Seeding and Growth of β-Amyloid Aggregates upon Interaction with Neuronal Cell Membranes

 ,
,  ,
,  , and
, and 
Abstract
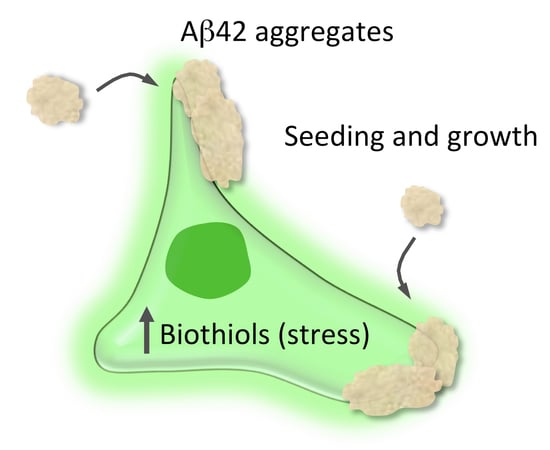
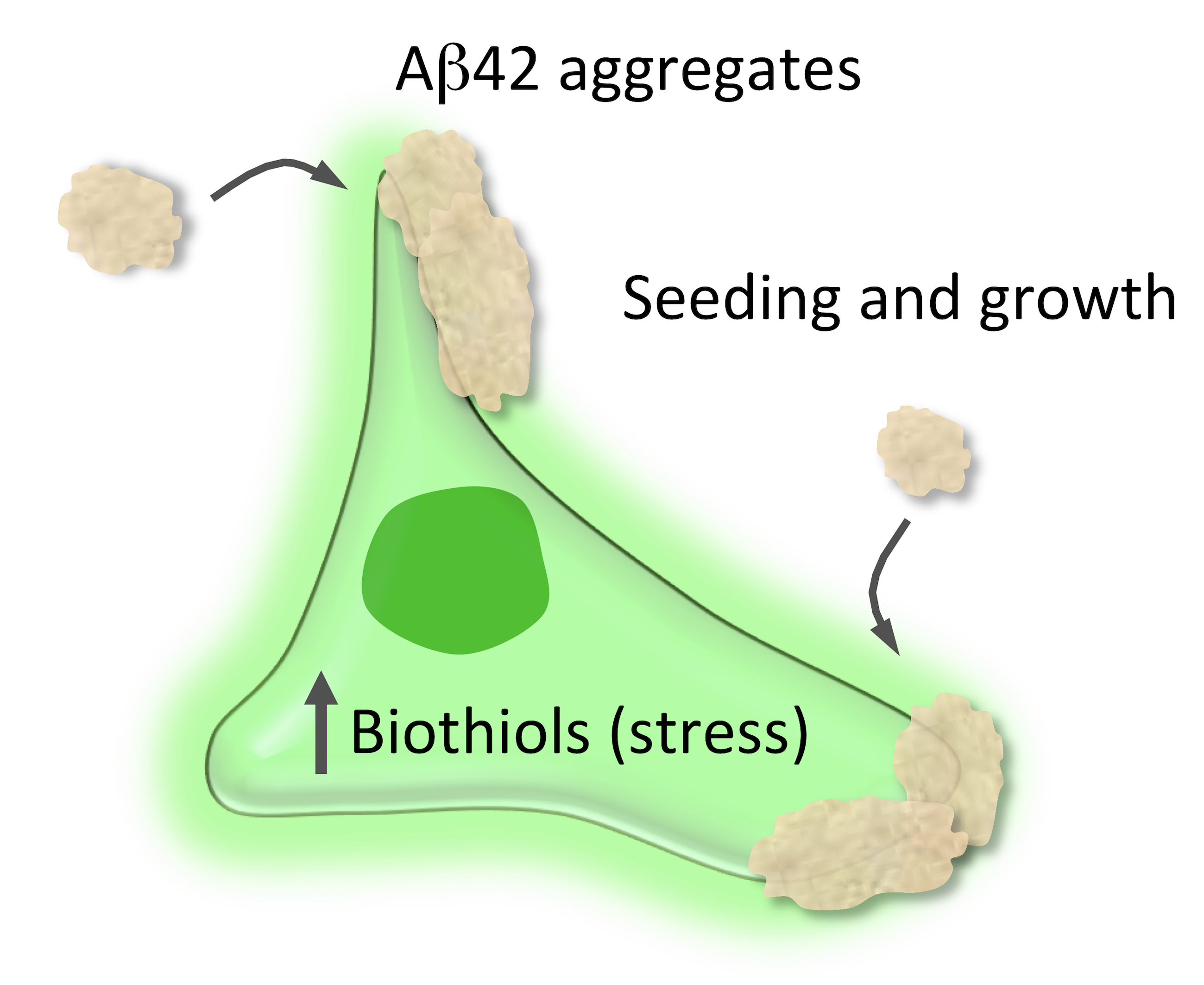{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
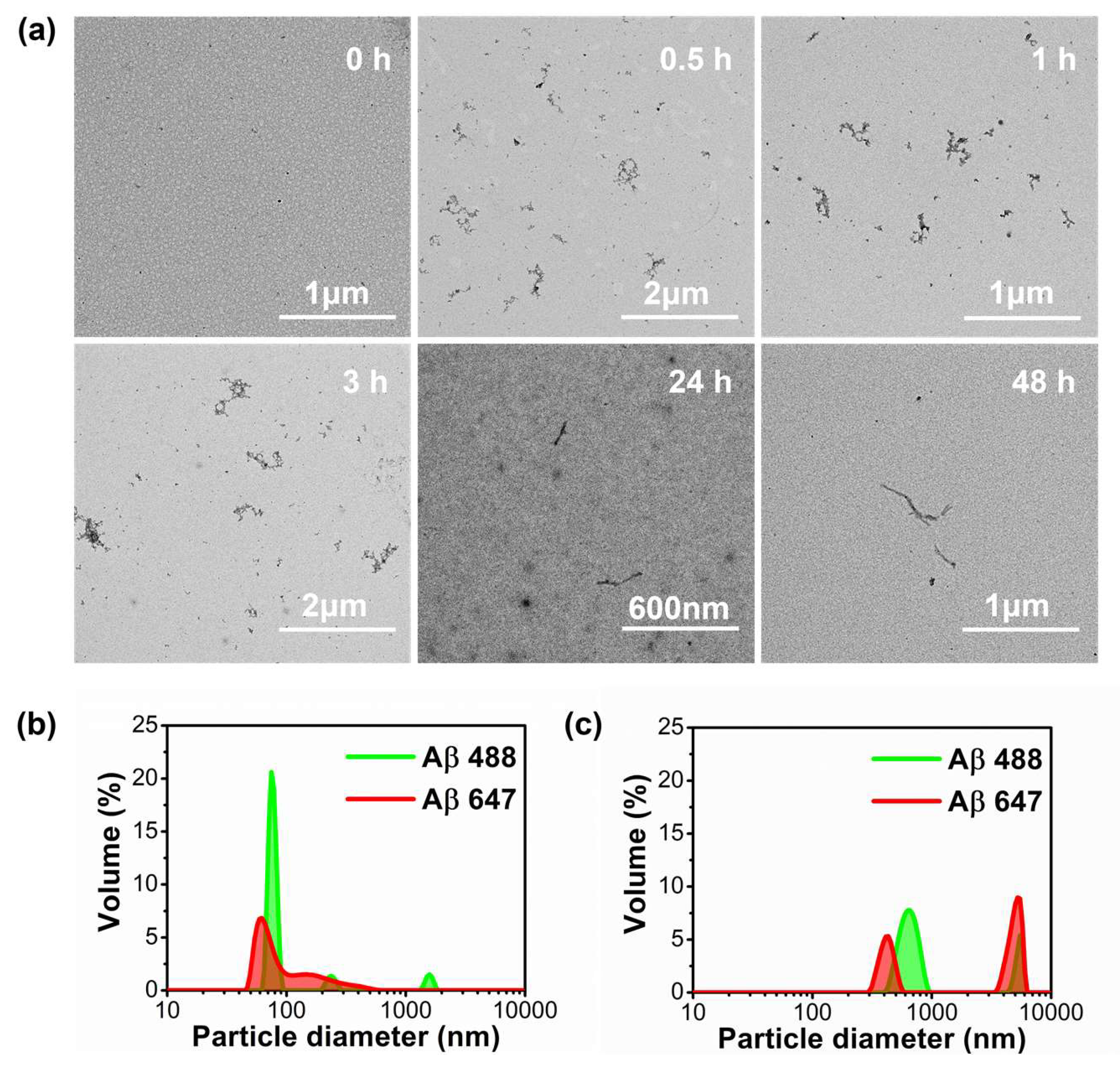
2. Results
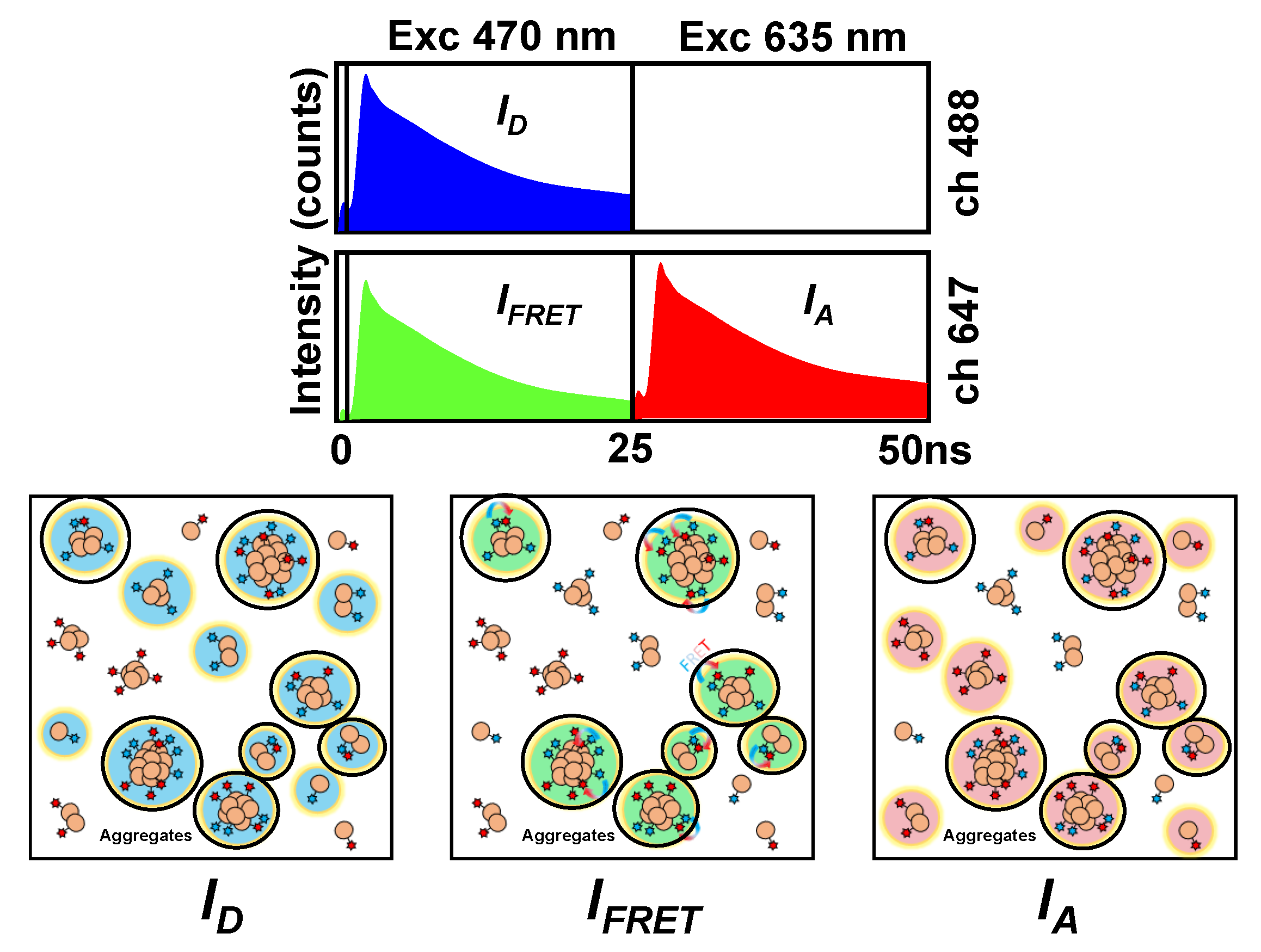
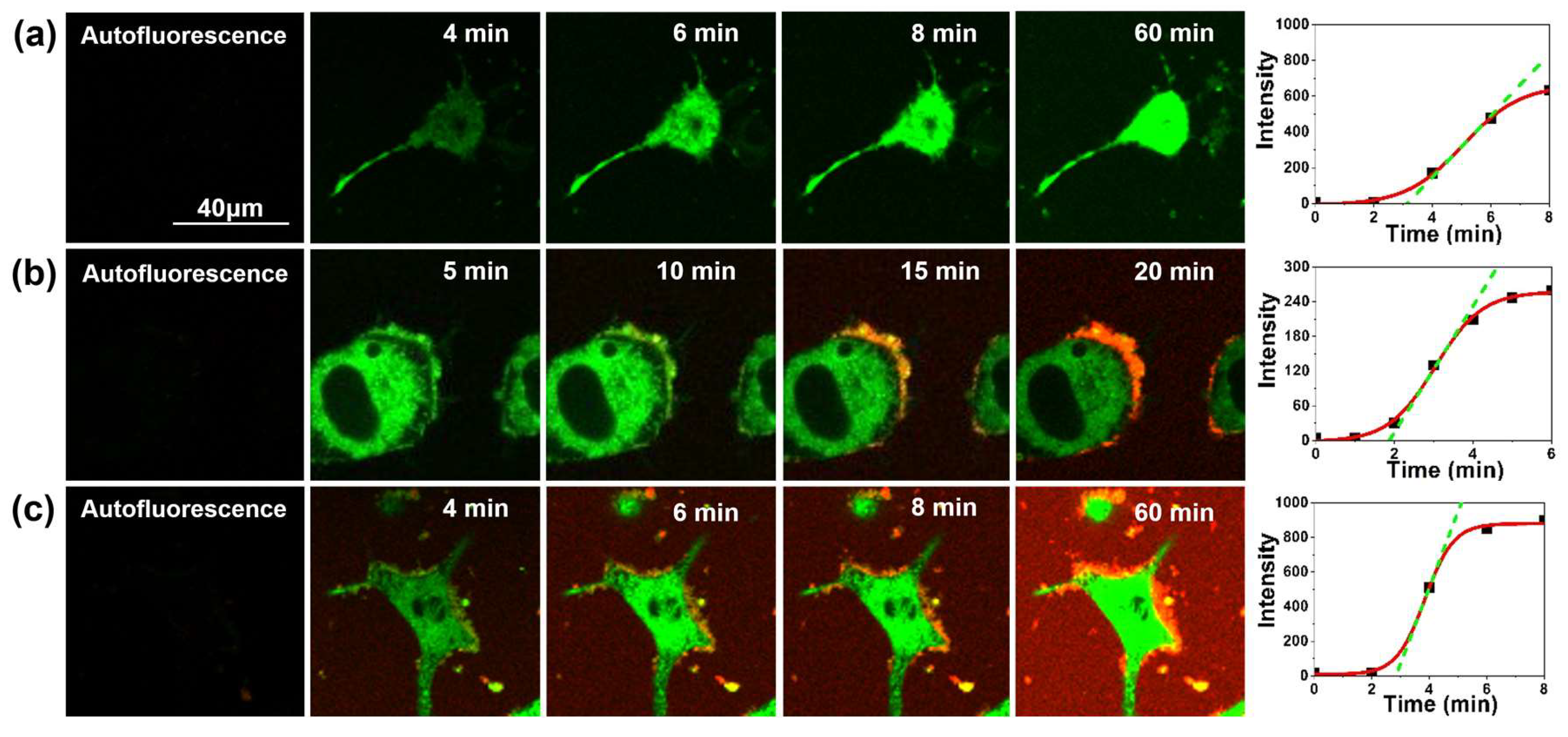
2.1. Interaction between Aβ42 Aggregates and N2a Cells
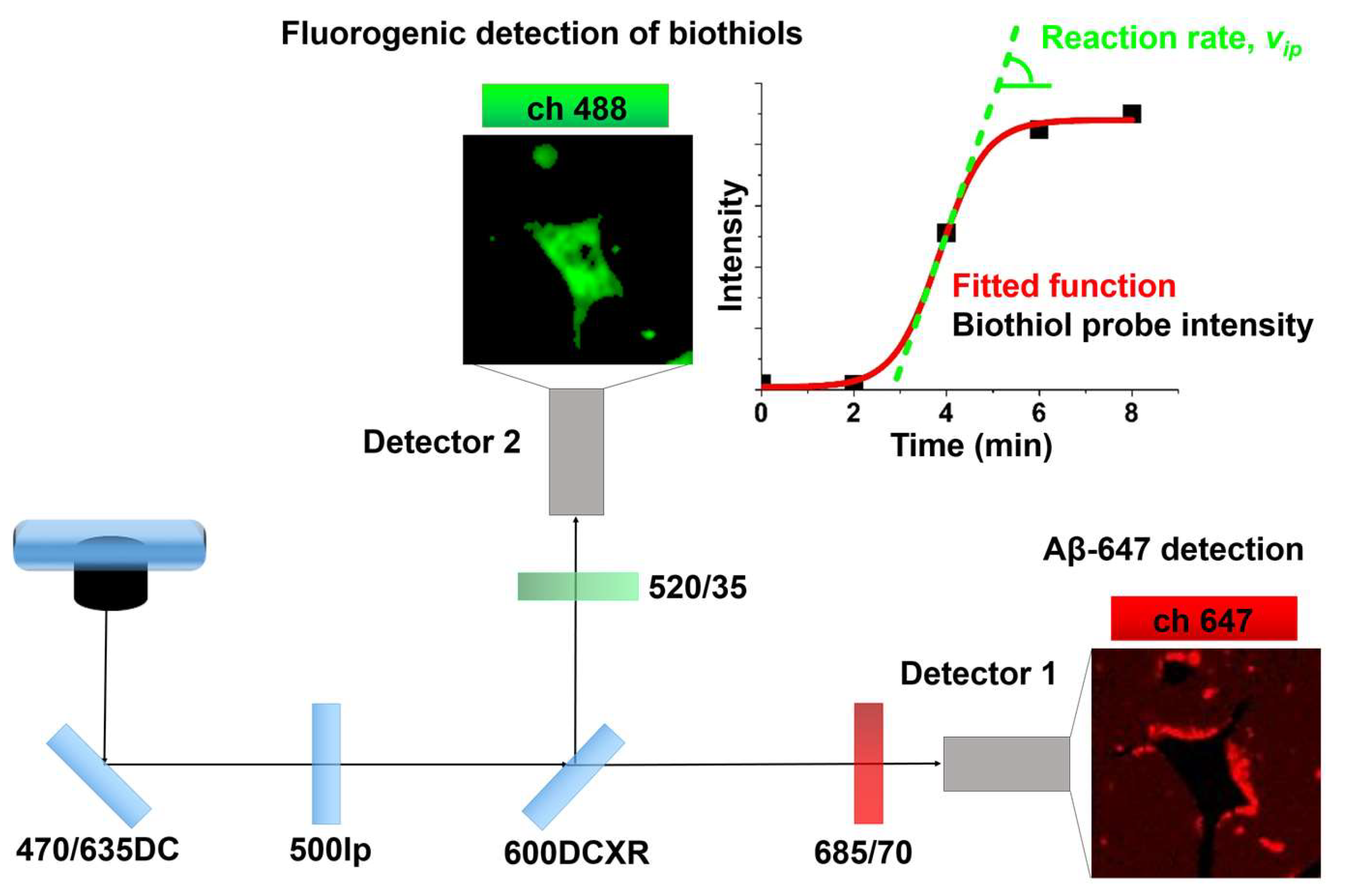
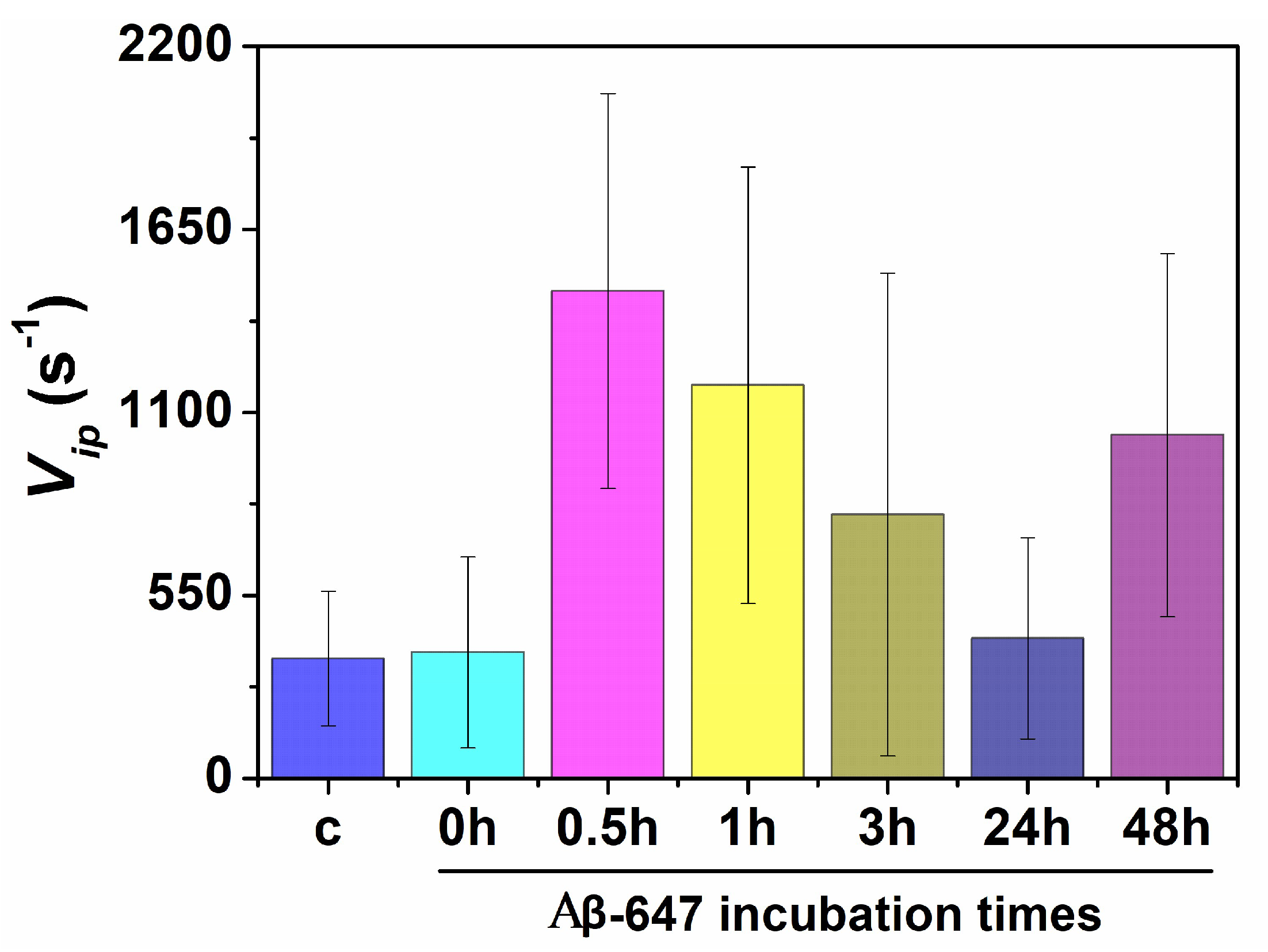
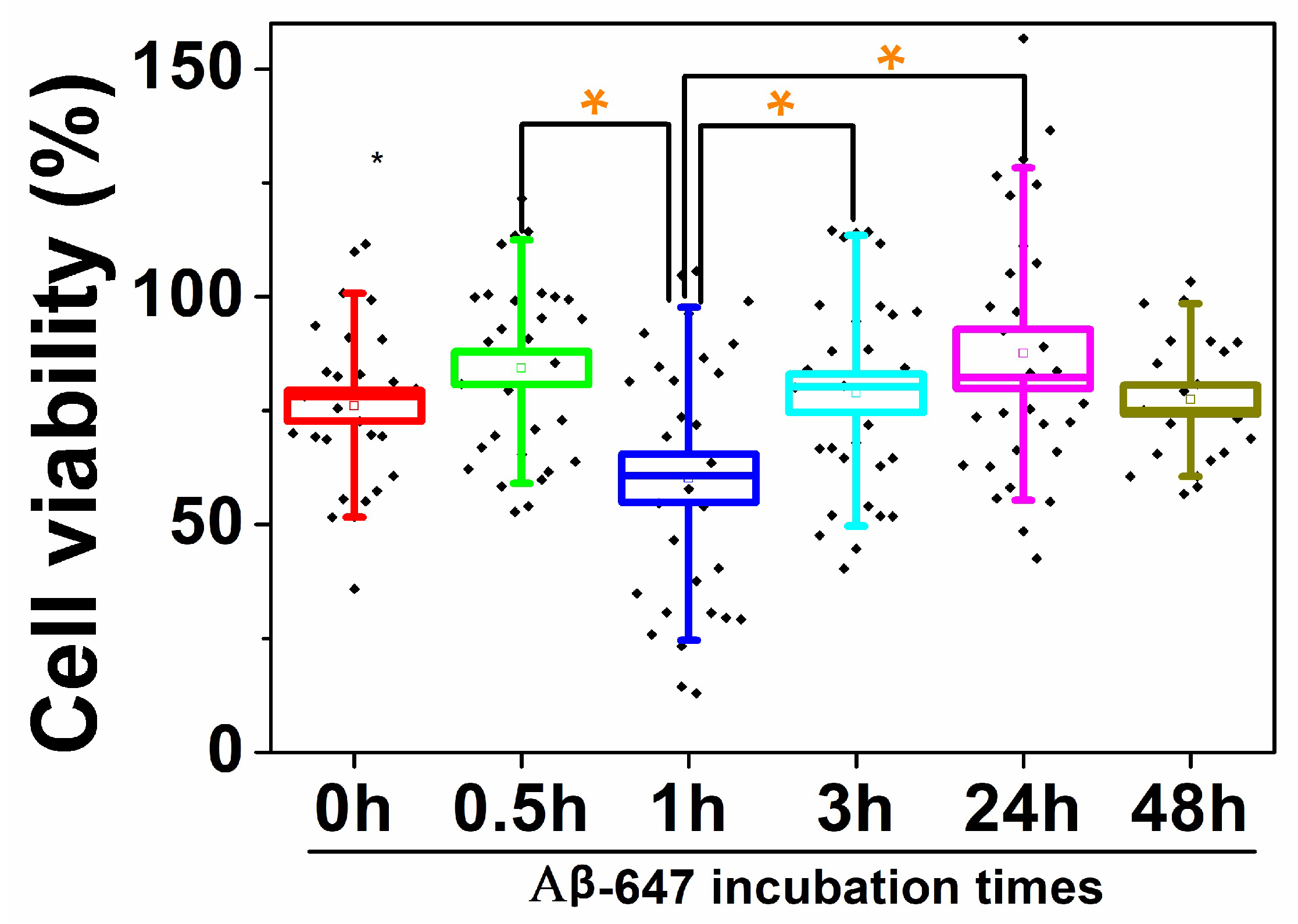
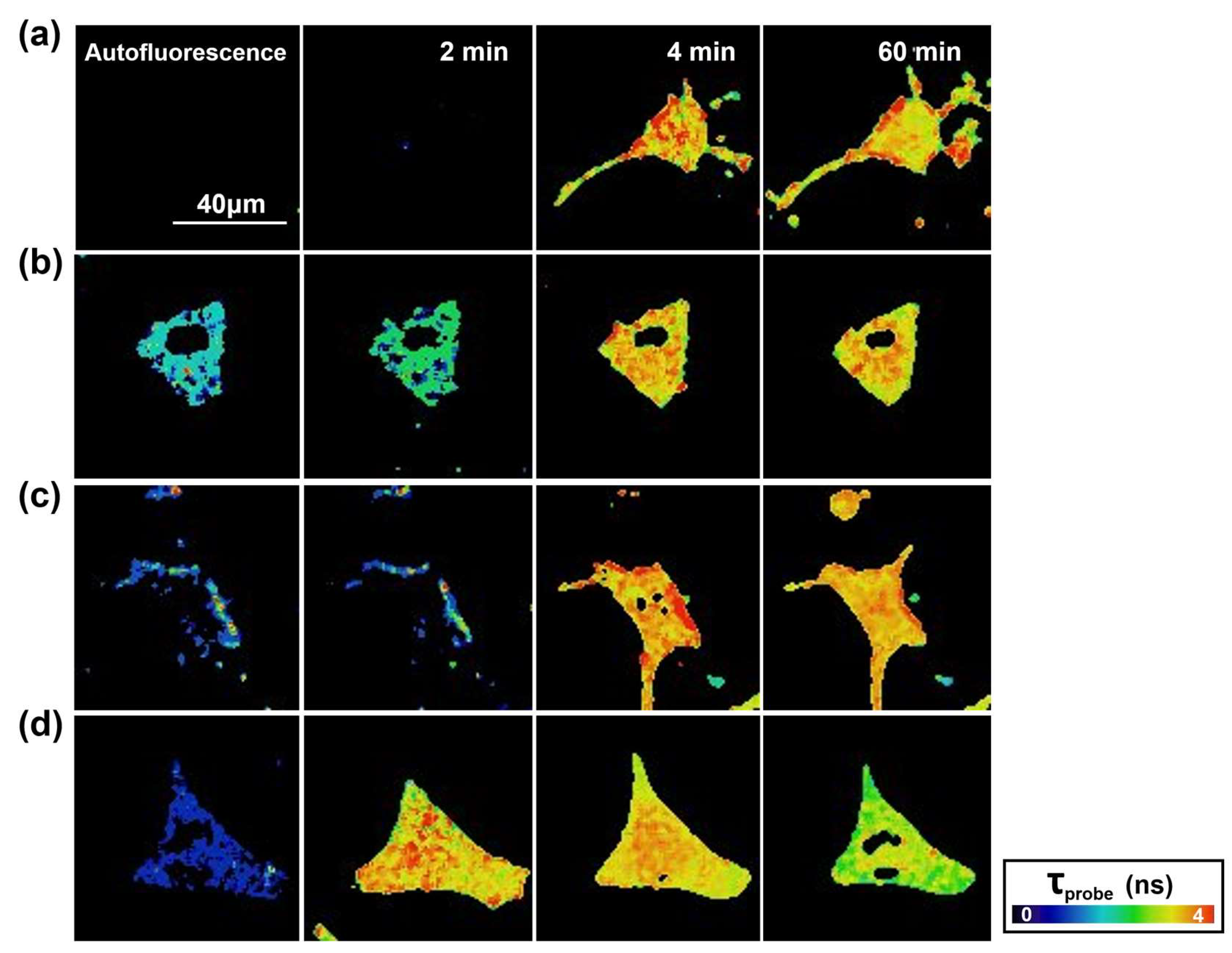
2.2. Study of Cellular Stress Using a Biothiol Probe
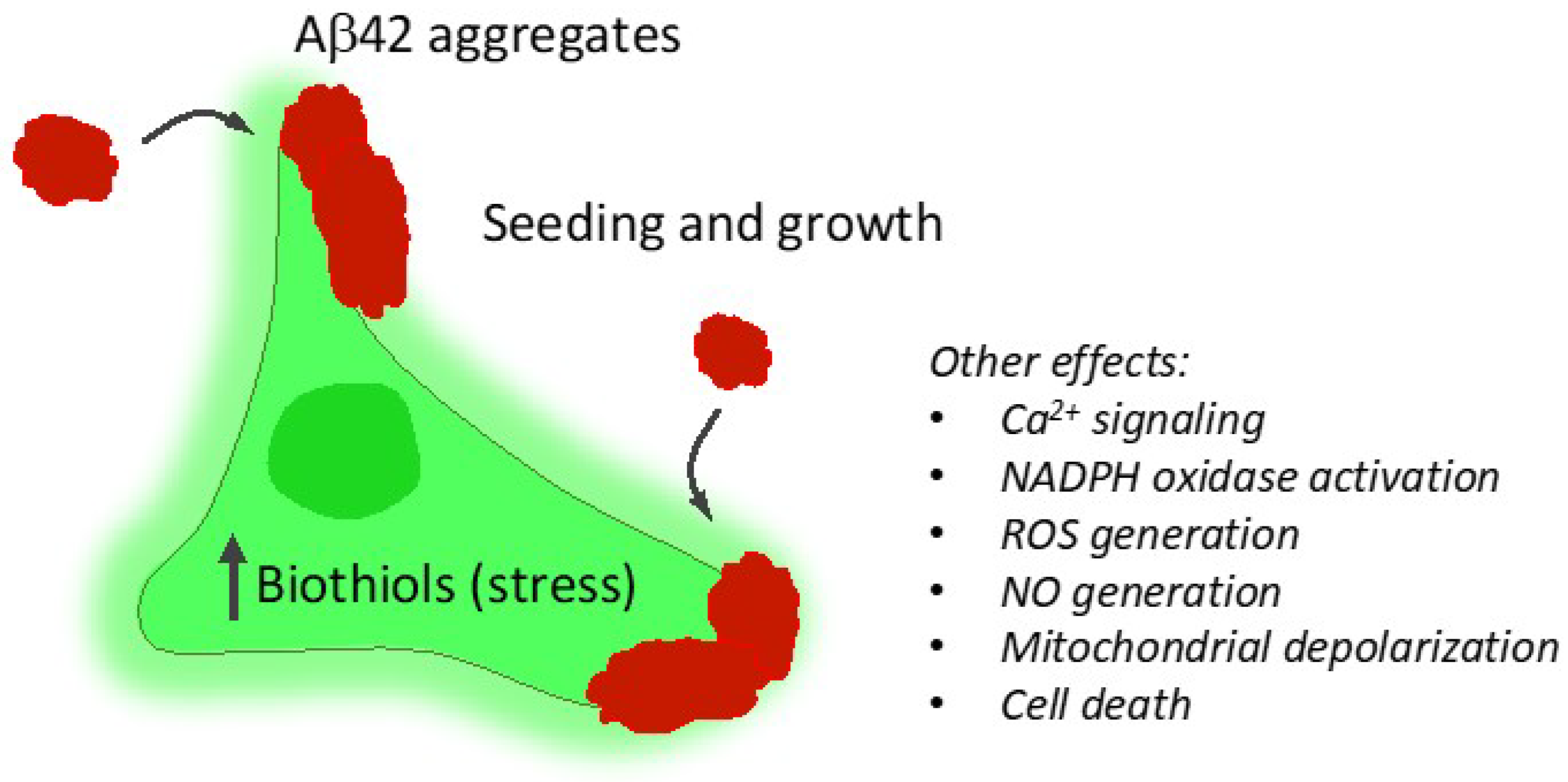
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Instruments
4.3. Aβ Amyloid Aggregation
4.4. Cell Viability Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FLIM | Fluorescence lifetime imaging microscopy |
| FRET | Fluorescence resonance energy transfer |
| PIE | Pulsed interleaved excitation |
| Aβ42 | β-Amyloid (1-42) peptide |
| Aβ-488 | β-Amyloid (1-42) peptide labeled with HiLyte Fluor 488 |
| Aβ-647 | β-Amyloid (1-42) peptide labeled with HiLyte Fluor 647 |
| GG-DNBS | Granada Green dinitrobenzene sulfonate |
| TEM | Transmission electron microscopy |
| DLS | Dynamic light scattering |
| TCSPC | Time-correlated single-photon counting |
References
- Temussi, P.A.; Masino, L.; Pastore, A. From Alzheimer to Huntington: Why is a structural understanding so difficult? EMBO J. 2003, 22, 355–361. [Google Scholar] [CrossRef]
- Relja, M. Pathophysiology and Classification of Neurodegenerative Diseases. EJIFCC 2004, 15, 97–99. [Google Scholar] [PubMed]
