Current and Future Role of Tyrosine Kinases Inhibition in Thyroid Cancer: From Biology to Therapy

 , ,
, ,
Abstract

1. Introduction
1.1. General Aspects
1.2. Histological Subtypes
1.2.1. Follicular-Derived Thyroid Cancers
1.2.2. Neuroendocrine C-Cell-Derived Thyroid Cancers
2. Molecular Biology
2.1. Differentiated TC (TCGA)
2.1.1. Papillary Thyroid Cancer
2.1.2. Follicular Thyroid Cancer
2.1.3. Hürthle Thyroid Cancer
2.1.4. Poorly Differentiated Thyroid Cancer
2.2. Medullary Thyroid Cancer
2.2.1. Germ Line RET (Rearranged during Transfection) Mutations
2.2.2. Somatic RET (Rearranged during Transfection) Mutations
2.2.3. Somatic RAS Mutations
2.2.4. Other Pathways
2.3. Anaplastic Thyroid Cancer
3. From Molecular Knowledge to Clinical Benefit: Approved Drugs
3.1. Differentiated Thyroid Cancer
3.2. Medullary Thyroid Cancer
3.3. Anaplastic Thyroid Cancer
4. Future Landscape
4.1. RET (Rearranged during Transfection) Inhibition
4.1.1. Differentiated Thyroid Cancer
4.1.2. Medullary Thyroid Cancer
4.2. Immunotherapy
4.2.1. Differentiated Thyroid Cancer
4.2.2. Medullary Thyroid Cancer
4.2.3. Anaplastic Thyroid Cancer
4.3. NTRK Inhibitors
4.4. ALK (Anaplastic Lymphoma Kinase) Inhibitors
4.4.1. Differentiated Thyroid Cancer
4.4.2. Medullary Thyroid Cancer
4.4.3. Anaplastic Thyroid Cancer
4.5. Radioactive Iodine Resensitization/Redifferentiation
4.6. Other Novel Therapeutic Approaches
4.6.1. Peptide Receptor Radionuclide Therapy (PRRT)
4.6.2. Epidermal Growth Factor Receptor 1 (EGFR) Epidermal Growth Factor Receptor 2/3 (HER2/3)
4.6.3. Mammalian Target of Rapamycin (mTOR)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019. [Google Scholar]
- Liu, J.W.; Chen, C.; Loh, E.W.; Chu, C.C.; Wang, M.Y.; Ouyang, H.J.; Chang, Y.T.; Zhuang, W.Z.; Chou, C.W.; Huang, D.J.; et al. Tyrosine Kinase Inhibitors for Advanced or Metastatic Thyroid Cancer: A Meta-Analysis of Randomized Controlled Trials. Curr. Med. Res. Opin. 2018, 34, 795–803. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Osamura, Y.R.; Kloppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs; WHO Press: Geneva, Switzerland, 2017. [Google Scholar]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High Prevalence of BRAF Mutations in Thyroid Cancer: Genetic Evidence for Constitutive Activation of the RET/PTC-RAS-BRAF Signaling Pathway in Papillary Thyroid Carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Shen, X.; Liu, R.; Xing, M. A Six-Genotype Genetic Prognostic Model for Papillary Thyroid Cancer. Endocr. Relat. Cancer 2017. [Google Scholar] [CrossRef]
- Song, Y.S.; Park, Y.J. Genomic Characterization of Differentiated Thyroid Carcinoma. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Bounacer, A.; Wicker, R.; Caillou, B.; Cailleux, A.F.; Sarasin, A.; Schlumberger, M.; Suárez, H.G. High Prevalence of Activating Ret Proto-Oncogene Rearrangements, in Thyroid Tumors from Patients Who Had Received External Radiation. Oncogene 1997. [Google Scholar] [CrossRef] [PubMed]
- Bongarzone, I.; Vigneri, P.; Mariani, L.; Collini, P.; Pilotti, S.; Pierotti, M.A. RET/NTRK1 Rearrangements in Thyroid Gland Tumors of the Papillary Carcinoma Family: Correlation with Clinicopathological Features. Clin. Cancer Res. 1998, 4, 223–228. [Google Scholar] [PubMed]
- Caronia, L.M.; Phay, J.E.; Shah, M.H. Role of BRAF in Thyroid Oncogenesis. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Westra, W.H.; Tufano, R.P.; Cohen, Y.; Rosenbaum, E.; Rhoden, K.J.; Carson, K.A.; Vasko, V.; Larin, A.; Tallini, G.; et al. BRAF Mutation Predicts a Poorer Clinical Prognosis for Papillary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2005. [Google Scholar] [CrossRef]
- Vasko, V.; Hu, S.; Wu, G.; Xing, J.C.; Larin, A.; Savchenko, V.; Trink, B.; Xing, M. High Prevalence and Possible de Novo Formation of BRAF Mutation in Metastasized Papillary Thyroid Cancer in Lymph Nodes. J. Clin. Endocrinol. Metab. 2005. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Viola, D.; Torregrossa, L.; Giannini, R.; Romei, C.; Ugolini, C.; Molinaro, E.; Agate, L.; Biagini, A.; Lupi, C.; et al. The BRAFV600E Mutation Is an Independent, Poor Prognostic Factor for the Outcome of Patients with Low-Risk Intrathyroid Papillary Thyroid Carcinoma: Single-Institution Results from a Large Cohort Study. J. Clin. Endocrinol. Metab. 2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, S.; Shen, X.; Zhu, G.; Liu, R.; Viola, D.; Elisei, R.; Puxeddu, E.; Fugazzola, L.; Colombo, C.; et al. BRAF V600E Confers Male Sex Disease-Specific Mortality Risk in Patients with Papillary Thyroid Cancer. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Zhu, Z.; Gandhi, M.; Nikiforova, M.N.; Fischer, A.H.; Nikiforov, Y.E. Molecular Profile and Clinical-Pathologic Features of the Follicular Variant of Papillary Thyroid Carcinoma: An Unusually High Prevalence of Ras Mutations. Am. J. Clin. Pathol. 2003. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. Ras Mutations Are Associated with Aggressive Tumor Phenotypes and Poor Prognosis in Thyroid Cancer. J. Clin. Oncol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Seethala, R.R.; Tallini, G.; Baloch, Z.W.; Basolo, F.; Thompson, L.D.R.; Barletta, J.A.; Wenig, B.M.; Ghuzlan, A.A.; Kakudo, K.; et al. Nomenclature Revision for Encapsulated Follicular Variant of Papillary Thyroid Carcinoma a Paradigm Shift to Reduce Overtreatment of Indolent Tumors. JAMA Oncol. 2016. [Google Scholar] [CrossRef]
- Moon, S.; Song, Y.S.; Kim, Y.A.; Lim, J.A.; Cho, S.W.; Moon, J.H.; Hahn, S.; Park, D.J.; Park, Y.J. Effects of Coexistent BRAFV600E and TERT Promoter Mutations on Poor Clinical Outcomes in Papillary Thyroid Cancer: A Meta-Analysis. Thyroid 2017. [Google Scholar] [CrossRef]
- Melo, M.; Da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, C.; Eloy, C.; et al. TERT Promoter Mutations Are a Major Indicator of Poor Outcome in Differentiated Thyroid Carcinomas. J. Clin. Endocrinol. Metab. 2014. [Google Scholar] [CrossRef]
- Jung, S.H.; Kim, M.S.; Jung, C.K.; Park, H.C.; Kim, S.Y.; Liu, J.; Bae, J.S.; Lee, S.H.; Kim, T.M.; Lee, S.H.; et al. Mutational Burdens and Evolutionary Ages of Thyroid Follicular Adenoma Are Comparable to Those of Follicular Carcinoma. Oncotarget 2016. [Google Scholar] [CrossRef]
- Parameswaran, R.; Brooks, S.; Sadler, G.P. Molecular Pathogenesis of Follicular Cell Derived Thyroid Cancers. Int. J. Surg. 2010. [Google Scholar] [CrossRef]
- Marques, A.R.; Espadinha, C.; Catarino, A.L.; Moniz, S.; Pereira, T.; Sobrinho, L.G.; Leite, V. Expression of PAX8-PPARγ1 Rearrangements in Both Follicular Thyroid Carcinomas and Adenomas. J. Clin. Endocrinol. Metab. 2002. [Google Scholar] [CrossRef]
