Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Improves Energetic Status and Cardiomyogenic Differentiation of Human Dilated Myocardium-Derived Primary Mesenchymal Cells

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Isolation and Identification of Healthy and Pathological hmMSCs
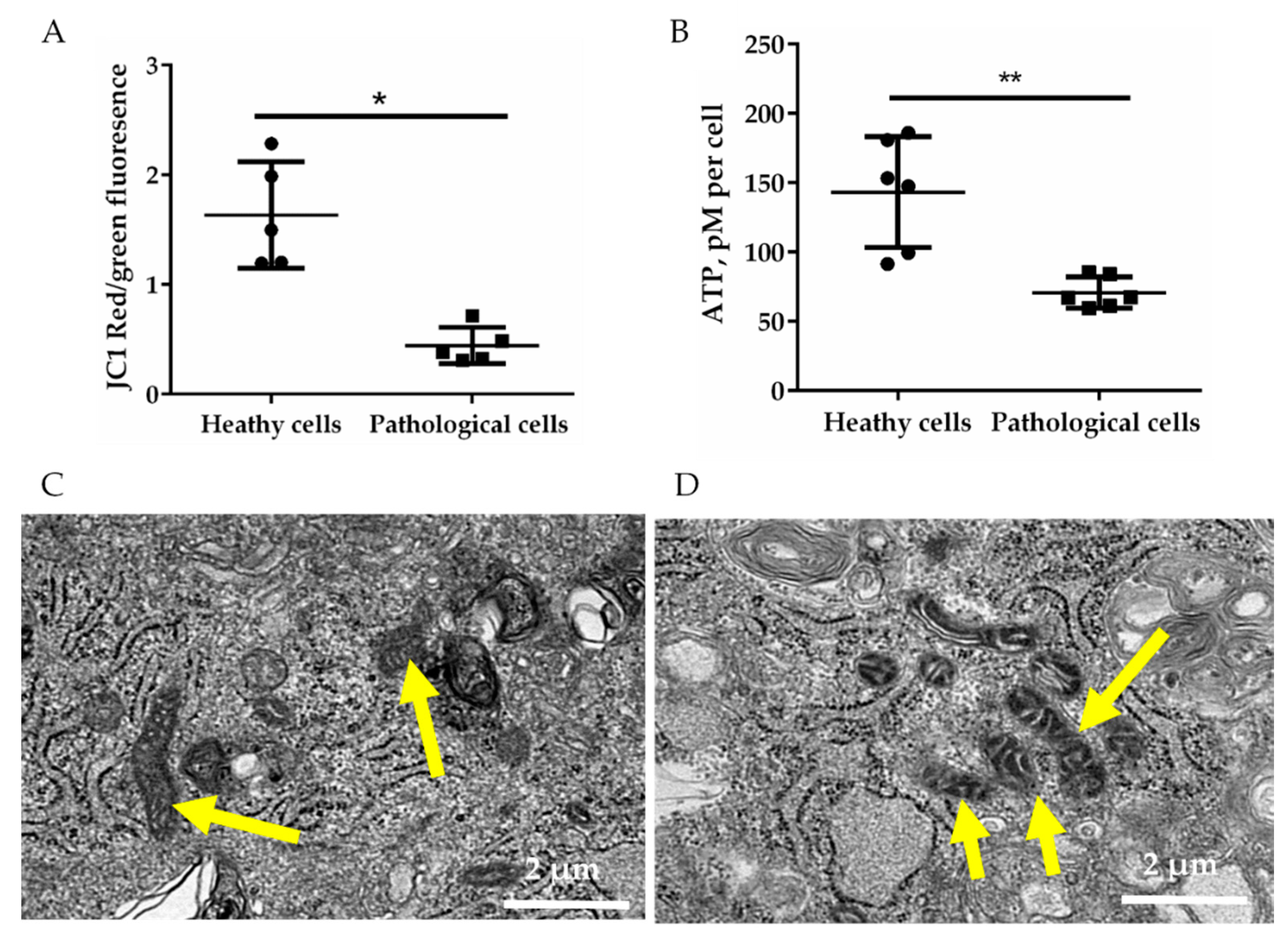
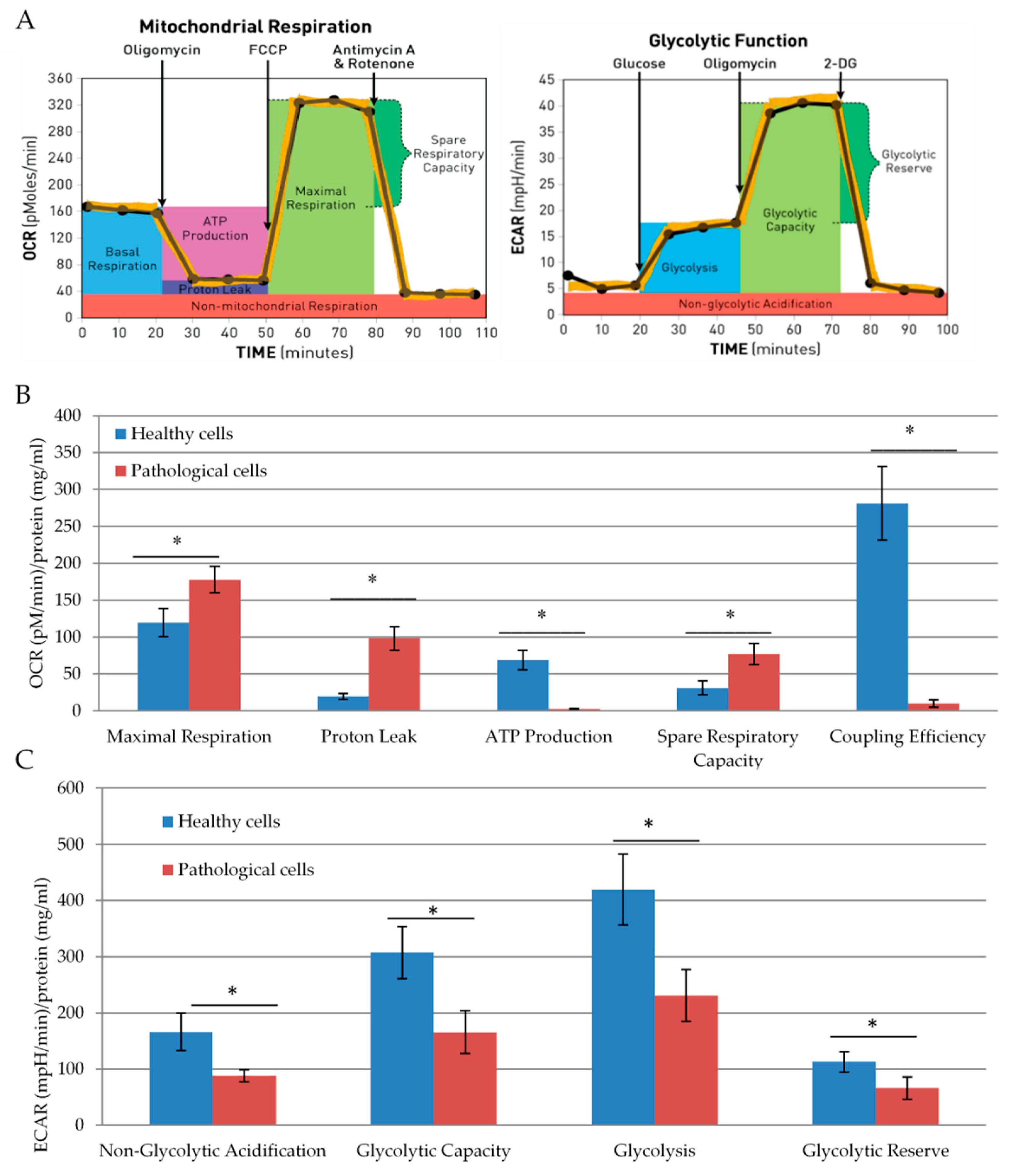
2.2. Energetic Profile of Healthy and Pathological hmMSCs
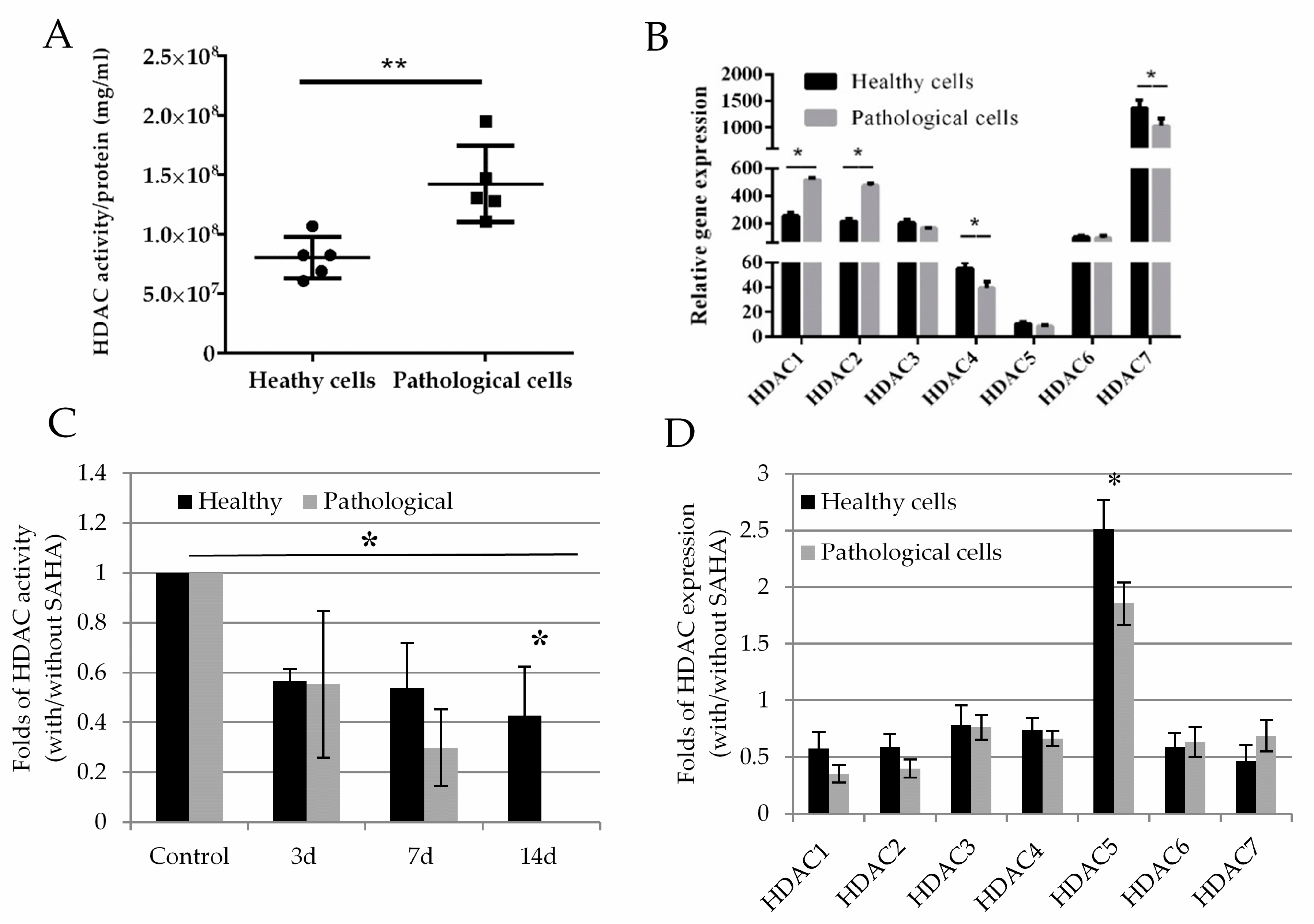
2.3. Histone Deacetylase Activity and Effect of SAHA on Healthy and Pathological hmMSC
2.4. The Impact of SAHA on Bioenergetics of Healthy and Pathological Cells
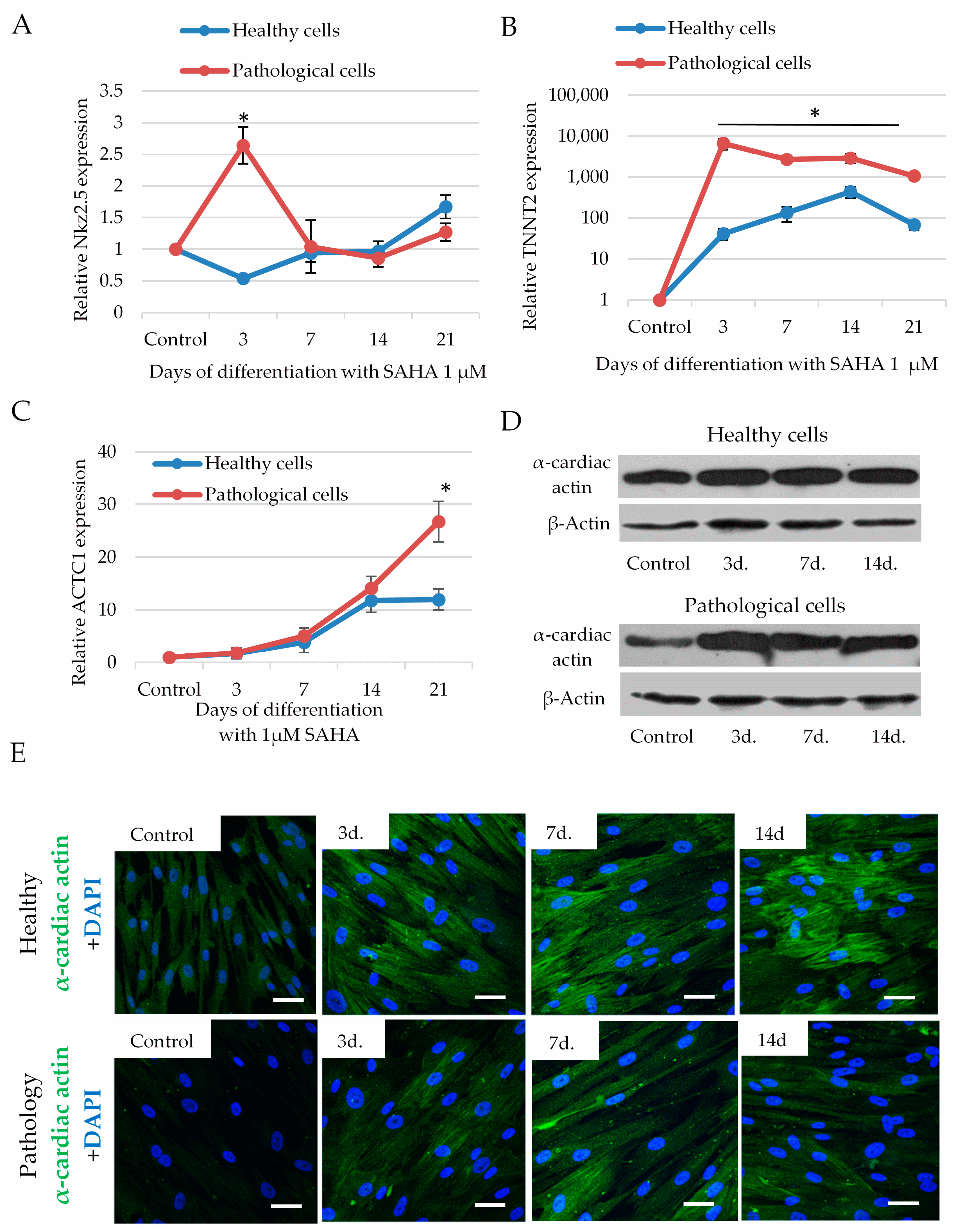
2.5. The Impact of SAHA on Cardiomyogenic Differentiation
3. Discussion
4. Materials and Methods
4.1. Isolation and Growth of hmMSC
4.2. Flow Cytometry
4.3. Measurement of Cell Proliferation
4.4. HDAC Activity Assay
4.5. ATP Activity Assay
4.6. Measurement of Mitochondrial Membrane Potential
4.7. Real Time PCR (RT-PCR)
4.8. Measurement of Metabolic Activity with Seahorse
4.9. Western Blotting
4.10. Immunocytochemistry
4.11. Electron Microscopy
4.12. Statistics
4.13. Ethical Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DCM | Dilated cardiomyopathy |
| HDAC | Histone deacetylase |
| JC1 | 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimi- dazolylcarbocyanine iodide |
| HDACi | Histone deacetylase inhibitor |
| SAHA | Suberoylanilide hydroxamic acid |
| ATP | Adenosine triphosphate |
| iPS | Induced pluripotent stem cells |
| RNA | Ribonucleic acid |
| ECAR | Extracellular acidification |
| OCR | Oxygen consumption rate |
| FCCP | Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone |
| 2-DG | 2-Deoxy-D-glucose |
| ACTC1 | Actin alpha cardiac muscle 1 |
| ACTB | Actin beta |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| DAPI | 4′,6-diamidino-2-phenylindole |
| ROS | Reactive oxygen species |
References