- Giannakopoulos, P.; Silhol, S.; Jallageas, V.; Mallet, J.; Bons, N.; Bouras, C.; Delaere, P. Quantitative analysis of tau protein-immunoreactive accumulations and beta amyloid protein deposits in the cerebral cortex of the mouse lemur, Microcebus murinus. Acta Neuropathol. 1997, 94, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Naslund, J.; Haroutunian, V.; Mohs, R.; Davis, K.L.; Davies, P.; Greengard, P.; Buxbaum, J.D. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 2000, 283, 1571–1577. [Google Scholar] [CrossRef]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Wirthensohn, D.C.; Flagmeier, P.; Hughes, C.; Aprile, F.A.; Ruggeri, F.S.; Whiten, D.R.; Emin, D.; Xia, Z.; Varela, J.A.; et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 2019, 10, 1541. [Google Scholar] [CrossRef]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef]
- Lv, Z.; Roychaudhuri, R.; Condron, M.M.; Teplow, D.B.; Lyubchenko, Y.L. Mechanism of amyloid β−protein dimerization determined using single−molecule AFM force spectroscopy. Sci. Rep. 2013, 3, 2880. [Google Scholar] [CrossRef]
- Bucciantini, M.; Calloni, G.; Chiti, F.; Formigli, L.; Nosi, D.; Dobson, C.M.; Stefani, M. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 2004, 279, 31374–31382. [Google Scholar] [CrossRef] [PubMed]
- Bronfman, F.C.; Soto, C.; Tapia, L.; Tapia, V.; Inestrosa, N.C. Extracellular matrix regulates the amount of the β-amyloid precursor protein and its amyloidogenic fragments. J. Cell. Physiol. 1996, 166, 360–369. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef]
- Stine, W.B., Jr.; LaDu, M.J. Apolipoprotein E: Structural and Functional Interactions with Amyloid β. Amyloid Proteins 2005, 211–243. [Google Scholar] [CrossRef]
