 29, 1293–1304. [Google Scholar] [CrossRef]
- Daniel, J.T.; Clark, R.J. G-protein coupled receptors targeted by analgesic venom peptides. Toxins 2017, 9, 372. [Google Scholar] [CrossRef]
- Maldifassi, M.C.; Wongsamitkul, N.; Baur, R.; Sigel, E. Xenopus oocytes: Optimized methods for microinjection, removal of follicular cell layers, and fast solution changes in electrophysiological experiments. J. Vis. Exp. 2016, 118, e55034. [Google Scholar] [CrossRef]
- Kobayashi, T.; Washiyama, K.; Ikeda, K. Modulators of G protein-activated inwardly rectifying K+ channels: Potentially therapeutic agents for addictive drug users. Ann. New York Acad. Sci. 2004, 1025, 590–594. [Google Scholar] [CrossRef]
- Pertwee, R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997, 74, 129–180. [Google Scholar] [CrossRef]
- Giblin, G.M.P.; Billinton, A.; Briggs, M.; Brown, A.J.; Chessell, I.P.; Clayton, N.M.; Eatherton, A.J.; Goldsmith, P.; Haslam, C.; Johnson, M.R.; et al. Discovery of 1-[4-(3-chlorophenylamino)-1-methyl-1H-pyrrolo[3,2-c]pyridin- 7-yl]-1-morpholin-4-ylmethanone (GSK554418A), a brain penetrant 5-azaindole CB2 agonist for the treatment of chronic pain. J. Med. Chem. 2009, 52, 5785–5788. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, S.; Ligresti, A.; Mugnaini, C.; Semeraro, T.; Cicione, L.; De Rosa, M.; Guida, F.; Luongo, L.; De Chiaro, M.; Cascio, M.G.; et al. Investigations on the 4-Quinolone-3-carboxylic acid motif. 3. Synthesis, structure-affinity relationships, and pharmacological characterization of 6-substituted 4-quinolone-3-carboxamides as highly selective cannabinoid-2 receptor ligands. J. Med. Chem. 2010, 53, 5915–5928. [Google Scholar] [CrossRef] [PubMed]
- Hollinshead, S.P.; Tidwell, M.W.; Palmer, J.; Guidetti, R.; Sanderson, A.; Johnson, M.P.; Chambers, M.G.; Oskins, J.; Stratford, R.; Astles, P.C. Selective cannabinoid receptor type 2 (CB2) agonists: Optimization of a series of purines leading to the identification of a clinical candidate for the treatment of osteoarthritic pain. J. Med. Chem. 2013, 56, 5722–5733. [Google Scholar] [CrossRef] [PubMed]
- Dolles, D.; Hoffmann, M.; Gunesch, S.; Marinelli, O.; Möller, J.; Santoni, G.; Chatonnet, A.; Lohse, M.J.; Wittmann, H.J.; Strasser, A.; et al. Structure-activity relationships and computational investigations into the development of potent and balanced dual-acting butyrylcholinesterase inhibitors and human cannabinoid receptor 2 ligands with pro-cognitive in vivo profiles. J. Med. Chem. 2018, 61, 1646–1663. [Google Scholar] [CrossRef] [PubMed]
- Synthetic cannabinoids in Europe (Perspectives on drugs). Available online: https://www.emcdda.europa.eu/system/files/publications/2753/POD_Synthetic%20cannabinoids_0.pdf (accessed on 29 June 2020).




