Role of Extracellular Matrix in Pathophysiology of Patent Ductus Arteriosus: Emphasis on Vascular Remodeling



Abstract
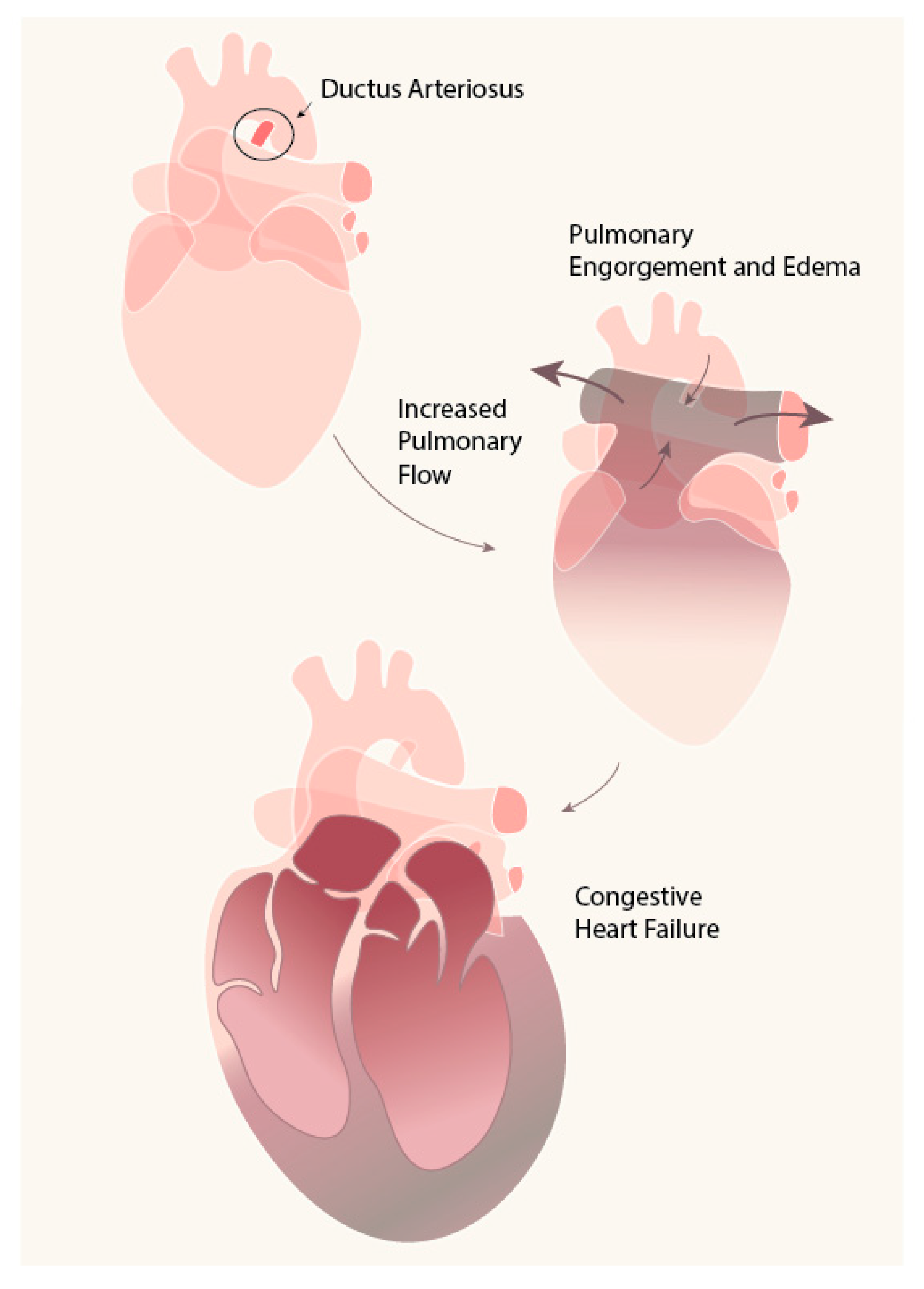
1. Introduction
2. DA: Mechanism of Closure
2.1. Functional Closure
2.2. Anatomical Closure
3. Role of ECM in the Cardiovascular System
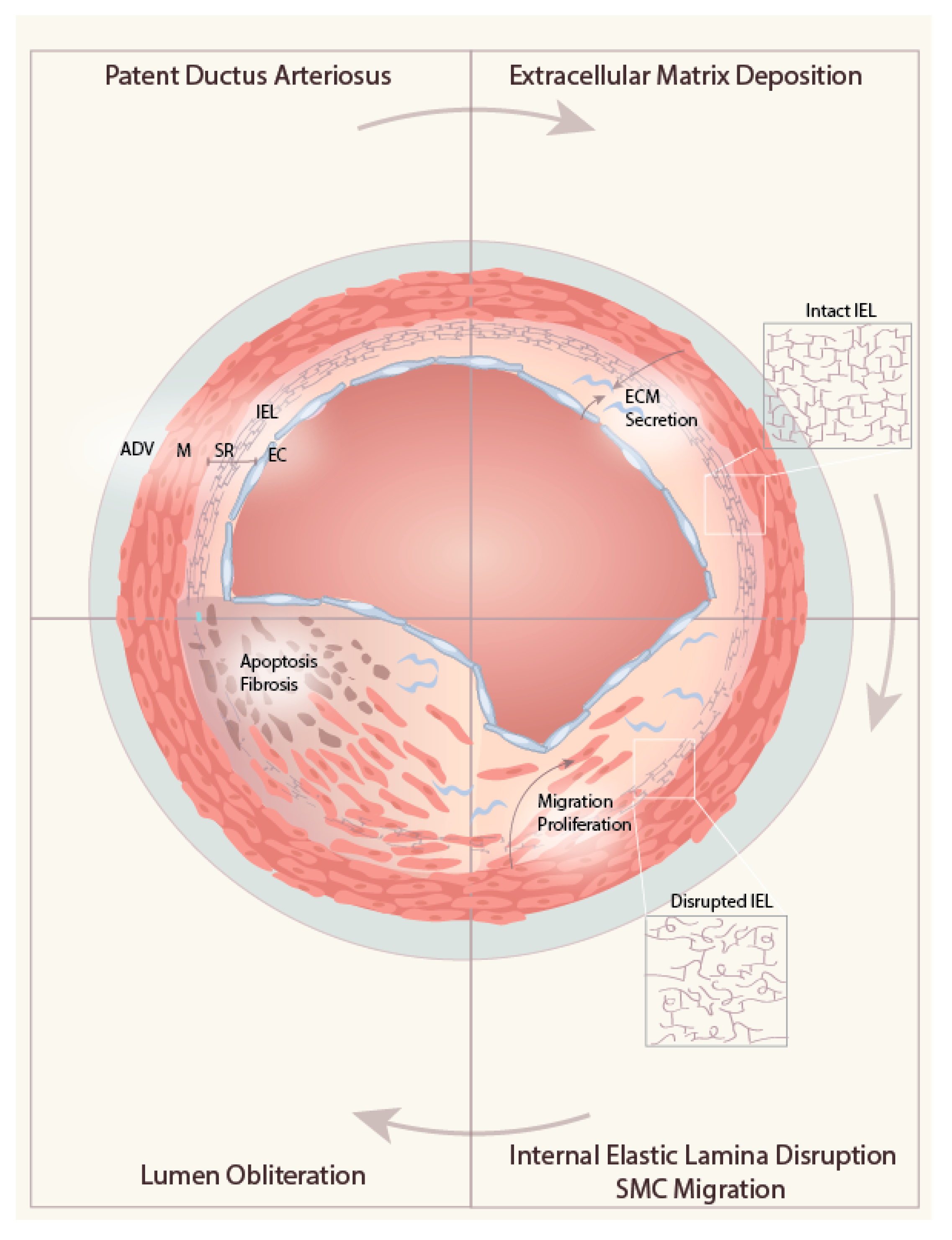
4. Role of ECM in DA Remodeling
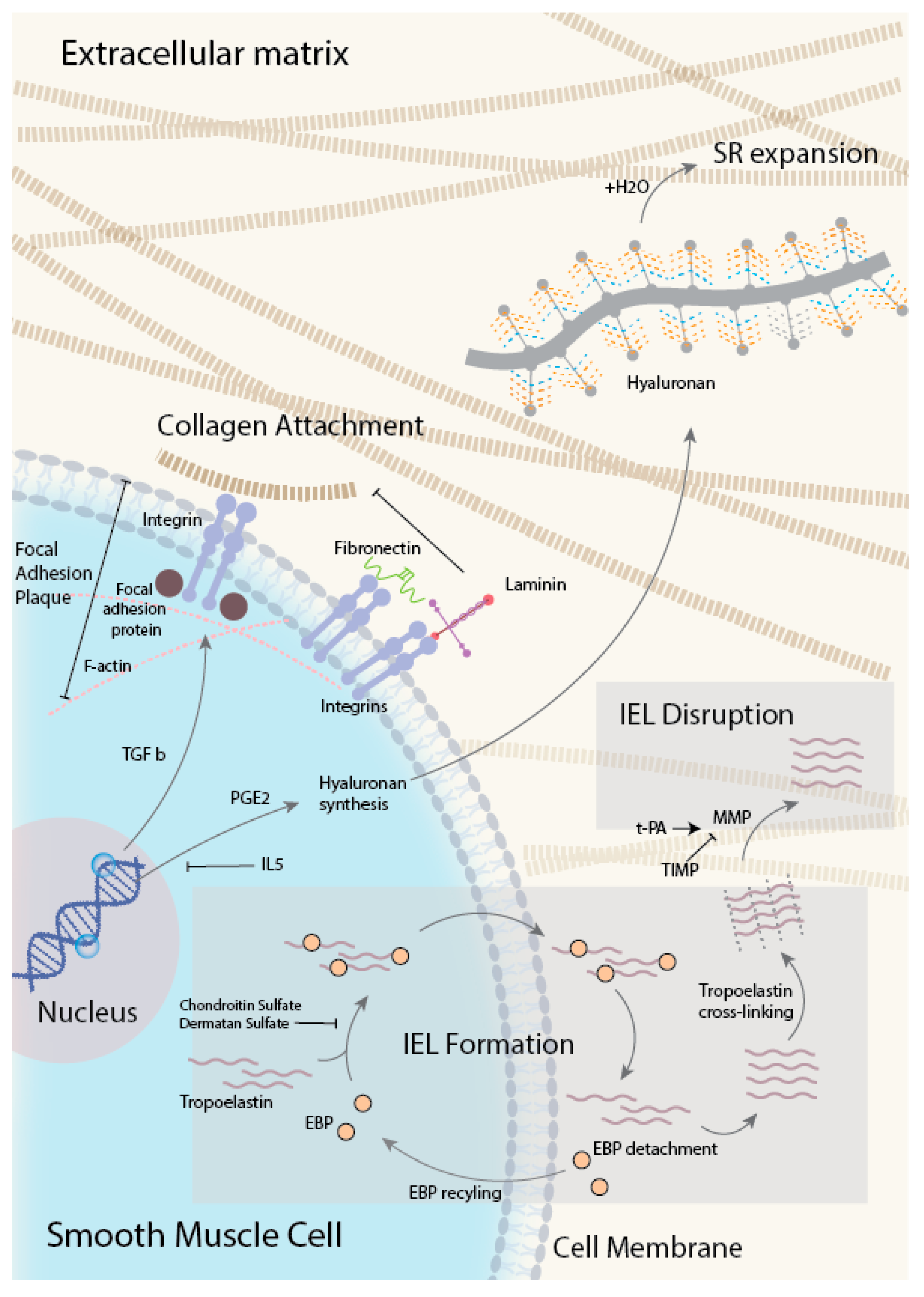
4.1. ECM Deposition in the Subendothelial Region
4.1.1. Hyaluronan (HA)
4.1.2. Fibronectin (FN)
4.2. Internal Elastic Lamina Disruption
4.2.1. LOX
4.2.2. t-PA
4.2.3. Carbohydrate and Its Modifications
Chondroitin Sulfate
Biglycan
Perlecan
Gal-1
4.3. ECM-induced Migration of Medial SMC into the SR
4.3.1. Hyaluronan Binding Protein
4.3.2. Integrin
4.3.3. TGF- β
4.3.4. Peroxidasin
4.3.5. Focal Adhesion
4.4. Obliteration of the Lumen
5. Conclusion and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DA | Ductus arteriosus |
| PDA | Patent ductus arteriosus |
| Ao | Aorta |
| PA | Pulmonary artery |
| SMC | Smooth muscle cells |
| DASMC | Ductus arteriosus smooth muscle cells |
| VSMC | Vascular smooth muscle cells |
| EC | Endothelial cells |
| ECM | Extracellular matrix |
| HA | Hyaluronan acid |
| GP | Glycoprotein |
| GAG | Glucoaminoglycan |
| SR | Subendothelial region |
| ADV | Adventitia |
| SM | Smooth muscle |
| IEL | Internal elastic lamina |
| LOX | Lysyl oxidase |
| HABP | Hyaluronan binding protein |
| EBP | Elastin binding protein |
| CS | Chondroitin sulfate |
| DS | Dermatan sulfate |
| FN | Fibronectin |
| LN | Laminin |
| VN | Vitronectin |
| FA | Focal adhesion |
References
- Clyman, R.I. Patent ductus arteriosus, its treatments, and the risks of pulmonary morbidity. Semin. Perinatol. 2018, 42, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Abu-Shaweesh, J.M.; Almidani, E. PDA: Does it matter? Int. J. Pediatr. Adolesc. Med. 2020, 7, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, A.; Walia, R.; Shah, S.S. Ibuprofen for the treatment of patent ductus arteriosus in preterm or low birth weight (or both) infants. Cochrane Database Syst. Rev. 2020, 2, CD003481. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.H.; Yang, S.N.; Chen, H.L.; Tseng, H.I.; Dai, Z.K.; Wu, J.R. B-type natriuretic peptide predicts responses to indomethacin in premature neonates with patent ductus arteriosus. J. Pediatr. 2010, 157, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.C.; Yeh, J.L.; Hsu, J.H. Molecular Mechanisms for Regulating Postnatal Ductus Arteriosus Closure. Int J. Mol. Sci. 2018, 19, 1861. [Google Scholar] [CrossRef]
- Shelton, E.L.; Singh, G.K.; Nichols, C.G. Novel drug targets for ductus arteriosus manipulation: Looking beyond prostaglandins. Semin. Perinatol. 2018, 42, 221–227. [Google Scholar] [CrossRef]
- Yokoyama, U. Prostaglandin E-mediated molecular mechanisms driving remodeling of the ductus arteriosus. Pediatr. Int. 2015, 57, 820–827. [Google Scholar] [CrossRef]