- Relini, A.; Marano, N.; Gliozzi, A. Misfolding of amyloidogenic proteins and their interactions with membranes. Biomolecules 2013, 4, 20–55. [Google Scholar] [CrossRef]
- Dyall, S.C. Amyloid-Beta Peptide, Oxidative Stress and Inflammation in Alzheimer’s Disease: Potential Neuroprotective Effects of Omega-3 Polyunsaturated Fatty Acids. Int. J. Alzheimer’s Dis. 2010, 2010, 274128. [Google Scholar] [CrossRef]
- Di Scala, C.; Yahi, N.; Boutemeur, S.; Flores, A.; Rodriguez, L.; Chahinian, H.; Fantini, J. Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 2016, 6, 28781. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef]
- Berezin, M.Y.; Achilefu, S. Fluorescence Lifetime Measurements and Biological Imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar] [CrossRef]
- Ruedas-Rama, M.J.; Alvarez-Pez, J.M.; Crovetto, L.; Paredes, J.M.; Orte, A. FLIM Strategies for Intracellular Sensing BT-Advanced Photon Counting: Applications, Methods, Instrumentation; Kapusta, P., Wahl, M., Erdmann, R., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 191–223. ISBN 978-3-319-15636-1. [Google Scholar]
- Aliyan, A.; Cook, N.P.; Martí, A.A. Interrogating Amyloid Aggregates using Fluorescent Probes. Chem. Rev. 2019, 119, 11819–11856. [Google Scholar] [CrossRef] [PubMed]
- Aprile, F.A.; Sormanni, P.; Podpolny, M.; Chhangur, S.; Needham, L.-M.; Ruggeri, F.S.; Perni, M.; Limbocker, R.; Heller, G.T.; Sneideris, T.; et al. Rational design of a conformation-specific antibody for the quantification of Aβ oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 13509–13518. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Aliyan, A.; Cook, N.P.; Augustine, A.; Bhak, G.; Maldonado, R.; Smith McWilliams, A.D.; Flores, E.M.; Mendez, N.; Shahnawaz, M.; et al. Monitoring the Formation of Amyloid Oligomers Using Photoluminescence Anisotropy. J. Am. Chem. Soc. 2019, 141, 15605–15610. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.; Sun, A.; Wang, M.; Wei, P.; Li, R.; Yi, T. A novel near-infrared fluorescent probe for detection of early-stage Aβ protofibrils in Alzheimer’s disease. Chem. Commun. 2020, 56, 1625–1628. [Google Scholar] [CrossRef] [PubMed]
- González-Vera, J.A.; Fueyo-González, F.; Alkorta, I.; Peyressatre, M.; Morris, M.C.; Herranz, R. Highly solvatochromic and tunable fluorophores based on a 4,5-quinolimide scaffold: Novel CDK5 probes. Chem. Commun. 2016, 52, 9652–9655. [Google Scholar] [CrossRef] [PubMed]
- Fueyo-González, F.; González-Vera, J.A.; Alkorta, I.; Infantes, L.; Jimeno, M.L.; Aranda, P.; Acuña-Castroviejo, D.; Ruiz-Arias, A.; Orte, A.; Herranz, R. Environment-Sensitive Probes for Illuminating Amyloid Aggregation in vitro and in Zebrafish. ACS Sens. 2020. [Google Scholar] [CrossRef]
- Jurado, R.; Castello, F.; Bondia, P.; Casado, S.; Flors, C.; Cuesta, R.; Domínguez-Vera, J.M.; Orte, A.; Gálvez, N. Apoferritin fibers: A new template for 1D fluorescent hybrid nanostructures. Nanoscale 2016, 8, 9648–9656. [Google Scholar] [CrossRef]
- Castello, F.; Casares, S.; Ruedas-Rama, M.J.; Orte, A. The First Step of Amyloidogenic Aggregation. J. Phys. Chem. B 2015, 119, 8260–8267. [Google Scholar] [CrossRef]