- Lloyd-Jones, D.; Adams, R.J.; Brown, T.M.; Carnethon, M.; Dai, S.; De Simone, G.; Ferguson, T.B.; Ford, E.; Furie, K.; Gillespie, C.; et al. Executive summary: Heart disease and stroke statistics-2010 update: A report from the american heart association. Circulation 2010, 121, e46–e215. [Google Scholar] [CrossRef] [PubMed]
- Kirk, R.; Naftel, D.; Hoffman, T.M.; Almond, C.; Boyle, G.; Caldwell, R.L.; Kirklin, J.K.; White, K.; Dipchand, A.I. Outcome of Pediatric Patients With Dilated Cardiomyopathy Listed for Transplant: A Multi-institutional Study. J. Heart Lung Transplant. 2009, 28, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Roura, S.; Galvez-Monton, C.; Lupon, J.; Bayes-Genis, A. Idiopathic Dilated Cardiomyopathy: Molecular Basis and Distilling Complexity to Advance. In Cardiomyopathies —Types and Treatments; Kirali, K., Ed.; InTech: Rijeka, Croatia, 2017. [Google Scholar]
- Frazier, A.H.; Ramirez-Correa, G.A.; Murphy, A.M. Molecular mechanisms of sarcomere dysfunction in dilated and hypertrophic cardiomyopathy. Prog. Pediatr. Cardiol. 2011, 31, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Guertl, B.; Noehammer, C.; Hoefler, G. Metabolic cardiomyopathies. Int. J. Exp. Pathol. 2000, 81, 349–372. [Google Scholar] [CrossRef]
- Dadson, K.; Hauck, L.; Billia, F. Molecular mechanisms in cardiomyopathy. Clin. Sci. 2017, 131, 1375–1392. [Google Scholar] [CrossRef]
- Ingwall, J.S.; Weiss, R.G. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond atp production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Henning, S.L.; Wambolt, R.B.; Schönekess, B.O.; Lopaschuk, G.D.; Allard, M.F. Contribution of glycogen to aerobic myocardial glucose utilization. Circulation 1996, 93, 1549–1555. [Google Scholar] [CrossRef]
- Opie, L.H.; Knuuti, J. The Adrenergic-Fatty Acid Load in Heart Failure. J. Am. Coll. Cardiol. 2009, 54, 1637–1646. [Google Scholar] [CrossRef]
- Heo, H.J.; Kim, H.K.; Youm, J.B.; Cho, S.W.; Song, I.S.; Lee, S.Y.; Ko, T.H.; Kim, N.; Ko, K.S.; Rhee, B.D.; et al. Mitochondrial pyruvate dehydrogenase phosphatase 1 regulates the early differentiation of cardiomyocytes from mouse embryonic stem cells. Exp. Mol. Med. 2016, 48, e254. [Google Scholar] [CrossRef] [PubMed]