- Clyman, R.I.; Chan, C.Y.; Mauray, F.; Chen, Y.Q.; Cox, W.; Seidner, S.R.; Lord, E.M.; Weiss, H.; Waleh, N.; Evans, S.M.; et al. Permanent anatomic closure of the ductus arteriosus in newborn baboons: The roles of postnatal constriction, hypoxia, and gestation. Pediatr. Res. 1999, 45, 19–29. [Google Scholar] [CrossRef]
- Momma, K.; Monma, M.; Toyoshima, K.; Hayama, E.; Nakanishi, T. Fetal and Neonatal Ductus Arteriosus Is Regulated with ATP-Sensitive Potassium Channel. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer: Tokyo, Japan, 2016; pp. 263–265. [Google Scholar]
- Yokoyama, U.; Minamisawa, S.; Ishikawa, Y. Regulation of vascular tone and remodeling of the ductus arteriosus. J. Smooth Muscle Res. 2010, 46, 77–87. [Google Scholar] [CrossRef]
- Daley, W.P.; Peters, S.B.; Larsen, M. Extracellular matrix dynamics in development and regenerative medicine. J. Cell Sci. 2008, 121, 255–264. [Google Scholar] [CrossRef]
- Jain, M.; Dhanesha, N.; Doddapattar, P.; Chorawala, M.R.; Nayak, M.K.; Cornelissen, A.; Guo, L.; Finn, A.V.; Lentz, S.R.; Chauhan, A.K. Smooth muscle cell-specific fibronectin-EDA mediates phenotypic switching and neointimal hyperplasia. J. Clin. Invest. 2020, 130, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Ji, Y.; Luo, Y.; Li, R.; Fay, W.P.; Wu, J. Plasminogen activator inhibitor-1 regulates the vascular expression of vitronectin. J. Thromb. Haemost. 2017, 15, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Au, Y.P.; Dobrowolska, G.; Morris, D.R.; Clowes, A.W. Heparin decreases activator protein-1 binding to DNA in part by posttranslational modification of Jun B. Circ. Res. 1994, 75, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kovanen, P.T. Heparin proteoglycans released from rat serosal mast cells inhibit proliferation of rat aortic smooth muscle cells in culture. Circ. Res. 1999, 84, 74–83. [Google Scholar] [CrossRef]
- Chang, Y.T.; Tseng, C.N.; Tannenberg, P.; Eriksson, L.; Yuan, K.; de Jesus Perez, V.A.; Lundberg, J.; Lengquist, M.; Botusan, I.R.; Catrina, S.B.; et al. Perlecan heparan sulfate deficiency impairs pulmonary vascular development and attenuates hypoxic pulmonary hypertension. Cardiovasc. Res. 2015, 107, 20–31. [Google Scholar] [CrossRef]
- Mead, T.J.; Du, Y.; Nelson, C.M.; Gueye, N.A.; Drazba, J.; Dancevic, C.M.; Vankemmelbeke, M.; Buttle, D.J.; Apte, S.S. ADAMTS9-Regulated Pericellular Matrix Dynamics Governs Focal Adhesion-Dependent Smooth Muscle Differentiation. Cell Rep. 2018, 23, 485–498. [Google Scholar] [CrossRef]
- Choi, E.T.; Khan, M.F.; Leidenfrost, J.E.; Collins, E.T.; Boc, K.P.; Villa, B.R.; Novack, D.V.; Parks, W.C.; Abendschein, D.R. Beta3-integrin mediates smooth muscle cell accumulation in neointima after carotid ligation in mice. Circulation 2004, 109, 1564–1569. [Google Scholar] [CrossRef]
- Lutshumba, J.; Liu, S.; Zhong, Y.; Hou, T.; Daugherty, A.; Lu, H.; Guo, Z.; Gong, M.C. Deletion of BMAL1 in Smooth Muscle Cells Protects Mice From Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1063–1075. [Google Scholar] [CrossRef]