- Castello, F.; Paredes, J.M.; Ruedas-rama, M.J.; Martin, M.; Roldan, M.; Casares, S.; Orte, A. Two-Step Amyloid Aggregation: Sequential Lag Phase Intermediates. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Ward, R.V.; Jennings, K.H.; Jepras, R.; Neville, W.; Owen, D.E.; Hawkins, J.; Christie, G.; Davis, J.B.; George, A.; Karran, E.H.; et al. Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of beta-amyloid peptide. Biochem. J. 2000, 348 Pt. 1, 137–144. [Google Scholar] [CrossRef]
- Narayan, P.; Holmstrom, K.M.; Kim, D.-H.; Whitcomb, D.J.; Wilson, M.R.; St George-Hyslop, P.; Wood, N.W.; Dobson, C.M.; Cho, K.; Abramov, A.Y.; et al. Rare individual amyloid-β oligomers act on astrocytes to initiate neuronal damage. Biochemistry 2014, 53, 2442–2453. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, K.; Williams, T.L.; Morris, K.L.; Jonckheere, W.; Vandersteen, A.; Kelly, G.; Schymkowitz, J.; Rousseau, F.; Pastore, A.; Serpell, L.C.; et al. Structural basis for increased toxicity of pathological aβ42:aβ40 ratios in Alzheimer disease. J. Biol. Chem. 2012, 287, 5650–5660. [Google Scholar] [CrossRef]
- Qiu, T.; Liu, Q.; Chen, Y.-X.; Zhao, Y.-F.; Li, Y.-M. Aβ42 and Aβ40: Similarities and differences. J. Pept. Sci. 2015, 21, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.K.; Ullman, O.; Stultz, C.M. Comparative studies of disordered proteins with similar sequences: Application to Aβ40 and Aβ. Biophys. J. 2013, 104, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.P.; Bhatnagar, A. Role of Thiols in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 133–139. [Google Scholar] [CrossRef]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Béringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef]