{kind=link}
{kind=link}
{kind=link}
| Phytocannabinoids | Binding Type/CB | Ki (nM)/CB | EC50/IC50 (nM)/CB | Bioactivity |
|---|---|---|---|---|
 (a) Δ9-THC | Partial agonist/CB1, CB2 | 5.00~80.0/CB1 1.70~75.0/CB2 [14] | 13.0~87.0/CB1 41.8, 61.0/CB2 [14] | Analgesic, antiemetic, orexigenic [53]; relief from muscle spasms/spasticity in multiple sclerosis [61] |
 (b) CBD | Antagonist/inverse agonist, negative allosteric modulator/CB1 Partial agonist/CB2 | 73.0~>10,000/CB1 370~>10,000/CB2 [14] | 3860/CB1 503, 2270/CB2 [14] | Anti-inflammatory, anti-nociceptive, anti-oxidant, anti-ischemic, neuroprotective, immunosuppressive [62]; anxiolytic [43,62] |
 (c) Δ9-THCP | Agonist/CB1, CB2 | 1.20/CB1 6.20/CB2 [39] | NA | Analgesic [39] |
 (d) CBDP | NA | NA | NA | NA |
| Synthetic Cannabinoids | Chemical Type | Ki (nM)/CB | EC50 (nM)/CB | Bioactivity |
|---|---|---|---|---|
 (a) HU-210 | Classical | 0.0608~0.730/CB1 0.170~0.524/CB2 [66] | 0.0702/CB1 [71] NA/CB2 | Analgesic [53]; neuroprotective [72,73] |
 (b) CP-55,940 | Nonclassical | 0.500~5.00/CB1 0.690~2.80/CB2 [66] | 0.0462~31.0/CB1 [24,71,74] NA/CB2 | Anti-nociceptive, anti-emetic [53] |
 (c) WIN-55,212-2 | Amino-alkylindole | 1.89~123/CB1 0.280~16.2/CB2 [66] | 5.50~3000/CB1 [71,74,75,76,77,78] NA/CB2 | Analgesic, anti-inflammatory [53]; neuroprotective [79] |
 (d) Nabilone | Classical | 1.84/CB1 2.19/CB2 [66] | NA | Analgesic, antiemetic, anti-inflammatory [53]; neuroprotective [80,81] |
| Synthetic Cannabinoids | Chemical Type | Ki (nM)/CB | EC50 (nM)/CB1 | Bioactivity |
|---|---|---|---|---|
 (a) ACEA | Eicosanoid | 1.40, 5.29/CB1 195, >2000/CB2 [66] | 0.0317, 51.0 [74] | Anti-depressant [86]; anti-nociceptive [53]; anti-ulcer [84]; neuroprotective [91,92]; potentiating activity of antiepileptic drugs [87]; reducing cognitive impairment [91] |
 (b) ACPA | Eicosanoid | 2.20/CB1 715/CB2 [66] | 0.0551, 37.0 [74] | Anti-depressant, anxiolytic [83]; anti-nociceptive [53]; |
 (c) Methanandamide | Eicosanoid | 17.9~28.3/CB1 815~868/CB2 [66] | 1000 [77] | Analgesic, anti-emetic, orexigenic, anti-proliferation, anti-migration [53] |
 (d) O-1812 | Eicosanoid | 3.40/CB1 3870/CB2 [66] | NA | Anti-nociceptive, suppressing spontaneous activity and catalepsy [93]; anti-convulsant [94] |
| Synthetic Cannabinoids | Chemical Type | Ki (nM)/CB | EC50 (nM)/CB2 | Bioactivity |
|---|---|---|---|---|
 (a) JWH-133 | Classical | 677/CB1 3.40/CB2 [104] | 63.0 [105] | Attenuating neurodegenerative and spatial memory impairment [27]; improving cerebral infarction [106]; anti-inflammatory, ameliorating sepsis [107]; anti-cancer [53]; anti-nociceptive [108]; protective effects on renal ischemia-reperfusion injury [109] and against cardiotoxicity [110] |
 (b) JWH-015 | Amino-alkylindole | 383/CB1 13.8/CB2 [104] | NA | Attenuating neurodegenerative, neuroprotective [106]; immunomodulatory, anti-inflammatory [53]; anti-nociceptive [111]; anti-cancer [112]; anti-obesity [25] |
 (c) PM-226 | Classical | >40,000/CB1 12.8/CB2 [95] | 38.7 [95] | Neuroprotective [95]; anti-neuroinflammatory [78] |
 (d) (d)HU-308 | Nonclassical | >10,000/CB1 22.7/CB2 [66] | 5.57 [23] | Anti-convulsant, neuroprotective [106]; anti-inflammatory [96,97]; anti-nociceptive [96]; anti-dyskinesia [98]; osteoprotective [97]; alleviating septic lung injury [113] |
 (e) GW-405833 | Amino-alkylindole | 4772/CB1 3.92/CB2 [24] | 0.650 [24] | Anti-nociceptive, anti-inflammatory [99]; protective effects on acute liver injury [114] |
 (f) L-759,633 | Classical | 1043, 15850/CB1 6.40, 20.0/CB2 [66] | 8.10 [115] | Analgesic [53] |
 (g) L-759,656 | Classical | 529~>20000/CB1 11.8~57.0 [66] | 3.10 [115] | Analgesic [53] |
| Synthetic Cannabinoids | Chemical Type | Ki (nM)/CB | IC50 (nM)/CB1 | Bioactivity |
|---|---|---|---|---|
 (a) Rimonabant (SR141716) | Others | 1.80~12.3/CB1 702~13200/CB2 [66] | 5.60~48.0 [116,122] | Anti-obesity, smoking cessation [53]; protective effects of retinal degeneration [118]; neuroprotective [119] |