- Hoque, A.; Sivakumaran, P.; Bond, S.T.; Ling, N.X.Y.; Kong, A.M.; Scott, J.W.; Bandara, N.; Hernández, D.; Liu, G.S.; Wong, R.C.B.; et al. Mitochondrial fission protein Drp1 inhibition promotes cardiac mesodermal differentiation of human pluripotent stem cells. Cell Death Discov. 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, B.; Bishi, D.K.; Rallapalli, S.; Arumugam, S.; Cherian, K.M.; Guhathakurta, S. Ischemic cardiac tissue conditioned media induced differentiation of human mesenchymal stem cells into early stage cardiomyocytes. Cytotechnology 2012, 64, 563–575. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kolwicz, S.C.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef]
- Whittington, H.J.; Ostrowski, P.J.; McAndrew, D.J.; Cao, F.; Shaw, A.; Eykyn, T.R.; Lake, H.A.; Tyler, J.; Schneider, J.E.; Neubauer, S.; et al. Over-expression of mitochondrial creatine kinase in the murine heart improves functional recovery and protects against injury following ischaemia-reperfusion. Cardiovasc. Res. 2018, 114, 858–869. [Google Scholar] [CrossRef]
- Sharov, V.G.; Todor, A.V.; Silverman, N.; Goldstein, S.; Sabbah, H.N. Abnormal mitochondrial respiration in failed human myocardium. J. Mol. Cell. Cardiol. 2000, 32, 2361–2367. [Google Scholar] [CrossRef] [PubMed]
- Scheubel, R.J.; Tostlebe, M.; Simm, A.; Rohrbach, S.; Prondzinsky, R.; Gellerich, F.N.; Silber, R.E.; Holtz, J. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J. Am. Coll. Cardiol. 2002, 40, 2174–2181. [Google Scholar] [CrossRef]
- Quigley, A.F.; Kapsa, R.M.I.; Esmore, D.; Hale, G.; Byrne, E. Mitochondrial respiratory chain activity in idiopathic dilated cardiomyopathy. J. Card. Fail. 2000, 6, 47–55. [Google Scholar] [CrossRef]
- Conway, M.A.; Bottomley, P.A.; Ouwerkerk, R.; Radda, G.K.; Rajagopalan, B.; Cooper, R. Mitral regurgitation: Impaired systolic function, eccentric hypertrophy, and increased severity are linked to lower phosphocreatine/ATP ratios in humans. Circulation 1998, 97, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Horn, M.; Cramer, M.; Harre, K.; Newell, J.B.; Pabst, T.; Ertl, G.; Hahn, D.; Ingwall, J.S.; Kochsiek, K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation 1997, 96, 2190–2196. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A. Therapeutic Potential for HDAC Inhibitors in the Heart. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 303–319. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. MEF2: A calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 2002, 27, 40–47. [Google Scholar] [CrossRef]
- Devereux, R.B.; Wachtell, K.; Gerdts, E.; Boman, K.; Nieminen, M.S.; Papademetriou, V.; Rokkedal, J.; Harris, K.; Aurup, P.; Dahlöf, B. Prognostic significance of left ventricular mass change during treatment of hypertension. J. Am. Med. Assoc. 2004, 292, 2350–2356. [Google Scholar] [CrossRef]
- Nural-guvener, H.F.; Zakharova, L.; Nimlos, J.; Popovic, S.; Mastroeni, D. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenes. Tissue Repair 2014, 7, 1–14. [Google Scholar] [CrossRef]
- Xie, M.; Kong, Y.; Tan, W.; May, H.; Battiprolu, P.K.; Pedrozo, Z.; Wang, Z.V.; Morales, C.; Luo, X.; Cho, G.; et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 2014, 129, 1139–1151. [Google Scholar] [CrossRef]
- Rafehi, H.; Balcerczyk, A.; Lunke, S.; Kaspi, A.; Ziemann, M.; Harikrishnan, K.N.; Okabe, J.; Khurana, I.; Ooi, J.; Khan, A.W.; et al. Vascular histone deacetylation by pharmacological HDAC inhibition. Genome Res. 2014, 24, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Dunlay, S.M.; Pereira, N.L.; Kushwaha, S.S. Contemporary strategies in the diagnosis and management of heart failure. Mayo Clin. Proc. 2014, 89, 662–676. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Olson, E.N. Toward transcriptional therapies for the failing heart: Chemical screens to modulate genes. J. Clin. Investig. 2005, 115, 538–546. [Google Scholar] [CrossRef]
- Hantson, P. Mechanisms of toxic cardiomyopathy. Clin. Toxicol. 2019, 57, 1–9. [Google Scholar] [CrossRef]