- Mummidi, S.; Das, N.A.; Carpenter, A.J.; Yoshida, T.; Yariswamy, M.; Mostany, R.; Izadpanah, R.; Higashi, Y.; Sukhanov, S.; Noda, M.; et al. RECK suppresses interleukin-17/TRAF3IP2-mediated MMP-13 activation and human aortic smooth muscle cell migration and proliferation. J. Cell Physiol. 2019, 234, 22242–22259. [Google Scholar] [CrossRef]
- Shin, S.S.; Ko, M.C.; Noh, D.H.; Hwang, B.; Park, Y.; Park, S.L.; Kim, W.J.; Moon, S.K. Morin inhibits PDGF-induced proliferation, migration, and invasion of vascular smooth muscle cells via modulating p27KIP1, AKT, and MMP-9 activities. Gen. Physiol. Biophys. 2018, 37, 633–645. [Google Scholar] [CrossRef]
- Ji, Y.; Weng, Z.; Fish, P.; Goyal, N.; Luo, M.; Myears, S.P.; Strawn, T.L.; Chandrasekar, B.; Wu, J.; Fay, W.P. Pharmacological Targeting of Plasminogen Activator Inhibitor-1 Decreases Vascular Smooth Muscle Cell Migration and Neointima Formation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Minamisawa, S.; Ishikawa, Y. The Multiple Roles of Prostaglandin E2 in the Regulation of the Ductus Arteriosus. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer: Tokyo, Japan, 2016; pp. 253–258. [Google Scholar]
- Shi, R.; Hu, C.; Yuan, Q.; Yang, T.; Peng, J.; Li, Y.; Bai, Y.; Cao, Z.; Cheng, G.; Zhang, G. Involvement of vascular peroxidase 1 in angiotensin II-induced vascular smooth muscle cell proliferation. Cardiovasc. Res. 2011, 91, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Nakamura, F.; Fukunaga, S. Perlecan Diversely Regulates the Migration and Proliferation of Distinct Cell Types in vitro. Cells Tissues Organs 2015, 200, 374–393. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, F.; Kramer, M.C.; Woudstra, P.; Yahagi, K.; Ladich, E.; Finn, A.V.; de Winter, R.J.; Kolodgie, F.D.; Wight, T.N.; Davis, H.R.; et al. Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: A pathology study. Atherosclerosis 2015, 241, 772–782. [Google Scholar] [CrossRef]
- Tsai, M.S.; Chiang, M.T.; Tsai, D.L.; Yang, C.W.; Hou, H.S.; Li, Y.R.; Chang, P.C.; Lin, H.H.; Chen, H.Y.; Hwang, I.S.; et al. Galectin-1 Restricts Vascular Smooth Muscle Cell Motility Via Modulating Adhesion Force and Focal Adhesion Dynamics. Sci. Rep. 2018, 8, 11497. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef]
- Smiljanic, K.; Obradovic, M.; Jovanovic, A.; Djordjevic, J.; Dobutovic, B.; Jevremovic, D.; Marche, P.; Isenovic, E.R. Thrombin stimulates VSMC proliferation through an EGFR-dependent pathway: Involvement of MMP-2. Mol. Cell Biochem. 2014, 396, 147–160. [Google Scholar] [CrossRef]
- Engur, D.; Kaynak-Turkmen, M.; Deveci, M.; Yenisey, C. Platelets and platelet-derived growth factor in closure of the ductus arteriosus. Turk. J. Pediatr. 2015, 57, 242–247. [Google Scholar]