- Yoo, S.K.; Lee, S.; Kim, S.J.; Jee, H.G.; Kim, B.A.; Cho, H.; Song, Y.S.; Cho, S.W.; Won, J.K.; Shin, J.Y.; et al. Comprehensive Analysis of the Transcriptional and Mutational Landscape of Follicular and Papillary Thyroid Cancers. PLoS Genet. 2016, 12, e1006239. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Teresi, R.E.; Broelsch, C.E.; Frilling, A.; Eng, C. A Limited Set of Human MicroRNA Is Deregulated in Follicular Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2006. [Google Scholar] [CrossRef] [PubMed]
- McHenry, C.R.; Phitayakorn, R. Follicular Adenoma and Carcinoma of the Thyroid Gland. Oncologist 2011. [Google Scholar] [CrossRef] [PubMed]
- Máximo, V.; Soares, P.; Lima, J.; Cameselle-Teijeiro, J.; Sobrinho-Simões, M. Mitochondrial DNA Somatic Mutations (Point Mutations and Large Deletions) and Mitochondrial DNA Variants in Human Thyroid Pathology: A Study with Emphasis on Hürthle Cell Tumors. Am. J. Pathol. 2002. [Google Scholar] [CrossRef]
- Roque, L.; Serpa, A.; Clode, A.; Castedo, S.; Soares, J. Significance of Trisomy 7 and 12 in Thyroid Lesions with Follicular Differentiation: A Cytogenetic and in Situ Hybridization Study. Lab. Investig. 1999, 79, 369–378. [Google Scholar]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and Transcriptomic Hallmarks of Poorly Differentiated and Anaplastic Thyroid Cancers. J. Clin. Investig. 2016. [Google Scholar] [CrossRef]
- Agrawal, N.; Jiao, Y.; Sausen, M.; Leary, R.; Bettegowda, C.; Roberts, N.J.; Bhan, S.; Ho, A.S.; Khan, Z.; Bishop, J.; et al. Exomic Sequencing of Medullary Thyroid Cancer Reveals Dominant and Mutually Exclusive Oncogenic Mutations in RET and RAS. J. Clin. Endocrinol. Metab. 2013. [Google Scholar] [CrossRef]
- Arighi, E.; Borrello, M.G.; Sariola, H. RET Tyrosine Kinase Signaling in Development and Cancer. Cytokine Growth Factor Rev. 2005. [Google Scholar] [CrossRef]
- Romei, C.; Ciampi, R.; Elisei, R. A Comprehensive Overview of the Role of the RET Proto-Oncogene in Thyroid Carcinoma. Nat. Rev. Endocrinol. 2016. [Google Scholar] [CrossRef]
- Eng, C.; Clayton, D.; Schuffenecker, I.; Lenoir, G.; Cote, G.; Gagel, R.F.; van Amstel, H.K.; Lips, C.J.; Nishisho, I.; Takai, S.I.; et al. The Relationship between Specific Ret Proto-Oncogene Mutations and Disease Phenotype in Multiple Endocrine Neoplasia Type 2: International RET Mutation Consortium Analysis. J. Am. Med. Assoc. 1996. [Google Scholar] [CrossRef]
- Romei, C.; Elisei, R.; Pinchera, A.; Ceccherini, I.; Molinaro, E.; Mancusi, F.; Martino, E.; Romeo, G.; Pacini, F. Somatic Mutations of the Ret Protooncogene in Sporadic Medullary Thyroid Carcinoma Are Not Restricted to Exon 16 and Are Associated with Tumor Recurrence. J. Clin. Endocrinol. Metab. 1996. [Google Scholar] [CrossRef]
- Elisei, R.; Cosci, B.; Romei, C.; Bottici, V.; Renzini, G.; Molinaro, E.; Agate, L.; Vivaldi, A.; Faviana, P.; Basolo, F.; et al. Prognostic Significance of Somatic RET Oncogene Mutations in Sporadic Medullary Thyroid Cancer: A 10-Year Follow-up Study. J. Clin. Endocrinol. Metab. 2008. [Google Scholar] [CrossRef] [PubMed]
- Mian, C.; Pennelli, G.; Barollo, S.; Cavedon, E.; Nacamulli, D.; Vianello, F.; Negro, I.; Pozza, G.; Boschin, I.M.; Pelizzo, M.R.; et al. Combined RET and Ki-67 Assessment in Sporadic Medullary Thyroid Carcinoma: A Useful Tool for Patient Risk Stratification. Eur. J. Endocrinol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, S.; Vaclavikova, E.; Sykorova, V.; Vcelak, J.; Novak, Z.; Duskova, J.; Ryska, A.; Laco, J.; Cap, J.; Kodetova, D.; et al. Somatic Mutations in the RET Proto-Oncogene in Sporadic Medullary Thyroid Carcinomas. Mol. Cell. Endocrinol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Moura, M.M.; Cavaco, B.M.; Pinto, A.E.; Domingues, R.; Santos, J.R.; Cid, M.O.; Bugalho, M.J.; Leite, V. Correlation of RET Somatic Mutations with Clinicopathological Features in Sporadic Medullary Thyroid Carcinomas. Br. J. Cancer 2009. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Yang, D.; Velcheti, V.; Drilon, A.; Meric-Bernstam, F. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Cote, G.J. Advances in Targeting RET-Dependent Cancers. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Ciampi, R.; Romei, C.; Ramone, T.; Prete, A.; Tacito, A.; Cappagli, V.; Bottici, V.; Viola, D.; Torregrossa, L.; Ugolini, C.; et al. Genetic Landscape of Somatic Mutations in a Large Cohort of Sporadic Medullary Thyroid Carcinomas Studied by Next-Generation Targeted Sequencing. iScience 2019. [Google Scholar] [CrossRef] [PubMed]
- Cote, G.J.; Evers, C.; Hu, M.I.; Grubbs, E.G.; Williams, M.D.; Hai, T.; Duose, D.Y.; Houston, M.R.; Bui, J.H.; Mehrotra, M.; et al. Prognostic Significance of Circulating RET M918T Mutated Tumor DNA in Patients with Advanced Medullary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2017. [Google Scholar] [CrossRef]
- Moura, M.M.; Cavaco, B.M.; Pinto, A.E.; Leite, V. High Prevalence of RAS Mutations in RET-Negative Sporadic Medullary Thyroid Carcinomas. J. Clin. Endocrinol. Metab. 2011. [Google Scholar] [CrossRef]
- Moura, M.M.; Cavaco, B.M.; Leite, V. RAS Proto-Oncogene in Medullary Thyroid Carcinoma. Endocrine-Relat. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, E.G.; Williams, M.D.; Scheet, P.; Vattathil, S.; Perrier, N.D.; Lee, J.E.; Gagel, R.F.; Hai, T.; Feng, L.; Cabanillas, M.E.; et al. Role of CDKN2C Copy Number in Sporadic Medullary Thyroid Carcinoma. Thyroid 2016. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.E.; Gule-monroe, M.K.; Subbiah, V.; Hu, M.; Perrier, N.D.; Cabanillas, M.E.; Lee, J.E.; Graham, P.H.; Cote, G.J.; Busaidy, N.L.; et al. Novel Use of a CLIA-Certified CDKN2C Loss Assay in Sporadic Medullary Thyroid Carcinoma. Surgery 2019. [Google Scholar] [CrossRef]
- Barbieri, R.B.; Bufalo, N.E.; Secolin, R.; Assumpção, L.V.M.; Maciel, R.M.B.; Cerutti, J.M.; Ward, L.S. Polymorphisms of Cell Cycle Control Genes Influence the Development of Sporadic Medullary Thyroid Carcinoma. Eur. J. Endocrinol. 2014. [Google Scholar] [CrossRef] [PubMed]
- El Naofal, M.; Kim, A.; Yon, H.Y.; Baity, M.; Ming, Z.; Bui-Griffith, J.; Tang, Z.; Robinson, M.; Grubbs, E.G.; Cote, G.J.; et al. Role of CDKN2C Fluorescence in Situ Hybridization in the Management of Medullary Thyroid Carcinoma. Ann. Clin. Lab. Sci. 2017, 47, 523–528. [Google Scholar] [PubMed]
- Lyra, J.; Vinagre, J.; Batista, R.; Pinto, V.; Prazeres, H.; Rodrigues, F.; Eloy, C.; Sobrinho-Simõesm, M.; Soares, P. MTOR Activation in Medullary Thyroid Carcinoma with RAS Mutation. Eur. J. Endocrinol. 2014. [Google Scholar] [CrossRef]
- Park, M.S.; Rosai, J.; Nguyen, H.T.; Capodieci, P.; Cordon-Cardo, C.; Koff, A. P27 and Rb Are on Overlapping Pathways Suppressing Tumorigenesis in Mice. Proc. Natl. Acad. Sci. USA 1999. [Google Scholar] [CrossRef]
- Van Veelen, W.; Klompmaker, R.; Gloerich, M.; Van Gasteren, C.J.R.; Kalkhoven, E.; Berger, R.; Lips, C.J.; Medema, R.H.; Höppener, J.W.; Acton, D.S. P18 Is a Tumor Suppressor Gene Involved in Human Medullary Thyroid Carcinoma and Pheochromocytoma Development. Int. J. Cancer 2009. [Google Scholar] [CrossRef]