- Harvey, P.A.; Leinwand, L.A. Cellular mechanisms of cardiomyopathy. J. Cell Biol. 2011, 194, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Blakeslee, W.W.; Demos-Davies, K.M.; Lemon, D.D.; Lutter, K.M.; Cavasin, M.A.; Payne, S.; Nunley, K.; Long, C.S.; McKinsey, T.A.; Miyamoto, S.D. Histone deacetylase adaptation in single ventricle heart disease and a young animal model of right ventricular hypertrophy. Pediatr. Res. 2017, 82, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Pu, W.T. Recounting cardiac cellular composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Heart, H.; Bergmann, O.; Zdunek, S.; Felker, A.; Jovinge, S.; Druid, H.; Frise, J.; Bergmann, O.; Zdunek, S.; Felker, A.; et al. Dynamics of Cell Generation and Turnover in the Article Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef]
- Aghila Rani, K.G.; Jayakumar, K.; Srinivas, G.; Nair, R.R.; Kartha, C.C. Isolation of ckit-positive cardiosphere-forming cells from human atrial biopsy. Asian Cardiovasc. Thorac. Ann. 2008, 16, 50–56. [Google Scholar] [CrossRef]
- Mount, S.; Kanda, P.; Parent, S.; Khan, S.; Michie, C.; Davila, L.; Chan, V.; Davies, R.A.; Haddad, H.; Courtman, D.; et al. Physiologic expansion of human heart-derived cells enhances therapeutic repair of injured myocardium. Stem Cell Res. Ther. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal stem cells: Time to change the name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef]
- Kee, H.J.; Eom, G.H.; Joung, H.; Shin, S.; Kim, J.R.; Cho, Y.K.; Choe, N.; Sim, B.W.; Jo, D.; Jeong, M.H.; et al. Activation of histone deacetylase 2 by inducible heat shock protein 70 in cardiac hypertrophy. Circ. Res. 2008, 103, 1259–1269. [Google Scholar] [CrossRef]
- Kee, H.J.; Sohn, I.S.; Nam, K.I.; Park, J.E.; Qian, Y.R.; Yin, Z.; Ahn, Y.; Jeong, M.H.; Bang, Y.J.; Kim, N.; et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 2006, 113, 51–59. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Zhao, Y.; Wang, J.; Dubielecka, P.M.; Zhuang, S.; Qin, G.; Chin, Y.E.; Kao, R.L.; Zhao, T.C. Myocyte-specific overexpressing HDAC4 promotes myocardial ischemia/reperfusion injury. Mol. Med. 2018, 24, 1–10. [Google Scholar] [CrossRef]