 (b) AM-251 | Others | 7.49/CB1 2290/CB2 [66] | 3.00 [122] | Anti-obesity; anti-depressant [53]; potentiating activity of antidepressant drugs [123]; improving albuminuria and renal tubular structure [124] as well as recognition memory [125] |
 (c) AM-281 | Others | 12.0/CB1 4200/CB2 [66] | 9.91 [122] | Improving cognitive deficits [53]; facilitatory effect on recognition memory [126]; ameliorating spatial learning and memory impairment [127]; protective effects against cardiotoxicity [110] |
 (d) Taranabant (MK-0364) | Others | 0.130, 0.270/CB1 170, 310/CB2 [104] | 0.290 [128] | Anti-obesity [129]; smoking cessation [130]; anti-nociceptive [131]; |
 (e) Otenabant (CP-945,598) | Others | 0.120~0.700/CB1 [132,133] 7663/CB2 [132] | 13.1 [122] | Anti-obesity [120] |
 (f) Surinabant (SR147778) | Others | 3.50/CB1 442/CB2 [134] | 9.60 [134] | Anti-obesity, smoking cessation, suppressing alcohol preference [53] |
| Synthetic Cannabinoids | Chemical Type | Ki (nM)/CB | IC50 (nM)/CB2 | Bioactivity |
|---|---|---|---|---|
 (a) SR144528 | Others | 50.3~>10,000/CB1 0.280~5.60/CB2 [66] | 39.0 [135] | Anti-nociceptive [53] |
 (b) AM-630 | Amino-alkylindole | 5152/CB1 31.2/CB2 [66] | 12.3 [122] | Inhibiting inflammatory osteolysis [136]; potentiating activity of antidepressant drugs [26]; improving memory, anti-oxidant [137] |
| Synthetic Cannabinoids | Chemical Type | Bioactivity |
|---|---|---|
 (a) Org29647 | Amino-alkylindole | NA |
 (b) Org27759 | Amino-alkylindole | NA |
 (c) Org27569 | Amino-alkylindole | Anti-obesity [12] |
 (d) GAT211 | Amino-alkylindole | Reducing the signs and symptoms of Huntington’s disease [146]; augmenting the pharmacological effects of the CB1 orthosteric agonists [12]; anti-nociceptive, anti-inflammatory [142] |
 (e) GAT229 | Amino-alkylindole | Anti-nociceptive and anti-inflammatory in combination with the orthosteric CB1 agonist [143]; reducing the signs and symptoms of Huntington’s disease [146] |
 (f) ZCZ011 | Amino-alkylindole | Anti-nociceptive, anti-inflammatory [139] |
 (g) RTI-371 | Others | Blocking cocaine-induced locomotor stimulation [147] |
 (h) PSNCBAM-1 | Others | Anti-obesity [139] |
 (i) CBD-DMH (HU-219) | Nonclassical | Anti-inflammatory [148], anxiolytic [149] |
 (j) Compound C2 | Others | Anti-nociceptive [145] |
| Peptides | Sequence | Bioactivity | Source |
|---|---|---|---|
| (a) Hemopressin (Hp) | PVNFKFLSH | Neuromodulatory, anorexigenic, alleviating liver fibrosis, antinociceptive [36] | α-Hemoglobin in rat |
| (b) VD-Hpα (pepcan-11) | VDPVNFKLLSH | Neuromodulatory, antinociceptive, inhibiting gastrointestinal mobility, inducing tolerance to thermal antinociception, orexigenic [36] | α-Hemoglobin in human and mouse |
| (c) RVD-Hpα (pepcan-12) | RVDPVNFKLLSH | Neuromodulatory, anxiolytic, anti-depressant [36] | α-Hemoglobin in human and mouse |
| (d) Pep19 | DIIADDEPLT | Reducing body weight, improving metabolic parameters [35] | Peptidyl-prolyl cis-trans isomerase A in human |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, D.; Peigneur, S.; Hendrickx, L.A.; Tytgat, J. Targeting Cannabinoid Receptors: Current Status and Prospects of Natural Products. Int. J. Mol. Sci. 2020, 21, 5064. https://doi.org/10.3390/ijms21145064
An D, Peigneur S, Hendrickx LA, Tytgat J. Targeting Cannabinoid Receptors: Current Status and Prospects of Natural Products. International Journal of Molecular Sciences. 2020; 21(14):5064. https://doi.org/10.3390/ijms21145064
Chicago/Turabian StyleAn, Dongchen, Steve Peigneur, Louise Antonia Hendrickx, and Jan Tytgat. 2020. "Targeting Cannabinoid Receptors: Current Status and Prospects of Natural Products" International Journal of Molecular Sciences 21, no. 14: 5064. https://doi.org/10.3390/ijms21145064
APA StyleAn, D., Peigneur, S., Hendrickx, L. A., & Tytgat, J. (2020). Targeting Cannabinoid Receptors: Current Status and Prospects of Natural Products. International Journal of Molecular Sciences, 21(14), 5064. https://doi.org/10.3390/ijms21145064