- Valenciaga, A.; Grubbs, E.G.; Porter, K.; Wakely, P.E.; Williams, M.D.; Cote, G.J.; Vasko, V.V.; Saji, M.; Ringel, M.D. Reduced Retinoblastoma Protein Expression Is Associated with Decreased Patient Survival in Medullary Thyroid Cancer. Thyroid 2017. [Google Scholar] [CrossRef]
- Valenciaga, A.; Saji, M.; Yu, L.; Zhang, X.; Bumrah, C.; Yilmaz, A.S.; Knippler, C.M.; Miles, W.; Giordano, T.J.; Cote, G.J.; et al. Transcriptional Targeting of Oncogene Addiction in Medullary Thyroid Cancer. JCI Insight 2018. [Google Scholar] [CrossRef]
- Capp, C.; Wajner, S.M.; Siqueira, D.R.; Brasil, B.A.; Meurer, L.; Maia, A.L. Increased Expression of Vascular Endothelial Growth Factor and Its Receptors, VEGFR-1 and VEGFR-2, in Medullary Thyroid Carcinoma. Thyroid 2010. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Antona, C.; Pallares, J.; Montero-Conde, C.; Inglada-Pérez, L.; Castelblanco, E.; Landa, I.; Leandro-García, L.J.; López-Jiménez, E.; Letón, R.; Cascón, A.; et al. Overexpression and Activation of EGFR and VEGFR2 in Medullary Thyroid Carcinomas Is Related to Metastasis. Endocr. Relat. Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Papotti, M.; Olivero, M.; Volante, M.; Negro, F.; Prat, M.; Comoglio, P.M.; DiRenzo, M.F. Expression of Hepatocyte Growth Factor (HGF) and Its Receptor (MET) in Medullary Carcinoma of the Thyroid. Endocr. Pathol. 2000. [Google Scholar] [CrossRef]
- Ezzat, S.; Huang, P.; Dackiw, A.; Asa, S.L. Dual Inhibition of RET and FGFR4 Restrains Medullary Thyroid Cancer Cell Growth. Clin. Cancer Res. 2005, 11, 1336–1341. [Google Scholar]
- Wei, S.; LiVolsi, V.A.; Montone, K.T.; Morrissette, J.J.D.; Baloch, Z.W. Detection of Molecular Alterations in Medullary Thyroid Carcinoma Using Next-Generation Sequencing: An Institutional Experience. Endocr. Pathol. 2016. [Google Scholar] [CrossRef]
- Chang, Y.S.; Chang, C.C.; Huang, H.Y.; Lin, C.Y.; Yeh, K.T.; Chang, J.G. Detection of Molecular Alterations in Taiwanese Patients with Medullary Thyroid Cancer Using Whole-Exome Sequencing. Endocr. Pathol. 2018. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Yoo, S.K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; Kim, Y.A.; Kim, S.J.; Cho, S.W.; Won, J.K.; et al. Integrative Analysis of Genomic and Transcriptomic Characteristics Associated with Progression of Aggressive Thyroid Cancer. Nat. Commun. 2019. [Google Scholar] [CrossRef]
- Fekete, C.A.; Mitchell, S.F.; Cherkasova, V.A.; Applefield, D.; Algire, M.A.; Maag, D.; Saini, A.K.; Lorsch, J.R.; Hinnebusch, A.G. N- and C-Terminal Residues of EIF1A Have Opposing Effects on the Fidelity of Start Codon Selection. EMBO J. 2007. [Google Scholar] [CrossRef]
- Bell, R.J.A.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. The Transcription Factor GABP Selectively Binds and Activates the Mutant TERT Promoter in Cancer. Science 2015. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Nocera, M.; Baudin, E.; Pellegriti, G.; Cailleux, A.F.; Mechelany-Corone, C.; Schlumberger, M. Treatment of Advanced Medullary Thyroid Cancer with an Alternating Combination of Doxorubicin-Streptozocin and 5 FU-Dacarbazine. Br. J. Cancer 2000. [Google Scholar] [CrossRef] [PubMed]
- Filetti, S.; Durante, C.; Hartl, D.; Leboulleux, S.; Locati, L.D.; Newbold, K.; Papotti, M.G.; Berruti, A. Thyroid Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019. [Google Scholar] [CrossRef]
- Riesco-Eizaguirre, G.; Galofré, J.C.; Grande, E.; Zafón Llopis, C.; Ramón y Cajal Asensio, T.; Navarro González, E.; Jiménez-Fonseca, P.; Santamaría Sandi, J.; Gómez Sáez, J.M.; Capdevila, J. Spanish Consensus for the Management of Patients with Advanced Radioactive Iodine Refractory Differentiated Thyroid Cancer. Endocrinol. Nutr. 2016. [Google Scholar] [CrossRef]
- Medina, E.G.J.; Viúdez, J.C.S.A.; Porras, E.G.I.; Ramón, T.; Trigo, C.J. SEOM Clinical Guideline Thyroid Cancer (2019). Clin. Transl. Oncol. 2020, 22, 223–235. [Google Scholar] [CrossRef]
- Sabra, M.M.; Sherman, E.J.; Tuttle, R.M. Tumor Volume Doubling Time of Pulmonary Metastases Predicts Overall Survival and Can Guide the Initiation of Multikinase Inhibitor Therapy in Patients with Metastatic, Follicular Cell-Derived Thyroid Carcinoma. Cancer 2017. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in Radioactive Iodine-Refractory, Locally Advanced or Metastatic Diff Erentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 3 Trial. Lancet 2014. [Google Scholar] [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer. N. Engl. J. Med. 2015. [Google Scholar] [CrossRef]
- Lenvatinib and Sorafenib for Treating Differentiated Thyroid Cancer after Radioactive Iodine. Health Technol. Assess. 2020, 24, 1.
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.W.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in Patients with BRAFV600E-Positive Metastatic or Unresectable Papillary Thyroid Cancer Refractory to Radioactive Iodine: A Non-Randomised, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2016. [Google Scholar] [CrossRef]
- Locati, L.D.; Licitra, L.; Agate, L.; Ou, S.H.I.; Boucher, A.; Jarzab, B.; Qin, S.; Kane, M.A.; Wirth, L.J.; Chen, C.; et al. Treatment of Advanced Thyroid Cancer with Axitinib: Phase 2 Study with Pharmacokinetic/Pharmacodynamic and Quality-of-Life Assessments. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.W.; Tortorici, M.; Kim, S.; Ingrosso, A.; Pithavala, Y.K.; Bycott, P. A Phase Ii Trial of Axitinib in Patients with Various Histologic Subtypes of Advanced Thyroid Cancer: Long-Term Outcomes and Pharmacokinetic/Pharmacodynamic Analyses. Cancer Chemother. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Bass, M.B.; Sherman, S.I.; Schlumberger, M.J.; Davis, M.T.; Kivman, L.; Khoo, H.M.; Notari, K.H.; Peachm, M.; Hei, Y.J.; Patterson, S.D. Biomarkers as Predictors of Response to Treatment with Motesanib in Patients with Progressive Advanced Thyroid Cancer. J. Clin. Endocrinol. Metab. 2010. [Google Scholar] [CrossRef] [PubMed]
- Carr, L.L.; Mankoff, D.A.; Goulart, B.H.; Eaton, K.D.; Capell, P.T.; Kell, E.M.; Bauman, J.E.; Martins, R.G. Phase II Study of Daily Sunitinib in FDG-PET-Positive, Iodine-Refractory Differentiated Thyroid Cancer and Metastatic Medullary Carcinoma of the Thyroid with Functional Imaging Correlation. Clin. Cancer Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Gupta-Abramson, V.; Troxel, A.B.; Nellore, A.; Puttaswamy, K.; Redlinger, M.; Ransone, K.; Mandel, S.J.; Flaherty, K.T.; Loevner, L.A.; O’Dwyer, P.J.; et al. Phase II Trial of Sorafenib in Advanced Thyroid Cancer. J. Clin. Oncol. 2008. [Google Scholar] [CrossRef]
- Hayes, D.N.; Lucas, A.S.; Tanvetyanon, T.; Krzyzanowska, M.K.; Chung, C.H.; Murphy, B.A.; Gilbert, J.; Mehra, R.; Moore, D.T.; Sheikh, A.; et al. Phase II Efficacy and Pharmacogenomic Study of Selumetinib (AZD6244; ARRY-142886) in Iodine-131 Refractory Papillary Thyroid Carcinoma with or without Follicular Elements. Clin. Cancer Res. 2012. [Google Scholar] [CrossRef]