- Puente-Muñoz, V.; Paredes, J.M.; Resa, S.; Vílchez, J.D.; Zitnan, M.; Miguel, D.; Girón, M.D.; Cuerva, J.M.; Salto, R.; Crovetto, L. New Thiol-Sensitive Dye Application for Measuring Oxidative Stress in Cell Cultures. Sci. Rep. 2019, 9, 1659. [Google Scholar] [CrossRef]
- Novo, M.; Freire, S.; Al-Soufi, W. Critical aggregation concentration for the formation of early Amyloid-β (1–42) oligomers. Sci. Rep. 2018, 8, 1783. [Google Scholar] [CrossRef]
- Morel, B.; Carrasco, M.P.; Jurado, S.; Marco, C.; Conejero-Lara, F. Dynamic micellar oligomers of amyloid beta peptides play a crucial role in their aggregation mechanisms. Phys. Chem. Chem. Phys. 2018, 20, 20597–20614. [Google Scholar] [CrossRef]
- Herrero-Foncubierta, P.; Paredes, J.M.; Giron, M.D.; Salto, R.; Cuerva, J.M.; Miguel, D.; Orte, A. A Red-Emitting, Multidimensional Sensor for the Simultaneous Cellular Imaging of Biothiols and Phosphate Ions. Sensors 2018, 18, 161. [Google Scholar] [CrossRef]
- Daniels, W.; Hendricks, J.; Salie, R.; Taljaard, J. The Role of the MAP-Kinase Superfamily in β-Amyloid Toxicity. Metab. Brain Dis. 2001, 16, 175–185. [Google Scholar] [CrossRef]
- Castelletto, V.; Ryumin, P.; Cramer, R.; Hamley, I.W.; Taylor, M.; Allsop, D.; Reza, M.; Ruokolainen, J.; Arnold, T.; Hermida-Merino, D.; et al. Self-Assembly and Anti-Amyloid Cytotoxicity Activity of Amyloid beta Peptide Derivatives. Sci. Rep. 2017, 7, 43637. [Google Scholar] [CrossRef]
- Resa, S.; Orte, A.; Miguel, D.; Paredes, J.M.; Puente-Muñoz, V.; Salto, R.; Giron, M.D.; Ruedas-Rama, M.J.; Cuerva, J.M.; Alvarez-Pez, J.M.; et al. New Dual Fluorescent Probe for Simultaneous Biothiol and Phosphate Bioimaging. Chem. Eur. J. 2015, 21, 14772–14779. [Google Scholar] [CrossRef] [PubMed]