- Leng, Y.; Wu, Y.; Lei, S.; Zhou, B.; Qiu, Z.; Wang, K.; Xia, Z. Inhibition of HDAC6 activity alleviates myocardial ischemia/reperfusion injury in diabetic rats: Potential role of peroxiredoxin 1 acetylation and redox regulation. Oxid. Med. Cell. Longev. 2018, 2018, 9494052. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; McKinsey, T.A.; Zhang, C.L.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Histone Deacetylases 5 and 9 Govern Responsiveness of the Heart to a Subset of Stress Signals and Play Redundant Roles in Heart Development. Mol. Cell. Biol. 2004, 24, 8467–8476. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Schreiter, F.C.; Bagchi, R.A.; Tatman, P.D.; Hannink, M.; McKinsey, T.A. HDAC5 catalytic activity suppresses cardiomyocyte oxidative stress and NRF2 target gene expression. J. Biol. Chem. 2019, 294, 8640–8652. [Google Scholar] [CrossRef]
- Ha, C.H.; Kim, J.Y.; Zhao, J.; Wang, W.; Jhun, B.S.; Wong, C.; Jin, Z.G. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 15467–15472. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Young, B.D.; Li, S.; Qi, X.; Richardson, J.A.; Olson, E.N. Histone Deacetylase 7 Maintains Vascular Integrity by Repressing Matrix Metalloproteinase 10. Cell 2006, 126, 321–334. [Google Scholar] [CrossRef]
- De Ruijter, A.J.M.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B.P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Bertos, N.R.; Wang, A.H.; Yang, X.J. Class II histone deacetylases: Structure, function, and regulation. Biochem. Cell Biol. 2001, 79, 243–252. [Google Scholar] [CrossRef]
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D.; et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 2013, 62, 732–742. [Google Scholar] [CrossRef]
- Marks, P.A. Discovery and development of SAHA as an anticancer agent. Oncogene 2007, 26, 1351–1356. [Google Scholar] [CrossRef]