- Lim, S.M.; Chang, H.; Yoon, M.J.; Hong, Y.K.; Kim, H.; Chung, W.Y.; Park, C.S.; Nam, K.H.; Kang, S.W.; Kim, M.K.; et al. A Multicenter, Phase II Trial of Everolimus in Locally Advanced or Metastatic Thyroid Cancer of All Histologic Subtypes. Ann. Oncol. 2013. [Google Scholar] [CrossRef]
- Sherman, E.J.; Dunn, L.A.; Ho, A.L.; Baxi, S.S.; Ghossein, R.A.; Fury, M.G.; Haque, S.; Sima, C.S.; Cullen, G.; Fagin, J.A.; et al. Phase 2 Study Evaluating the Combination of Sorafenib and Temsirolimus in the Treatment of Radioactive Iodine-Refractory Thyroid Cancer. Cancer 2017. [Google Scholar] [CrossRef]
- Droz, J.P.; Rougier, P.; Goddefroy, V. Chemotherapy for Medullary Thyroid Carcinoma (Phase I Trial of Monochemotherapy with Adriamycin and Cis-Platinum). Bull. Cancer 1984, 71, 195–199. [Google Scholar]
- Shimaoka, K.; Schoenfeld, D.A.; Dewys, W.D.; Creech, R.H.; Deconti, R. A Randomized Trial of Doxorubicin versus Doxorubicin plus Cisplatin in Patients with Advanced Thyroid Carcinoma. Cancer 1985. [Google Scholar] [CrossRef]
- Wells, S.A.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, E.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Schoffski, P.; Elisei, R.; Müller, S.; Brose, M.S.; Shah, M.H.; Licitra, L.F.; Jarzab, B.; Medvedev, V.; Kreissi, M.; Niederle, B.; et al. An International, Double-Blind, Randomized, Placebo-Controlled Phase III Trial (EXAM) of Cabozantinib (XL184) in Medullary Thyroid Carcinoma (MTC) Patients (Pts) with Documented RECIST Progression at Baseline. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef]
- Ravaud, A.; de la Fouchardière, C.; Caron, P.; Doussau, A.; Do Cao, C.; Asselineau, J.; Rodien, P.; Pouessel, D.; Nicolli-Sire, P.; Klein, M.; et al. A Multicenter Phase II Study of Sunitinib in Patients with Locally Advanced or Metastatic Differentiated, Anaplastic or Medullary Thyroid Carcinomas: Mature Data from the THYSU Study. Eur. J. Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Jarzab, B.; Cabanillas, M.E.; Robinson, B.; Pacini, F.; Ball, D.W.; McCaffrey, J.; Newbold, K.; Allison, R.; Martins, R.G.; et al. A Phase II Trial of the Multitargeted Tyrosine Kinase Inhibitor Lenvatinib (E7080) in Advanced Medullary Thyroid Cancer. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef]
- Thomas, L.; Lai, S.Y.; Dong, W.; Feng, L.; Dadu, R.; Regone, R.M.; Cabanillas, M.E. Sorafenib in Metastatic Thyroid Cancer: A Systematic Review. Oncologist 2014. [Google Scholar] [CrossRef]
- Bible, K.C.; Suman, V.J.; Molina, J.R.; Smallridge, R.C.; Maples, W.J.; Menefee, M.E.; Rubin, J.; Karlin, N.; Sideras, K.; Morris, J.C.; et al. A Multicenter Phase 2 Trial of Pazopanib in Metastatic and Progressive Medullary Thyroid Carcinoma: MC057H. J. Clin. Endocrinol. Metab. 2014. [Google Scholar] [CrossRef] [PubMed]
- Appels, N.M.G.M.; Beijnen, J.H.; Schellens, J.H.M. Development of Farnesyl Transferase Inhibitors: A Review. Oncologist 2005. [Google Scholar] [CrossRef]
- Ho, A.L.; Chau, N.G.; Wong, D.J.L.; Cabanillas, M.E.; Bauman, J.R.; Bible, K.C.; Brose, M.S.; Calvo, E.; Boni, V.; Burrows, F.; et al. An Open-Label, Phase II Study of Tipifarnib for the Treatment of HRAS Mutant Solid Tumors, Including Squamous Cell Carcinomas of the Head and Neck. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients with Locally Advanced or Metastatic BRAF V600–Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Smallridge, R.C.; Ain, K.B.; Asa, S.L.; Bible, K.C.; Brierley, J.D.; Burman, K.D.; Kebekew, E.; Lee, N.Y.; Nikiforov, Y.E.; Rosenthal, M.S.; et al. American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer. Thyroid 2012. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.K.; Zing, M. Highly Prevalent Genetic Alterations in Receptor Tyrosine Kinases and Phosphatidylinositol 3-Kinase/Akt and Mitogen-Activated Protein Kinase Pathways in Anaplastic and Follicular Thyroid Cancers. J. Clin. Endocrinol. Metab. 2008. [Google Scholar] [CrossRef] [PubMed]
- Dias-Santagata, D.; Lennerz, J.K.; Sadow, P.M.; Frazier, R.P.; Raju, S.G.; Henry, D.; Chung, T.; Kherani, J.; Rothenberg, S.M.; Wirth, L. Response to RET-Specific Therapy in RET Fusion-Positive Anaplastic Thyroid Carcinoma. Thyroid 2020. [Google Scholar] [CrossRef] [PubMed]
- Tahara, M.; Kiyota, N.; Yamazaki, T.; Chayahara, N.; Nakano, K.; Inagaki, L.; Toda, K.; Enokida, T.; Minami, H.; Imamura, Y.; et al. Lenvatinib for Anaplastic Thyroid Cancer. Front. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.M.; Bocci, G.; Di Desidero, T.; Ruffilli, I.; Elia, G.; Ragusa, F.; Fioravanti, A.; Orlandi, P.; Paparo, S.R.; Patrizio, A.; et al. Vandetanib Has Antineoplastic Activity in Anaplastic Thyroid Cancer, in Vitro and in Vivo. Oncol. Rep. 2018. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Centanni, M.; Virili, C.; Miccoli, M.; Ferrari, P.; Ruffilli, I.; Ragusa, F.; Antonelli, A.; Falahi, P. Sunitinib in the Treatment of Thyroid Cancer. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Antonelli, A.; Bocci, G.; La Motta, C.; Ferrari, S.M.; Fallahi, P.; Ruffilli, I.; Di Domenicantonio, A.; Fioravanti, A.; Sartini, S.; Minuto, M.; et al. CLM94, a Novel Cyclic Amide with Anti-VEGFR-2 and Antiangiogenic Properties, Is Active against Primary Anaplastic Thyroid Cancer in Vitro and in Vivo. J. Clin. Endocrinol. Metab. 2012. [Google Scholar] [CrossRef]
- Marten, K.A.; Gudena, V.K. Use of Vemurafenib in Anaplastic Thyroid Carcinoma: A Case Report. Cancer Biol. Ther. 2015. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamong, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Dadu, R.; Iyer, P.; Wanland, K.B.; Busaidy, N.L.; Ying, A.; Gule-Monroe, M.; Wang, J.R.; Zafereo, M.; Hofmann, M.C. Acquired Secondary RAS Mutation in BRAF V600E -Mutated Thyroid Cancer Patients Treated with BRAF Inhibitors. Thyroid 2020. [Google Scholar] [CrossRef]
- Wirth, L.J.; Kohno, T.; Udagawa, H.; Matsumoto, S.; Ishii, G.; Ebata, K.; Tuch, B.B.; Zhu, E.Y.; Nguyen, M.; Smith, S.; et al. Emergence and Targeting of Acquired and Hereditary Resistance to Multikinase RET Inhibition in Patients With RET-Altered Cancer. JCO Precis. Oncol. 2019. [Google Scholar] [CrossRef]
- Taylor, M.H.; Gainor, J.F.; Hu, M.I.-N.; Zhu, V.W.; Lopes, G.; Leboulleux, S.; Brose, M.S.; Schuler, M.H.; Bowles, D.W.; Kim, D.W.; et al. Activity and Tolerability of BLU-667, a Highly Potent and Selective RET Inhibitor, in Patients with Advanced RET-Altered Thyroid Cancers. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Shah, M.H.; Sherman, E.J.; Robinson, B.; Solomon, B.J.; Kang, H.; Lorch, J.H.; Worden, F.P.; Brose, M.S.; Leboulleux, S.; Godbert, Y.; et al. Selpercatinib (LOXO-292) in Patients with RET-Mutant Medullary Thyroid Cancer. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3594. [Google Scholar] [CrossRef]