- Clyman, R.I.; Tannenbaum, J.; Chen, Y.Q.; Cooper, D.; Yurchenco, P.D.; Kramer, R.H.; Waleh, N.S. Ductus arteriosus smooth muscle cell migration on collagen: Dependence on laminin and its receptors. J. Cell Sci. 1994, 107, 1007–1018. [Google Scholar]
- Boudreau, N.; Turley, E.; Rabinovitch, M. Fibronectin, hyaluronan, and a hyaluronan binding protein contribute to increased ductus arteriosus smooth muscle cell migration. Dev. Biol. 1991, 143, 235–247. [Google Scholar] [CrossRef]
- Bokenkamp, R.; Gittenberger-De Groot, A.C.; Van Munsteren, C.J.; Grauss, R.W.; Ottenkamp, J.; Deruiter, M.C. Persistent ductus arteriosus in the Brown-Norway inbred rat strain. Pediatr. Res. 2006, 60, 407–412. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Slomp, J.; van Munsteren, J.C.; Poelmann, R.E.; de Reeder, E.G.; Bogers, A.J.; Gittenberger-de Groot, A.C. Formation of intimal cushions in the ductus arteriosus as a model for vascular intimal thickening. An immunohistochemical study of changes in extracellular matrix components. Atherosclerosis 1992, 93, 25–39. [Google Scholar] [CrossRef]
- Hinek, A.; Mecham, R.P.; Keeley, F.; Rabinovitch, M. Impaired elastin fiber assembly related to reduced 67-kD elastin-binding protein in fetal lamb ductus arteriosus and in cultured aortic smooth muscle cells treated with chondroitin sulfate. J. Clin. Invest. 1991, 88, 2083–2094. [Google Scholar] [CrossRef] [PubMed]
- De Reeder, E.G.; Girard, N.; Poelmann, R.E.; Van Munsteren, J.C.; Patterson, D.F.; Gittenberger-De Groot, A.C. Hyaluronic acid accumulation and endothelial cell detachment in intimal thickening of the vessel wall. The normal and genetically defective ductus arteriosus. Am. J. Pathol. 1988, 132, 574–585. [Google Scholar] [PubMed]
- Saito, J.; Yokoyama, U.; Nicho, N.; Zheng, Y.W.; Ichikawa, Y.; Ito, S.; Umemura, M.; Fujita, T.; Ito, S.; Taniguchi, H.; et al. Tissue-type plasminogen activator contributes to remodeling of the rat ductus arteriosus. PLoS ONE 2018, 13, e0190871. [Google Scholar] [CrossRef]
- Yokoyama, U.; Minamisawa, S.; Quan, H.; Ghatak, S.; Akaike, T.; Segi-Nishida, E.; Iwasaki, S.; Iwamoto, M.; Misra, S.; Tamura, K.; et al. Chronic activation of the prostaglandin receptor EP4 promotes hyaluronan-mediated neointimal formation in the ductus arteriosus. J. Clin. Invest. 2006, 116, 3026–3034. [Google Scholar] [CrossRef]
- Iwasaki, S.; Minamisawa, S.; Yokoyama, U.; Akaike, T.; Quan, H.; Nagashima, Y.; Nishimaki, S.; Ishikawa, Y.; Yokota, S. Interleukin-15 inhibits smooth muscle cell proliferation and hyaluronan production in rat ductus arteriosus. Pediatr. Res. 2007, 62, 392–398. [Google Scholar] [CrossRef]
- Tannenbaum, J.E.; Waleh, N.S.; Mauray, F.; Breuss, J.; Pytela, R.; Kramer, R.H.; Clyman, R.I. Transforming growth factor beta 1 inhibits fetal lamb ductus arteriosus smooth muscle cell migration. Pediatr. Res. 1995, 37, 561–570. [Google Scholar] [CrossRef]
- Tannenbaum, J.E.; Waleh, N.S.; Mauray, F.; Gold, L.; Perkett, E.A.; Clyman, R.I. Transforming growth factor-beta protein and messenger RNA expression is increased in the closing ductus arteriosus. Pediatr. Res. 1996, 39, 427–434. [Google Scholar] [CrossRef]