- Crowther, D.C.; Kinghorn, K.J.; Miranda, E.; Page, R.; Curry, J.A.; Duthie, F.A.I.; Gubb, D.C.; Lomas, D.A. Intraneuronal Aβ, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 2005, 132, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Greeve, I.; Kretzschmar, D.; Tschäpe, J.-A.; Beyn, A.; Brellinger, C.; Schweizer, M.; Nitsch, R.M.; Reifegerste, R. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J. Neurosci. 2004, 24, 3899–3906. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Sergeant, N.; Itier, J.-M.; Blanchard, V.; Wirths, O.; van der Kolk, N.; Vingtdeux, V.; van de Steeg, E.; Ret, G.; Canton, T.; et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Aβ42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 2004, 165, 1289–1300. [Google Scholar] [CrossRef]
- Terzi, E.; Hölzemann, G.; Seelig, J. Interaction of Alzheimer β-Amyloid Peptide(1−40) with Lipid Membranes. Biochemistry 1997, 36, 14845–14852. [Google Scholar] [CrossRef]
- Verdier, Y.; Penke, B. Binding Sites of Amyloid β-Peptide in Cell Plasma Membrane and Implications for Alzheimers Disease. Curr. Protein Pept. Sci. 2004, 5, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N. Chapter 11—Alzheimer’s Disease. In Brain Lipids in Synaptic Function and Neurological Disease, 1st ed.; Academic Press: San Diego, CA, USA, 2015; pp. 253–277. ISBN 978-0-12-800111-0. [Google Scholar]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T. Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Kedia, N.; Illes-Toth, E.; Haralampiev, I.; Prisner, S.; Herrmann, A.; Wanker, E.E.; Bieschke, J. Amyloid-β(1-42) Aggregation Initiates Its Cellular Uptake and Cytotoxicity. J. Biol. Chem. 2016, 291, 19590–19606. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Hashemi, M.; Zagorski, K.; Lyubchenko, Y.L. Interaction of Aβ42 with Membranes Triggers the Self-Assembly into Oligomers. Int. J. Mol. Sci. 2020, 21, 1129. [Google Scholar] [CrossRef] [PubMed]