- Drilon, A.E.; Subbiah, V.; Oxnard, G.R.; Bauer, T.M.; Velcheti, V.; Lakhani, N.J.; Besse, B.; Park, K.; Patel, J.D.; Cabanillas, M.E.; et al. A Phase 1 Study of LOXO-292, a Potent and Highly Selective RET Inhibitor, in Patients with RET -Altered Cancers. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Wirth, L.; Sherman, E.; Drilon, A.; Solomon, B.; Robinson, B.; Lorch, J.; McCoach, C.; Patel, J.D.; Leboulleux, S.; Worden, F.; et al. Registrational Results of LOXO-292 in Patients with RET-Altered Thyroid Cancers. Ann. Oncol. 2019. [Google Scholar] [CrossRef]
- Blueprint Medicines Announces the Achievement of Key Portfolio Milestones [News Release]; Blueprint Medicine Coorporation: Cambridge, MA, USA, 2020; Available online: https://bit.ly/2R3cW9R (accessed on 2 June 2020).
- Li, D.; Tang, P.Z.; Chen, X.; Ge, M.; Zhang, Y.; Guo, Z.; Wang, J.; Shi, F.; Zhang, J.; Cheng, Y.; et al. Anlotinib Treatment in Locally Advanced or Metastatic Medullary Thyroid Carcinoma: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Phase IIB Trial. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Chen, J.; Ji, Q.; Cao, J.; Ji, D.; Bai, C.; Lin, Y.; Pan, B.; Sun, G.; Li, J.; Qi, C.; et al. A Phase II Multicenter Trial of the Multitargeted Kinase Inhibitor Sulfatinib in Advanced Medullary Thyroid Cancer (MTC) and Radioiodine (RAI)-Refractory Differentiated Thyroid Cancer (DTC). J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimiatriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018. [Google Scholar] [CrossRef]
- Varricchi, G.; Loffredo, S.; Marone, G.; Modestino, L.; Fallahi, P.; Ferrari, S.M.; de Paulis, A.; Antonelli, A.; Galdiero, M.R. The Immune Landscape of Thyroid Cancer in the Context of Immune Checkpoint Inhibition. Int. J. Mol. Sci. 2019, 20, 3934. [Google Scholar] [CrossRef]
- Naoum, G.E.; Morkos, M.; Kim, B.; Arafat, W. Novel Targeted Therapies and Immunotherapy for Advanced Thyroid Cancers. Mol. Cancer 2018. [Google Scholar] [CrossRef]
- Angell, T.E.; Lechner, M.G.; Jang, J.K.; Correa, A.J.; LoPresti, J.S.; Epstein, A.L. BRAFV600E in Papillary Thyroid Carcinoma Is Associated with Increased Programmed Death Ligand 1 Expression and Suppressive Immune Cell Infiltration. Thyroid 2014. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Varga, A.; Brose, M.S.; Aggarwal, R.R.; Lin, C.C.; Prawira, A.; de Brad, F.; Tamura, K.; Doi, T.; Piha-Paul, S.A.; et al. Safety and Antitumor Activity of the Anti-PD-1 Antibody Pembrolizumab in Patients with Advanced, PD-L1-Positive Papillary or Follicular Thyroid Cancer. BMC Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Lorch, J.; Barletta, J.A.; Nehs, M.; Uppaluri, R.; Alexander, E.K.; Haddad, R.I.; Hanna, G.J.; Margalit, D.N.; Tishler, R.B.; Schoenfeld, J.D.; et al. A Phase II Study of Nivolumab (N) plus Ipilimumab (I) in Radioiodine Refractory Differentiated Thyroid Cancer (RAIR DTC) with Exploratory Cohorts in Anaplastic (ATC) and Medullary Thyroid Cancer (MTC). J. Clin. Oncol. 2020, 38, 6513. [Google Scholar] [CrossRef]
- Haugen, B.R.; French, J.D.; Worden, F.; Konda, B.; Sherman, E.J.; Dadu, R.; Gianoukakis, A.G.; Wolfe, E.G.; Foster, N.R.; Bowles, D.W.; et al. Lenvatinib plus Pembrolizumab Combination Therapy in Patients with Radioiodine-Refractory (RAIR), Progressive Differentiated Thyroid Cancer (DTC): Results of a Multicenter Phase II International Thyroid Oncology Group Trial. J. Clin. Oncol. 2020, 38, 6512. [Google Scholar] [CrossRef]
- Bongiovanni, M.; Rebecchini, C.; Saglietti, C.; Bulliard, J.L.; Marino, L.; De Leval, L.; Sykiotis, G. Very Low Expression of Pd-L1 in Medullary Thyroid Carcinoma. Endocr. Relat. Cancer 2017. [Google Scholar] [CrossRef]
- Bi, Y.; Ren, X.; Bai, X.; Meng, Y.; Luo, Y.; Cao, J.; Zhang, Y.; Liang, Z. PD-1/PD-L1 Expressions in Medullary Thyroid Carcinoma: Clinicopathologic and Prognostic Analysis of Chinese Population. Eur. J. Surg. Oncol. 2019. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Erickson, T.; Zhang, L.; Ellison, K.; Rivard, C.; Sams, S.; Hirsch, F.R.; Haugen, B.R.; French, J.D. Comprehensive Immune Profiling of Medullary Thyroid Cancer. Thyroid 2020. [Google Scholar] [CrossRef]
- Shi, X.; Yu, P.C.; Lei, B.W.; Li, C.W.; Zhang, Y.; Tan, L.C.; Shi, R.L.; Wang, J.; Ma, B.; Xu, W.B.; et al. Association between Programmed Death-Ligand 1 Expression and Clinicopathological Characteristics, Structural Recurrence, and Biochemical Recurrence/Persistent Disease in Medullary Thyroid Carcinoma. Thyroid 2019. [Google Scholar] [CrossRef]
- Ingenwerth, M.; Goetz, M.; Schmid, K.W.; Theurer, S. The Mismatch Repair System Is Not Affected in Medullary Thyroid Carcinoma Independent of Stromal Desmoplasia or Ret Proto-Oncogene Mutation. Ann. Diagn. Pathol. 2020. [Google Scholar] [CrossRef]
- Caillou, B.; Talbot, M.; Weyemi, U.; Pioche-Durieu, C.; Ghuzlan, A.; Bidart, J.M.; Chouaib, S.; Schlumberger, M.; Dupuy, C. Tumor-Associated Macrophages (TAMs) Form an Interconnected Cellular Supportive Network in Anaplastic Thyroid Carcinoma. PLoS ONE 2011, 6, e22567. [Google Scholar] [CrossRef]
- Ryder, M.; Ghossein, R.A.; Ricarte-Filho, J.C.M.; Knauf, J.A.; Fagin, J.A. Increased Density of Tumor-Associated Macrophages Is Associated with Decreased Survival in Advanced Thyroid Cancer. Endocr. Relat. Cancer 2008. [Google Scholar] [CrossRef]
- Garg, M.; Okamoto, R.; Nagata, Y.; Kanojia, D.; Venkatesan, S.; Anand, M.T.; Braunstein, G.D.; Said, J.W.; Doan, N.B.; Ho, Q.; et al. Establishment and Characterization of Novel Human Primary and Metastatic Anaplastic Thyroid Cancer Cell Lines and Their Genomic Evolution over a Year as a Primagraft. J. Clin. Endocrinol. Metab. 2015. [Google Scholar] [CrossRef] [PubMed]
- Salajegheh, A.; Pakneshan, S.; Rahman, A.; Dolan-Evans, E.; Zhang, S.; Kwong, E.; Gopalan, V.; Lo, C.Y.; Smith, R.A.; Yin Lam, A.K. Co-Regulatory Potential of Vascular Endothelial Growth Factor-A and Vascular Endothelial Growth Factor-C in Thyroid Carcinoma. Hum. Pathol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Amar, S. P53 Suppresses CCL2-Induced Subcutaneous Tumor Xenograft. Tumor Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Lin, B.; Wang, M.; Liang, X.; Su, L.; Okose, O.; Lv, W.; Li, J. Immunotherapy in Anaplastic Thyroid Cancer. Am. J. Transl. Res. 2020. [Google Scholar]
- Passaro, C.; Borriello, F.; Vastolo, V.; Di Somma, S.; Scamardella, E.; Gigantino, V.; Franco, R.; Marone, G.; Portella, G. The Oncolytic Virus Dl922-947 Reduces IL-8/CXCL8 and MCP- 1/CCL2 Expression and Impairs Angiogenesis and Macrophage Infiltration in Anaplastic Thyroid Carcinoma. Oncotarget 2016. [Google Scholar] [CrossRef]
- Lin, S.F.; Price, D.L.; Chen, C.H.; Brader, P.; Li, S.; Gonzalez, L.; Zhang, Q.; Yu, Y.A.; Chen, N.; Szalay, A.A.; et al. Oncolytic Vaccinia Virotherapy of Anaplastic Thyroid Cancer in Vivo. J. Clin. Endocrinol. Metab. 2008. [Google Scholar] [CrossRef]
- Min, I.M.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Wyrwas, B.; Scognamiglio, T.; Moore, M.D.; Wang, W.; Park, S.; Park, S.; et al. CAR T Therapy Targeting ICAM-1 Eliminates Advanced Human Thyroid Tumors. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef]