- Levet, S.; Ouarne, M.; Ciais, D.; Coutton, C.; Subileau, M.; Mallet, C.; Ricard, N.; Bidart, M.; Debillon, T.; Faravelli, F.; et al. BMP9 and BMP10 are necessary for proper closure of the ductus arteriosus. Proc. Natl. Acad. Sci. USA 2015, 112, E3207–E3215. [Google Scholar] [CrossRef]
- De Reeder, E.G.; Poelmann, R.E.; van Munsteren, J.C.; Patterson, D.F.; Gittenberger-de Groot, A.C. Ultrastructural and immunohistochemical changes of the extracellular matrix during intimal cushion formation in the ductus arteriosus of the dog. Atherosclerosis 1989, 79, 29–40. [Google Scholar] [CrossRef]
- Segi, E.; Sugimoto, Y.; Yamasaki, A.; Aze, Y.; Oida, H.; Nishimura, T.; Murata, T.; Matsuoka, T.; Ushikubi, F.; Hirose, M.; et al. Patent ductus arteriosus and neonatal death in prostaglandin receptor EP4-deficient mice. Biochem. Biophys. Res. Commun. 1998, 246, 7–12. [Google Scholar] [CrossRef]
- Nguyen, M.; Camenisch, T.; Snouwaert, J.N.; Hicks, E.; Coffman, T.M.; Anderson, P.A.; Malouf, N.N.; Koller, B.H. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature 1997, 390, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Loftin, C.D.; Trivedi, D.B.; Tiano, H.F.; Clark, J.A.; Lee, C.A.; Epstein, J.A.; Morham, S.G.; Breyer, M.D.; Nguyen, M.; Hawkins, B.M.; et al. Failure of ductus arteriosus closure and remodeling in neonatal mice deficient in cyclooxygenase-1 and cyclooxygenase-2. Proc. Natl. Acad. Sci. USA 2001, 98, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Minamisawa, S.; Shioda, A.; Ishiwata, R.; Jin, M.H.; Masuda, M.; Asou, T.; Sugimoto, Y.; Aoki, H.; Nakamura, T.; et al. Prostaglandin E2 inhibits elastogenesis in the ductus arteriosus via EP4 signaling. Circulation 2014, 129, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Minamisawa, S.; Katayama, A.; Tang, T.; Suzuki, S.; Iwatsubo, K.; Iwasaki, S.; Kurotani, R.; Okumura, S.; Sato, M.; et al. Differential regulation of vascular tone and remodeling via stimulation of type 2 and type 6 adenylyl cyclases in the ductus arteriosus. Circ. Res. 2010, 106, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Lubas, M.; Lund, A.H. Emerging connections between RNA and autophagy. Autophagy 2017, 13, 3–23. [Google Scholar] [CrossRef]
- Mason, C.A.; Bigras, J.L.; O’Blenes, S.B.; Zhou, B.; McIntyre, B.; Nakamura, N.; Kaneda, Y.; Rabinovitch, M. Gene transfer in utero biologically engineers a patent ductus arteriosus in lambs by arresting fibronectin-dependent neointimal formation. Nat. Med. 1999, 5, 176–182. [Google Scholar] [CrossRef]
- Murphy, P.A.; Hynes, R.O. Alternative splicing of endothelial fibronectin is induced by disturbed hemodynamics and protects against hemorrhage of the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2042–2050. [Google Scholar] [CrossRef]
- Toda, T.; Tsuda, N.; Takagi, T.; Nishimori, I.; Leszczynski, D.; Kummerow, F. Ultrastructure of developing human ductus arteriosus. J. Anat. 1980, 131, 25–37. [Google Scholar]