- Dear, A.J.; Michaels, T.C.T.; Meisl, G.; Klenerman, D.; Wu, S.; Perrett, S.; Linse, S.; Dobson, C.M.; Knowles, T.P.J. Kinetic diversity of amyloid oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 12087–12094. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Arias, Á.; Paredes, J.M.; Di Biase, C.; Cuerva, J.M.; Giron, M.D.; Salto, R.; González-Vera, J.A.; Orte, A. Seeding and Growth of β-Amyloid Aggregates upon Interaction with Neuronal Cell Membranes. Int. J. Mol. Sci. 2020, 21, 5035. https://doi.org/10.3390/ijms21145035
Ruiz-Arias Á, Paredes JM, Di Biase C, Cuerva JM, Giron MD, Salto R, González-Vera JA, Orte A. Seeding and Growth of β-Amyloid Aggregates upon Interaction with Neuronal Cell Membranes. International Journal of Molecular Sciences. 2020; 21(14):5035. https://doi.org/10.3390/ijms21145035
Chicago/Turabian StyleRuiz-Arias, Álvaro, Jose M. Paredes, Chiara Di Biase, Juan M. Cuerva, María D. Giron, Rafael Salto, Juan A. González-Vera, and Angel Orte. 2020. "Seeding and Growth of β-Amyloid Aggregates upon Interaction with Neuronal Cell Membranes" International Journal of Molecular Sciences 21, no. 14: 5035. https://doi.org/10.3390/ijms21145035
APA StyleRuiz-Arias, Á., Paredes, J. M., Di Biase, C., Cuerva, J. M., Giron, M. D., Salto, R., González-Vera, J. A., & Orte, A. (2020). Seeding and Growth of β-Amyloid Aggregates upon Interaction with Neuronal Cell Membranes. International Journal of Molecular Sciences, 21(14), 5035. https://doi.org/10.3390/ijms21145035