- Cantara, S.; Bertelli, E.; Occhini, R.; Regoli, M.; Brilli, L.; Pacini, F.; Castagna, M.G.; Toti, P. Blockade of the Programmed Death Ligand 1 (PD-L1) as Potential Therapy for Anaplastic Thyroid Cancer. Endocrine 2019. [Google Scholar] [CrossRef]
- Brauner, E.; Gunda, V.; Vanden Borre, P.; Zurakowski, D.; Kim, Y.S.; Dennett, K.V.; Amin, S.; Freeman, G.J.; Parangi, S. Combining BRAF Inhibitor and Anti PD-L1 Antibody Dramatically Improves Tumor Regression and Anti Tumor Immunity in an Immunocompetent Murine Model of Anaplastic Thyroid Cancer. Oncotarget 2016. [Google Scholar] [CrossRef]
- Capdevila, J.; Wirth, L.J.; Ernst, T.; Ponce Aix, S.; Lin, C.-C.; Ramlau, R.; Butler, M.O.; Delord, J.P.; Gelderblom, H.; Ascierto, P.A.; et al. PD-1 Blockade in Anaplastic Thyroid Carcinoma. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Dadu, R.; Ferrarotto, R.; Liu, S.; Fellman, B.M.; Gross, N.D.; Gule-Monroe, M.; Lu, C.; Grosu, H.; Williams, M.D.; et al. Atezolizumab Combinations with Targeted Therapy for Anaplastic Thyroid Carcinoma (ATC). J. Clin. Oncol. 2020, 38, 6514. [Google Scholar] [CrossRef]
- Chintakuntlawar, A.V.; Yin, J.; Foote, R.L.; Kasperbauer, J.L.; Rivera, M.; Asmus, E.; Garces, N.I.; Janus, J.R.; Liu, M.; Ma, D.J.; et al. A Phase 2 Study of Pembrolizumab Combined with Chemoradiotherapy as Initial Treatment for Anaplastic Thyroid Cancer. Thyroid 2019. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Dubois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; Van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in Patients with TRK Fusion-Positive Solid Tumours: A Pooled Analysis of Three Phase 1/2 Clinical Trials. Lancet Oncol. 2020, 41, 1–10. [Google Scholar] [CrossRef]
- Cabanillas, O.M.E. Targeted Therapy for Advanced Thyroid Cancer: Kinase Inhibitors and Beyond. Endocr. Rev. 2019, 40, 1573–1604. [Google Scholar] [CrossRef] [PubMed]
- Lassen, U.N.; Albert, C.M.; Kummar, S.; Van Tilburg, C.M.; DuBois, S.G.; Geoerger, B.; Mascarenhas, L.; Federman, N.; Federman, N.; Basu-Mallick, F.; et al. Larotrectinib Efficacy and Safety in TRK Fusion Cancer: An Expanded Clinical Dataset Showing Consistency in an Age and Tumor Agnostic Approach. Ann Oncol. 2018, 29 (Suppl. 8). [Google Scholar] [CrossRef]
- Drilon, A.E.; Farago, A.F.; Tan, D.S.-W.; Kummar, S.; McDermott, R.S.; Berlin, J.; Patel, J.D.; Brose, M.S.; Leyvraz, S.; Tahara, M.; et al. Activity and Safety of Larotrectinib in Adult Patients with TRK Fusion Cancer: An Expanded Data Set. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3610. [Google Scholar] [CrossRef]
- Demetri, G.D.; Paz-Ares, L.; Farago, A.F.; Liu, S.V.; Chawla, S.P.; Tosi, D.; Kim, E.S.; Blakely, C.; Krauss, J.C.; Sigal, D.; et al. Efficacy and Safety of Entrectinib in Patients with NTRK Fusion-Positive (NTRK-Fp) Tumors: Pooled Analysis of STARTRK-2, STARTRK-1 and ALKA-372-001. In Proceedings of the Abstract B. 43rd ESMO Congress (ESMO 2018), Munich, Germany, 19–23 October 2018; Volume 29, p. viii713. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Brad, F.; Rolfo, C.; Ahn, M.J.; Wolf, J.; et al. Articles Entrectinib in ROS1 Fusion-Positive Non-Small-Cell Lung Cancer: Integrated Analysis of Three Phase 1–2 Trials. Lancet Oncol. 2019, 2045. [Google Scholar] [CrossRef]
- Demeure, M.J.; Aziz, M.; Rosenberg, R.; Gurley, S.D.; Bussey, K.J.; Carpten, J.D. Whole-Genome Sequencing of an Aggressive BRAF Wild-Type Papillary Thyroid Cancer Identified EML4-ALK Translocation as a Therapeutic Target. World J. Surg. 2014. [Google Scholar] [CrossRef]
- Chou, A.; Fraser, S.; Toon, C.W.; Clarkson, A.; Sioson, L.; Farzin, M.; Cussigh, C.; Aniss, A.; O´Neill, C.; Watson, N.; et al. A Detailed Clinicopathologic Study of ALK-Translocated Papillary Thyroid Carcinoma. Am. J. Surg. Pathol. 2015. [Google Scholar] [CrossRef]
- Ji, J.H.; Oh, Y.L.; Hong, M.; Yun, J.W.; Lee, H.W.; Kim, D.G.; Park, W.Y.; Shin, H.T.; Kim, K.M.; Ahn, M.J.; et al. Identification of Driving ALK Fusion Genes and Genomic Landscape of Medullary Thyroid Cancer. PLoS Genet. 2015, 11, e1005467. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Orlov, S.; Zhang, L.; Braiteh, F.; Huang, H.; Esaki, T.; Horibe, K.; Ahn, J.S.; Beck, J.T.; Edenfield, W.F.; et al. Long-Term Effects of Crizotinib in ALK-Positive Tumors (Excluding NSCLC): A Phase 1b Open-Label Study. Am. J. Hematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hillier, K.; Hughes, A.; Shamberger, R.C.; Shusterman, S.; Perez-Atayde, A.R.; Wassner, A.J.; Lafrate, A.J.; Dubuc, A.; Janeway, K.A.; Rothenberg, S.M.; et al. A Novel ALK Fusion in Pediatric Medullary Thyroid Carcinoma. Thyroid 2019. [Google Scholar] [CrossRef] [PubMed]
- Latteyer, S.; Tiedje, V.; König, K.; Ting, S.; Heukamp, L.C.; Meder, L.; Schmid, K.W.; Führer, D.; Moeller, L.C. Targeted Next-Generation Sequencing for TP53, RAS, BRAF, ALK and NF1 Mutations in Anaplastic Thyroid Cancer. Endocrine 2016. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Xing, M.M. Anaplastic Thyroid Cancers Harbor Novel Oncogenic Mutations of the ALK Gene. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Pérot, G.; Soubeyran, I.; Ribeiro, A.; Bonhomme, B.; Savagner, F.; Boutet-Bouzamondo, N.; Hostein, S.; Bonichon, F.; Godbert, Y.; Chibon, F. Identification of a Recurrent STRN/ALK Fusion in Thyroid Carcinomas. PLoS ONE 2014, 9, e87170. [Google Scholar] [CrossRef]
- Godbert, Y.; De Figueiredo, B.H.; Bonichon, F.; Chibon, F.; Hostein, I.; Pérot, G.; Dupin, C.; Daubech, A.; Belleannée, G.; Gros, A.; et al. Remarkable Response to Crizotinib in Woman with Anaplastic Lymphoma Kinase-Rearranged Anaplastic Thyroid Carcinoma. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef]
- Leroy, L.; Bonhomme, B.; Le Moulec, S.; Soubeyran, I.; Italiano, A.; Godbert, Y. Remarkable Response to Ceritinib and Brigatinib in an Anaplastic Lymphoma Kinase-Rearranged Anaplastic Thyroid Carcinoma Previously Treated with Crizotinib. Thyroid 2020. [Google Scholar] [CrossRef]
- Aashiq, M.; Silverman, D.A.; Na’ara, S.; Takahashi, H.; Amit, M. Radioiodine-Refractory Thyroid Cancer: Molecular Basis of Redifferentiation Therapies, Management, and Novel Therapies. Cancers 2019. [Google Scholar] [CrossRef]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-Enhanced Radioiodine Uptake in Advanced Thyroid Cancer. N. Engl. J. Med. 2013. [Google Scholar] [CrossRef]
- Brown, S.R.; Hall, A.; Buckley, H.L.; Flanagan, L.; Gonzalez De Castro, D.; Farnell, K.; Moss, L.; Gregory, R.; Newbold, K.; Du, Y.; et al. Investigating the Potential Clinical Benefit of Selumetinib in Resensitising Advanced Iodine Refractory Differentiated Thyroid Cancer to Radioiodine Therapy (SEL-I-METRY): Protocol for a Multicentre UK Single Arm Phase II Trial. BMC Cancer 2019. [Google Scholar] [CrossRef]