- Tada, T.; Wakabayashi, T.; Nakao, Y.; Ueki, R.; Ogawa, Y.; Inagawa, A.; Shibata, T.; Kishimoto, H. Human ductus arteriosus. A histological study on the relation between ductal maturation and gestational age. Acta Pathol. Jpn. 1985, 35, 23–34. [Google Scholar]
- Hinek, A.; Rabinovitch, M. The ductus arteriosus migratory smooth muscle cell phenotype processes tropoelastin to a 52-kDa product associated with impaired assembly of elastic laminae. J. Biol. Chem. 1993, 268, 1405–1413. [Google Scholar]
- Yokoyama, U.; Ichikawa, Y.; Minamisawa, S.; Ishikawa, Y. Pathology and molecular mechanisms of coarctation of the aorta and its association with the ductus arteriosus. J. Physiol. Sci. 2017, 67, 259–270. [Google Scholar] [CrossRef]
- Monea, S.; Lehti, K.; Keski-Oja, J.; Mignatti, P. Plasmin activates pro-matrix metalloproteinase-2 with a membrane-type 1 matrix metalloproteinase-dependent mechanism. J. Cell Physiol. 2002, 192, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, H.R. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb. Haemost. 2001, 86, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Okajima, T. Structure and function of extracellular O-GlcNAc. Curr. Opin. Struct. Biol. 2019, 56, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Marsico, G.; Russo, L.; Quondamatteo, F.; Pandit, A. Glycosylation and Integrin Regulation in Cancer. Trends Cancer 2018, 4, 537–552. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Fallon, J.R.; McNally, E.M. Non-Glycanated Biglycan and LTBP4: Leveraging the extracellular matrix for Duchenne Muscular Dystrophy therapeutics. Matrix Biol. 2018, 68–69, 616–627. [Google Scholar] [CrossRef]
- Nastase, M.V.; Young, M.F.; Schaefer, L. Biglycan: A multivalent proteoglycan providing structure and signals. J. Histochem. Cytochem. 2012, 60, 963–975. [Google Scholar] [CrossRef]
- Pietraszek-Gremplewicz, K.; Karamanou, K.; Niang, A.; Dauchez, M.; Belloy, N.; Maquart, F.X.; Baud, S.; Brezillon, S. Small leucine-rich proteoglycans and matrix metalloproteinase-14: Key partners? Matrix Biol. 2019, 75–76, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.A.; Sun, M.; Barnum, C.E.; Weiss, S.N.; Huegel, J.; Shetye, S.S.; Lin, L.; Saez, D.; Adams, S.M.; Iozzo, R.V.; et al. Decorin and biglycan are necessary for maintaining collagen fibril structure, fiber realignment, and mechanical properties of mature tendons. Matrix Biol. 2017, 64, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Panitch, A. Decorin mimic regulates platelet-derived growth factor and interferon-gamma stimulation of vascular smooth muscle cells. Biomacromolecules 2014, 15, 2090–2103. [Google Scholar] [CrossRef] [PubMed]