- Egler, V.; Korur, S.; Failly, M.; Boulay, J.L.; Imber, R.; Lino, M.M.; Merlo, A. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clin. Cancer Res. 2008, 14, 3132–3140. [Google Scholar] [CrossRef]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Fenning, A.; Lim, J.; Le, G.T.; Reid, R.C.; Halili, M.A.; Fairlie, D.P.; Brown, L. Antifibrotic activity of an inhibitor of histone deacetylases in DOCA-salt hypertensive rats: Research paper. Br. J. Pharmacol. 2010, 159, 1408–1417. [Google Scholar] [CrossRef]
- Feng, C.; Zhu, J.; Zhao, L.; Lu, T.; Zhang, W.; Liu, Z.; Tian, J. Suberoylanilide hydroxamic acid promotes cardiomyocyte differentiation of rat mesenchymal stem cells. Exp. Cell Res. 2009, 315, 3044–3051. [Google Scholar] [CrossRef]
- Sung, I.Y.; Son, H.N.; Ullah, I.; Bharti, D.; Park, J.M.; Cho, Y.C.; Byun, J.H.; Kang, Y.H.; Sung, S.J.; Kim, J.W.; et al. Cardiomyogenic differentiation of human dental follicle-derived stem cells by suberoylanilide hydroxamic acid and their in vivo homing property. Int. J. Med. Sci. 2016, 13, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Xia, Y.; Chen, Z.; Chen, A.; Wu, Y.; Jia, J.; Sun, A.; Zou, Y.; Qian, J.; Ge, J. Cardiac proteome profiling in ischemic and dilated cardiomyopathy mouse models. Front. Physiol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, A.; Till, H.; Unterberg, C.; Oberschmidt, R.; Figulla, H.R.; Wiegand, V. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur. Heart J. 1990, 11, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pu, W.T. Cardiomyocyte maturation: New phase in development. Circ. Res. 2020, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.L.; Graham, B.T.; Pabon, L.M.; Han, S.J.; Murry, C.E.; Sniadecki, N.J. Measuring the contractile forces of human induced pluripotent stem cell-derived cardiomyocytes with arrays of microposts. J. Biomech. Eng. 2014, 136. [Google Scholar] [CrossRef]
- Tu, C.; Chao, B.S.; Wu, J.C. Strategies for improving the maturity of human induced pluripotent stem cell-derived cardiomyocytes. Circ. Res. 2018, 123, 512–514. [Google Scholar] [CrossRef]
- Neglia, D.; De Caterina, A.; Marraccini, P.; Natali, A.; Ciardetti, M.; Vecoli, C.; Gastaldelli, A.; Ciociaro, D.; Pellegrini, P.; Testa, R.; et al. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3270-8. [Google Scholar] [CrossRef]