- Rothenberg, S.M.; McFadden, D.G.; Palmer, E.L.; Daniels, G.H.; Wirth, L.J. Redifferentiation of Iodine-Refractory BRAF V600E-Mutant Metastatic Papillary Thyroid Cancer with Dabrafenib. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef]
- Dunn, L.A.; Sherman, E.J.; Baxi, S.S.; Tchekmedyian, V.; Grewal, R.K.; Larson, S.M.; Pentlow, K.S.; Haque, S.; Tuttle, R.M.; Sabra, M.M.; et al. Vemurafenib Redifferentiation of BRAF Mutant, Rai-Refractory Thyroid Cancers. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Salavati, A.; Puranik, A. Peptide ReceptorRadionuclideTherapy (PRRT) of Medullary and Nonmedullary Thyroid Cancer Using Radiolabeled Somatostatin Analogues. Semin. Nucl. Med. 2016, 46, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Iten, F.; Müller, B.; Schindler, C.; Rochlitz, C.; Oertli, D.; Mäcke, H.R.; Müller-Brand, J.; Walter, M.A. Cancer Therapy: Clinical Response to [90 Yttrium-DOTA] -TOC T Reatment Is Associated with Long-Term Survival Benefit in Metastasized Medullary Thyroid Cancer: A Phase II Clinical Trial. Clin. Cancer Res. 2007, 13, 6696–6703. [Google Scholar] [CrossRef]
- Castellani, M.; Seregni, E.; Maccauro, M.; Chiesa, C.; Aliberti, G.; Orunesu, E.; Bombardieri, E. MIBG for Diagnosis and Therapy of Medullary Thyroid Carcinoma: Is There Still a Role? Q. J. Nucl. Med. Mol. Imaging 2008, 52, 430–440. [Google Scholar]
- Ilias, I.; Divgi, C.; Pacak, K. Current Role of MIBG in the Diagnosis of Pheochromocytoma and Medullary Thyroid Cancer. Semin. Nucl. Med. 2012, 41, 364–368. [Google Scholar] [CrossRef]
- Sherman, E.J.; Ho, A.L.; Fagin, J.A.; Haque, S.; Robinson, C.; Ghossein, R.A.; Chen, H.X.; Pfister, D.G. Combination of Dabrafenib (DAB) and Lapatinib (LAP) for the Treatment of BRAF-Mutant Thyroid Cancer. J. Clin. Oncol. 2018, 36 (Suppl. 15), 6087. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Ruiz-llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of Feedback Inhibition of HER3 Transcription by RAF and MEK Inhibitors Attenuates Their Antitumor Effects in BRAF -Mutant Thyroid Carcinomas. Cancer Discov. 2013. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Fu, S.; Hong, D.S.; Janku, F.; Karp, D.D.; Naing, A.; Pant, S.; Rodon, J.; Subbiah, V.; Tsimberidou, A.M.; et al. Phase I Study of the Pan-HER Inhibitor Neratinib given in Combination with Everolimus, Palbociclib or Trametinib in Advanced Cancer Subjects with EGFR Mutation/Amplification, HER2 Mutation/Amplification or HER3/4 Mutation. J. Clin. Oncol. 2018, 36 (Suppl. 15), TPS2611. [Google Scholar] [CrossRef]
- Schneider, T.C.; De Wit, D.; Links, T.P.; Van Erp, N.P.; Van Der Hoeven, J.J.M.; Gelderblom, H.; van Wezel, T.; van Eijk, R.; Morreau, H.; Guchelaar, H.J.; et al. Beneficial Effects of the MTOR Inhibitor Everolimus in Patients with Advanced Medullary Thyroid Carcinoma: Subgroup Results of a Phase II Trial. Int. J. Endocrinol. 2015. [Google Scholar] [CrossRef]



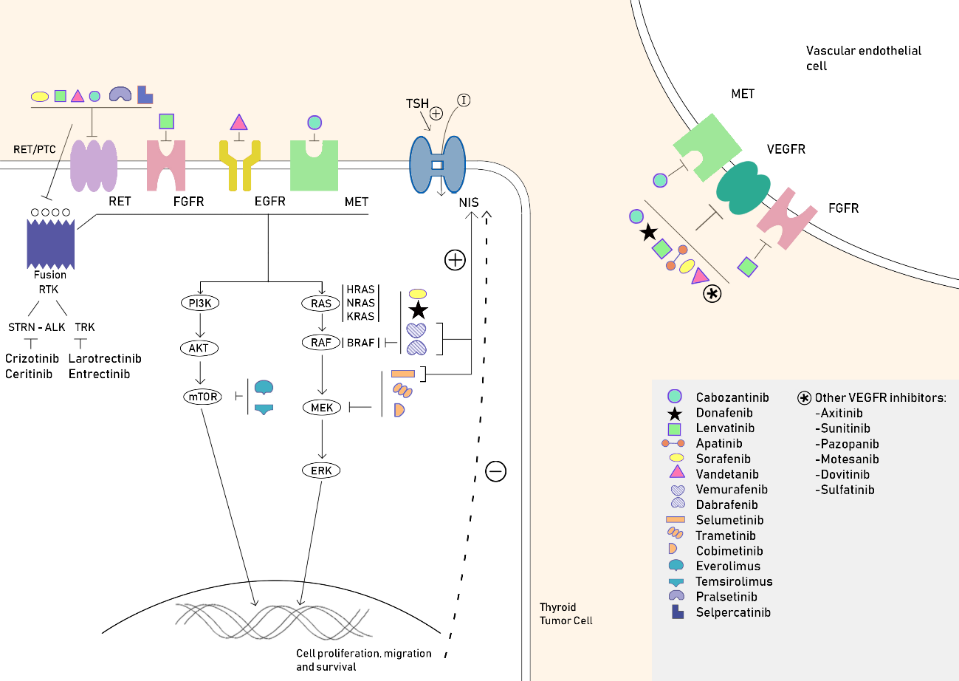{kind=link}
{kind=link}
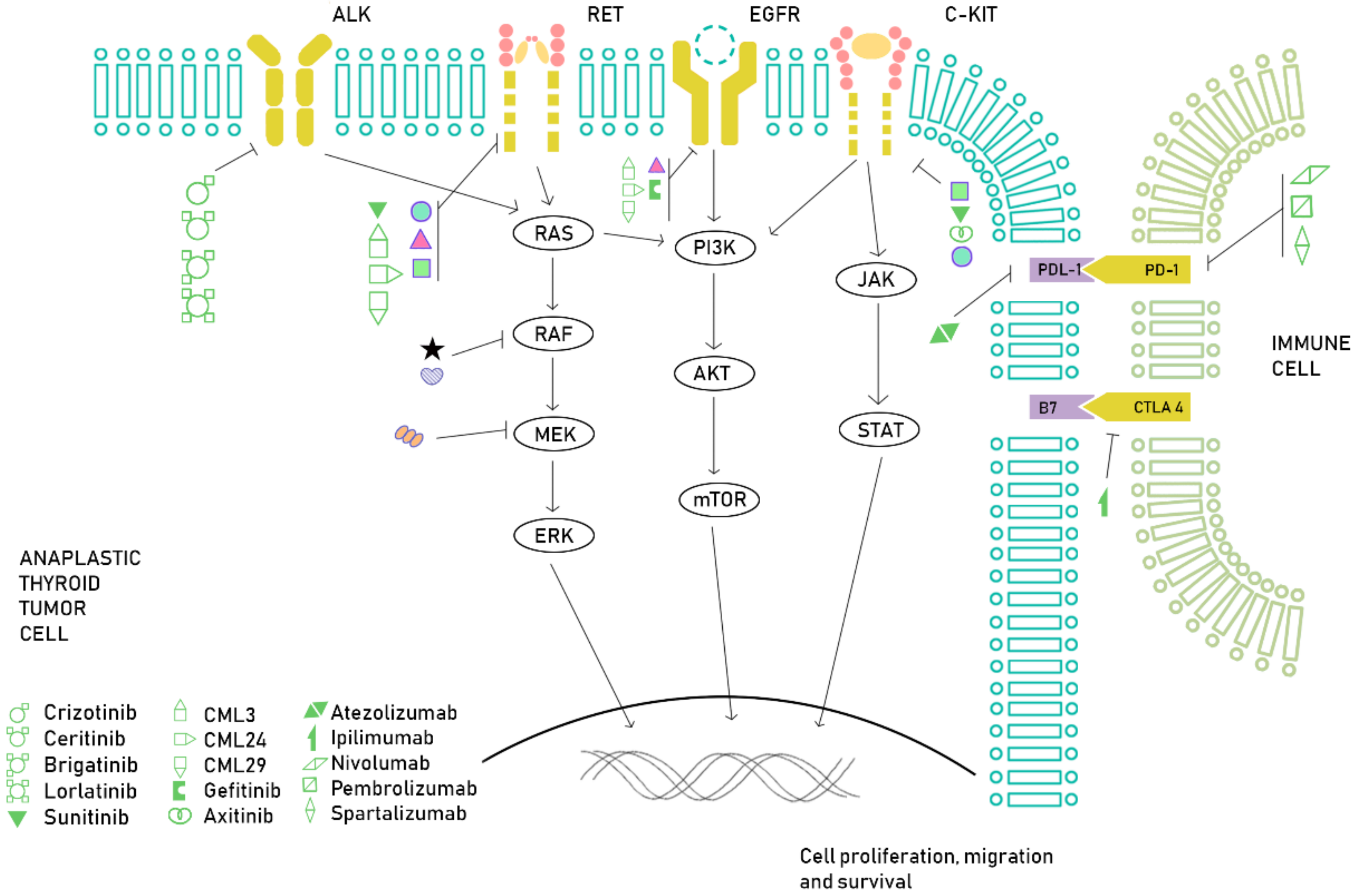{kind=link}
| Sorafenib (DECISION) * [69] | Lenvatinib (SELECT) * [70] | |
|---|---|---|
| N | 417 | 392 |
| Target | RAF, VEGFR 1–3, PDGFR, c-KIT and RET | FGFR 1–4, VEGFR 1–3, RET, c-KIT and PDGFR α |
| Treatment line | 1st line | 1st line or progressed to previous TKI (maximum one; 25%) |
| Population | RECIST Disease progression within the previous 14 months | IRR evidence of progression within the previous 13 months |
| Histology | Papillary (56.8%) Poorly differentiated (9.6%) Follicular (25.4%) | Papillary (50%) Poorly differentiated (10.7%) Follicular (20.3%) Hürthle cell (18.4%) |
| Metastatic location | Bone (27%) Only Lung (16.7%) | Bone (39.8%) Lung (86.6%) |
| Comparator arm | Placebo | Placebo |
| Crossover placebo-intervention | Allowed | Allowed |
| Randomization | 1:1 | 2:1 |
| Primary Endpoint | PFS | PFS |
| Results | 10.8 vs. 5.8 months (p < 0.0001) | 18.3 vs. 3.6 months (p < 0.001) |
| Secondary Endpoints | OS, TTP, DCR, ORR | ORR, OS |
| Results | OS: 39.4 vs. 42.8 months (52.8% vs. 54.8% deaths in 8 years) # ● Crossover correction: significance not reached | OS: 41.6 vs. 34.5 months + ● Subgroup analysis: improved OS in older population (>65 yo) ● Crossover correction: significance reached [71] |
| TTP: 337 vs. 175 days (p <0.0001) | ||
| DCR: 86.2% vs. 74.6% (p = 0.015) ORR (No CR): 12.2% vs. 0.5% (p < 0.0001) | DCR: 87.7% vs. 55.7% (p < 0.001) ORR (4 CR): 64.8% vs. 1.5% (p < 0.001) | |
| Adverse Effects G3/G4 | 37.2% (Hand-Foot syndrome) | 75.9% (hypertension) |
| Dose Reduction | 64.3% | 67% |
| Discontinuation | 19% | 14% |
| Drug | Combination | Drug Targets | Population | Phase | N | Primary Endpoint | Status | Results |
|---|---|---|---|---|---|---|---|---|
| Cabozantinib NCT03690388 | N/A | MET, VEGFR2, FLT3, c-KIT, and RET | RAI-R DTC Progressed to 1 or 2 previous AntiVEGFR | III | 300 | PFS ORR | Recruiting | Not available |
| Donafenib NCT03602495 | N/A | RAF, VEGFR, PDGFR | RAI-R DTC | III | 204 | PFS | Recruiting | Not available |
| Apatinib NCT03048877 | N/A | VEGFR2 | RAI-R DTC | III | 118 | PFS | Active, not recruiting | Not available |
| Axitinib NCT00389441 | N/A | VEGFR, PDGFR, c-kit | RAI-R DTC Unresectable Locally-Advanced Thyroid Cancer | II | 52 | ORR | Completed | 34.6% [73] |
| Axitinib NCT00094055 | N/A | VEGFR, PDGFR, c-kit | RAI-R DTC Metastatic ATC, MTC | II | 60 | ORR | Completed | 30% [74] |
| Motesanib NCT00121628 | N/A | VEGFR, PDGFR, c-kit | RAI-R DTC MTC | II | 184 | ORR | Completed | 14% [75] |
| Sulfatinib NCT02614495 | N/A | VEGFR, FGFR1 | RAI-R DTC MTC | II | 66 | ORR | Completed | Not available |
| Sunitinib NCT00519896 | N/A | PDGFR, FLT3, c-KIT, VEGFR, RET | RAI-R DTC MTC | II | 35 | ORR | Completed | 33.3% [76] |
| Pazopanib NCT01813136 | N/A | VEGFR, PDGFR, c-kit | RAI-R DTC | II | 168 | TTF | Completed | Not available |
| Dovitinib NCT01964144 | N/A | VEGFR, FGFR | RAI-R DTC MTC | II | 40 | ORR | Completed | Not available |
| Anlotinib NCT02586337 | N/A | VEGFR, FGFR, PDGFR, c-kit | RAI-R DTC | II | 113 | PFS | Active, not recruiting | Not available |
| Sorafenib NCT00654238 | N/A | VEGFR, PDGFR, BRAF | RAI-R DTC, ATC, MTC | II | 59 | ORR, SD | Completed | 86.4% in DTC; 50% in PDTC [77] |
| Selumetinib NCT00559949 | N/A | MEK 1–2 | RAI-R DTC | II | 39 | ORR | Completed | 3.1% [78] |
| Vemurafenib NCT01286753 | N/A | BRAF | BRAF V600E RAI-R DTC | II | 51 | ORR | Completed | 42.3% [72] |
| Dabrafenib NCT01947023 | Lapatinib | BRAF + EGFR/HER2 | BRAF V600E or V600K RAI-R DTC | I | 21 | MTD | Active, not recruiting | Not available |
| Dabrafenib NCT01723202 | Trametinib | BRAF +/- MEK | BRAF + RAI-R DTC | II | 53 | ORR | Active, not recruiting | Not available |
| Trametinib NCT03244956 | Dabrafenib | MEK +/- BRAF | RAS (NRAS or KRAS or HRAS) or BRAFV600E or K601E mutation RAI-R DTC | II | 87 | ORR | Recruiting | Not available |
| Everolimus NCT01164176 | N/A | mTOR | RAI-R DTC | II | 40 | DCR | Completed | 81% [79] |