- Scuruchi, M.; Poti, F.; Rodriguez-Carrio, J.; Campo, G.M.; Mandraffino, G. Biglycan and atherosclerosis: Lessons from high cardiovascular risk conditions. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158545. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, P.; Carroll, K.L.; Berceli, S.A.; Barnhill, S.; Wrenshall, L.E. Expression of a Functional IL-2 Receptor in Vascular Smooth Muscle Cells. J. Immunol. 2019, 202, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Sadowitz, B.; Seymour, K.; Gahtan, V.; Maier, K.G. The role of hyaluronic acid in atherosclerosis and intimal hyperplasia. J. Surg. Res. 2012, 173, e63–e72. [Google Scholar] [CrossRef]
- Clyman, R.I.; Mauray, F.; Kramer, R.H. Beta 1 and beta 3 integrins have different roles in the adhesion and migration of vascular smooth muscle cells on extracellular matrix. Exp. Cell Res. 1992, 200, 272–284. [Google Scholar] [CrossRef]
- Majesky, M.W.; Lindner, V.; Twardzik, D.R.; Schwartz, S.M.; Reidy, M.A. Production of transforming growth factor beta 1 during repair of arterial injury. J. Clin. Invest. 1991, 88, 904–910. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Z.; Xu, Q.; Peng, H.; Chen, L.; Huang, X.; Yang, T.; Yu, Z.; Cheng, G.; Zhang, G.; et al. Involvement of vascular peroxidase 1 in angiotensin II-induced hypertrophy of H9c2 cells. J. Am. Soc. Hypertens. 2017, 11, 519–529. [Google Scholar] [CrossRef]
- Yeh, J.L.; Wu, J.R.; Wu, B.N.; Yang, S.F.; Dai, Z.K.; Liou, S.F.; Hsu, J.H. B-type natriuretic peptide prevents postnatal closure of ductus arteriosus by both vasodilation and anti-remodeling in neonatal rats. Clin. Sci. 2018, 132, 2045–2058. [Google Scholar] [CrossRef]
- Bhave, G.; Cummings, C.F.; Vanacore, R.M.; Kumagai-Cresse, C.; Ero-Tolliver, I.A.; Rafi, M.; Kang, J.S.; Pedchenko, V.; Fessler, L.I.; Fessler, J.H.; et al. Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat. Chem. Biol. 2012, 8, 784–790. [Google Scholar] [CrossRef] [PubMed]
- McCall, A.S.; Cummings, C.F.; Bhave, G.; Vanacore, R.; Page-McCaw, A.; Hudson, B.G. Bromine is an essential trace element for assembly of collagen IV scaffolds in tissue development and architecture. Cell 2014, 157, 1380–1392. [Google Scholar] [CrossRef] [PubMed]
- Gotenstein, J.R.; Swale, R.E.; Fukuda, T.; Wu, Z.; Giurumescu, C.A.; Goncharov, A.; Jin, Y.; Chisholm, A.D. The C. elegans peroxidasin PXN-2 is essential for embryonic morphogenesis and inhibits adult axon regeneration. Development 2010, 137, 3603–3613. [Google Scholar] [CrossRef]
- Khan, K.; Rudkin, A.; Parry, D.A.; Burdon, K.P.; McKibbin, M.; Logan, C.V.; Abdelhamed, Z.I.; Muecke, J.S.; Fernandez-Fuentes, N.; Laurie, K.J.; et al. Homozygous mutations in PXDN cause congenital cataract, corneal opacity, and developmental glaucoma. Am. J. Hum. Genet. 2011, 89, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Lao, R.; Ling-Fung Tang, P.; Wan, E.; Mayer, W.; Bardakjian, T.; Shaw, G.M.; Kwok, P.Y.; Schneider, A.; Slavotinek, A. Novel mutations in PXDN cause microphthalmia and anterior segment dysgenesis. Eur. J. Hum. Genet. 2015, 23, 337–341. [Google Scholar] [CrossRef]