- Liu, Y.; Nguyen, P.; Baris, T.Z.; Poirier, M.C. Molecular analysis of mitochondrial compromise in rodent cardiomyocytes exposed long term to nucleoside reverse transcriptase inhibitors (NRTIs). Cardiovasc. Toxicol. 2012, 12, 123–134. [Google Scholar] [CrossRef]
- Messina, E.; De Angelis, L.; Frati, G.; Morrone, S.; Chimenti, S.; Fiordaliso, F.; Salio, M.; Battaglia, M.; Latronico, M.V.G.; Coletta, M.; et al. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ. Res. 2004, 95, 911–921. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miksiunas, R.; Rucinskas, K.; Janusauskas, V.; Labeit, S.; Bironaite, D. Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Improves Energetic Status and Cardiomyogenic Differentiation of Human Dilated Myocardium-Derived Primary Mesenchymal Cells. Int. J. Mol. Sci. 2020, 21, 4845. https://doi.org/10.3390/ijms21144845
Miksiunas R, Rucinskas K, Janusauskas V, Labeit S, Bironaite D. Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Improves Energetic Status and Cardiomyogenic Differentiation of Human Dilated Myocardium-Derived Primary Mesenchymal Cells. International Journal of Molecular Sciences. 2020; 21(14):4845. https://doi.org/10.3390/ijms21144845
Chicago/Turabian StyleMiksiunas, Rokas, Kestutis Rucinskas, Vilius Janusauskas, Siegfried Labeit, and Daiva Bironaite. 2020. "Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Improves Energetic Status and Cardiomyogenic Differentiation of Human Dilated Myocardium-Derived Primary Mesenchymal Cells" International Journal of Molecular Sciences 21, no. 14: 4845. https://doi.org/10.3390/ijms21144845
APA StyleMiksiunas, R., Rucinskas, K., Janusauskas, V., Labeit, S., & Bironaite, D. (2020). Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Improves Energetic Status and Cardiomyogenic Differentiation of Human Dilated Myocardium-Derived Primary Mesenchymal Cells. International Journal of Molecular Sciences, 21(14), 4845. https://doi.org/10.3390/ijms21144845