| Everolimus NCT01263951 | Sorafenib | mTOR + VEGFR, PDGFR, BRAF | RAI-R DTC progressed to sorafenib | II | 35 | PFS, ORR, SD | Completed | Not available |
| Temsirolimus NCT01025453 | Sorafenib | mTOR + VEGFR, PDGFR, BRAF | RAI-R DTC | II | 37 | ORR | Completed | 26.7% [80] |
| Everolimus NCT03139747 | Lenvatinib | mTOR + FGFR, VEGFR, RET, c-KIT and PDGFR α | RAI-R DTC progressed to lenvatinib | II | 5 | PFS | Completed | Not available |
| Sorafenib NCT02143726 | Everolimus | VEGFR, PDGFR, BRAF +/− mTOR | RAI hurthle cell thyroid cancer | II | 35 | PFS | Active, not recruiting | Not available |
| Neratinib NCT03065387 | Everolimus, palbociclib or trametinib | Pan-HER inhibitor + mTOR, CDK4/6, MEK | Refractory and Advanced or Metastatic Solid Tumors | I | 120 | MTD | Recruiting | Not available |
| ZETA [83] | EXAM [84] | |
|---|---|---|
| Study design | Randomized (2:1), double blind, placebo-controlled, phase III | Randomized (2:1), double blind, placebo-controlled, phase III |
| Experimental arm | Vandetanib 300mg/24h | Cabozantinib 140 mg/24 h |
| Target | VEGFR, EGFR, RET | MET, VEGFR2, FLT3, cKIT and RET |
| Number of patients | 331 | 330 |
| Tumor stage | Unresectable/Metastatic | Unresectable/Metastatic Documented RECIST progression |
| Previous treatment lines | 132 (40%) previously treated | 128 (40%) previously treated |
| RET mutational status: RET+/RET-/RET unknown | 56% (187)/2.4% (8)/41% (136) | 48% (159)/12% (41)/39% (130) |
| Primary endpoint | PFS | PFS |
| Progressive disease | Not mandatory | Yes |
| Overall Response Rate | 45% vs. 13% | 28% vs. 0% |
| Disease Control Rate | 87% vs. 71% | 55.3% vs. 13.5% |
| Progression Free Survival (PFS) | 30.5 m vs. 19.3 m (HR 0.27) | 11.2 m vs. 4 m (HR 0.28) |
| Drug | Combination | Drug Targets | Population | Phase | N | Primary Endpoint | Status | Results |
|---|---|---|---|---|---|---|---|---|
| Selpercatinib (LOXO-292) NCT03157128 | N/A | RET | Advanced solid tumors | I/II | 970 | MTD, RP2D, ORR | Recruiting | 56% [104] |
| Selpercatinib (LOXO-292) NCT04211337 | Cabozantinib Vandetanib | RET | RET-m MTC | III | 400 | TFFS | Recruiting | Not available |
| Pralsetinib (BLU-667) NCT03037385 | N/A | RET | Advanced solid tumors | I/II | 527 | MTD, ORR | Recruiting | 89% [107] |
| Anlotinib NCT02586350 | N/A | VEGFR, FGFR, PDGFR, c-kit, RET | MTC | IIB | 91 | PFS | Completed | 20.67 months [108] |
| Surufatinib NCT02614495 | N/A | VEGFR, FGFR, RET | RAI-R DTC, MTC | II | 66 | ORR | Completed | PR 17% [109] |
| Nintedanib NCT01788982 | N/A | VEGFR, FGFR, PDGFR, RET | DTC, MTC | II | 143 | PFS | Active, not recruiting | Not available |
| Regorafenib NCT02657551 | N/A | VEGFR, PDGFR, c-kit, RET | MTC | II | 33 | PFS | Recruiting | Not available |
| BOS172738 NCT03780517 | N/A | VEGFR, RET | RET-gene altered tumors | I | 144 | TEAE, MTD, RP2D | Recruiting | Not available |
| TPX-0046 NCT04161391 | N/A | SRC, RET | RET-gene altered tumors | I/II | 362 | DLTs, MTD, ORR | Recruiting | Not available |
| Target | Drug | Combination | Population | Phase | N | Primary Endpoint | Results |
|---|---|---|---|---|---|---|---|
| PD-1 | Pembrolizumab NCT02688608 | N/A | ATC | II | 20 | RR | Not available |
| Pembrolizumab NCT03072160 | N/A | MTC | II | 17 | CL #, PR/CR | Not available | |
| Pembrolizumab NCT03012620 | N/A | Rare cancers | II | 350 | ORR | Not available | |
| Pembrolizumab NCT02628067 | N/A | Advanced solid tumors | II | 1350 | ORR | Not available | |
| Pembrolizumab NCT02054806 | N/A | Advanced solid tumors | Ib | 477 | Best OR | ORR 9% [114] | |
| Pembrolizumab NCT03360890 | Docetaxel | TC and salivary gland tumors | I | 46 | ORR | Not available | |
| Pembrolizumab NCT03211117 | Docetaxel Doxorubicin | ATC | II | 3 | OSR | Terminated [135] | |
| Pembrolizumab NCT02973997 | Lenvatinib | RAI-R DTC | II | 60 | CRR > 15% | Primary endpoint not reached [116] | |
| Pembrolizumab NCT04171622 | Lenvatinib | ATC | II | 25 | OS | Not available | |
| Pembrolizumab NCT04234113 | SO-C101 | Solid Tumors | I/Ib | 96 | DLTs, AEs | Not available | |
| Nivolumab NCT04061980 | Encorafenib Binimetinib | RAIR BRAF-mutated DTC | II | 40 | ORR | Not available | |
| Spartalizumab NCT02404441 | N/A | Advanced solid tumors | I/II | 319 | RP2D, DLTs, ORR | 19% * [133] | |
| Cemiplimab NCT04238624 | DabrafenibTrametinib | BRAF V600E ATC | II | 15 | ORR | Not available | |
| PD-1 and CTLA-4 | Nivolumab Ipilimumab NCT03246958 | N/A | RAI-R DTC, ATC, MTC | II | 54 | RR (CR + PR) | 9.4% (DTC) 30% (ATC) 0% (MTC) [115] |
| Nivolumab Ipilimumab NCT03914300 | Cabozantinib | DTC | II | 24 | ORR | Not available | |
| Nivolumab Ipilimumab NCT02834013 | N/A | Rare tumors | II | 818 | ORR | Not available | |
| PD-L1 | Atezolizumab NCT03181100 | 1: Vemurafenib/Cobimetinib 2: Cobimetinib 3: Bevacizumab 4: Paclitaxel | PDTC, ATC Cohort selection depending driver mutation | II | 50 | OS | 1: not reached 2: 18.23 mo 3: 6.21 mo 4: 4.44 mo [134] |
| Atezolizumab NCT03170960 | Cabozantinib | Locally advanced or metastatic solid tumors | Ib | 1732 | MTD, ORR | Not available | |
| Atezolizumab NCT04400474 | Cabozantinib | Advanced and progressive tumors from endocrine system | II | 144 | ORR | Not available | |
| Durvalumab NCT03215095 | N/A | TC | I | 11 | DLTs | Not available | |
| Avelumab NCT03475953 | Regorafenib | RAI-R DTC | I/II | 362 | RP2D, OR | Not available | |
| PD-L1 and CTLA-4 | Durvalumab Tremelimumab NCT03122496 | SBRT | ATC | I | 13 | OS | Not available |
| Durvalumab Tremelimumab NCT03753919 | N/A | DTC, MTC, ATC | II | 46 | PFS, OS | Not available |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
San Román Gil, M.; Pozas, J.; Molina-Cerrillo, J.; Gómez, J.; Pian, H.; Pozas, M.; Carrato, A.; Grande, E.; Alonso-Gordoa, T. Current and Future Role of Tyrosine Kinases Inhibition in Thyroid Cancer: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 4951. https://doi.org/10.3390/ijms21144951
San Román Gil M, Pozas J, Molina-Cerrillo J, Gómez J, Pian H, Pozas M, Carrato A, Grande E, Alonso-Gordoa T. Current and Future Role of Tyrosine Kinases Inhibition in Thyroid Cancer: From Biology to Therapy. International Journal of Molecular Sciences. 2020; 21(14):4951. https://doi.org/10.3390/ijms21144951
Chicago/Turabian StyleSan Román Gil, María, Javier Pozas, Javier Molina-Cerrillo, Joaquín Gómez, Héctor Pian, Miguel Pozas, Alfredo Carrato, Enrique Grande, and Teresa Alonso-Gordoa. 2020. "Current and Future Role of Tyrosine Kinases Inhibition in Thyroid Cancer: From Biology to Therapy" International Journal of Molecular Sciences 21, no. 14: 4951. https://doi.org/10.3390/ijms21144951
APA StyleSan Román Gil, M., Pozas, J., Molina-Cerrillo, J., Gómez, J., Pian, H., Pozas, M., Carrato, A., Grande, E., & Alonso-Gordoa, T. (2020). Current and Future Role of Tyrosine Kinases Inhibition in Thyroid Cancer: From Biology to Therapy. International Journal of Molecular Sciences, 21(14), 4951. https://doi.org/10.3390/ijms21144951