- Soudi, M.; Paumann-Page, M.; Delporte, C.; Pirker, K.F.; Bellei, M.; Edenhofer, E.; Stadlmayr, G.; Battistuzzi, G.; Boudjeltia, K.Z.; Furtmuller, P.G.; et al. Multidomain human peroxidasin 1 is a highly glycosylated and stable homotrimeric high spin ferric peroxidase. J. Biol. Chem. 2015, 290, 10876–10890. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P. Vascular peroxidase 1/peroxidasin: A complex protein with a simple function? Cardiovasc. Res. 2011, 91, 1–2. [Google Scholar] [CrossRef]
- Francis, S.E.; Goh, K.L.; Hodivala-Dilke, K.; Bader, B.L.; Stark, M.; Davidson, D.; Hynes, R.O. Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 927–933. [Google Scholar] [CrossRef]
- Romer, L.H.; Birukov, K.G.; Garcia, J.G. Focal adhesions: Paradigm for a signaling nexus. Circ. Res. 2006, 98, 606–616. [Google Scholar] [CrossRef]
- Silver, M.M.; Freedom, R.M.; Silver, M.D.; Olley, P.M. The morphology of the human newborn ductus arteriosus: A reappraisal of its structure and closure with special reference to prostaglandin E1 therapy. Hum. Pathol. 1981, 12, 1123–1136. [Google Scholar] [CrossRef]
- Goldbarg, S.; Quinn, T.; Waleh, N.; Roman, C.; Liu, B.M.; Mauray, F.; Clyman, R.I. Effects of hypoxia, hypoglycemia, and muscle shortening on cell death in the sheep ductus arteriosus. Pediatr. Res. 2003, 54, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Slomp, J.; Gittenberger-de Groot, A.C.; Glukhova, M.A.; Conny van Munsteren, J.; Kockx, M.M.; Schwartz, S.M.; Koteliansky, V.E. Differentiation, dedifferentiation, and apoptosis of smooth muscle cells during the development of the human ductus arteriosus. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Hermes-DeSantis, E.R.; Clyman, R.I. Patent ductus arteriosus: Pathophysiology and management. J. Perinatol. 2006, 26 (Suppl. 1), S14–S18. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Matrix Element | Effect on SMC | Mechanism | Reference |
|---|---|---|---|
| Glycoprotein | |||
| Fibronectin | Proliferation | Cyclin activation | [12] |
| Vitronectin | Migration | plasminogen activator inhibitor-1 | [13] |
| Proteoglycan | |||
| Heparan sulfate | Anti-proliferation | Growth factor interaction | [14,15,16] |
| ECM Mediator | Effect on SMC | Mechanism | Reference |
|---|---|---|---|
| Matrix binding receptors | |||
| Focal Adhesion | Migration | Increased migration through ECM adhesion | [17] |
| Vitronectin receptor | Migration | Increased SMC accumulation | [18] |
| Matrix degrading enzymes | |||
| TIMP | Anti-migration | MMP inhibition | [19] |
| MMP | Migration | Matrix degradation | [20] |
| PDGF | Migration | MMP expression | [21] |
| t-PA | Migration | Matrix degradation | [22] |
| LOX | Anti-migration | Matrix crosslink | [23] |
| Peroxidasin | Anti-migration | Matrix crosslink | [24] |
| Carbohydrate Modification | |||
| Perlecan | Anti-proliferation | unknown | [25]. |
| Biglycan | Pro-inflammation | unknown | [26] |
| Gal-1 | Anti-proliferation | suppresses PDGF induced response | [27] |
| Matrix mediating cytokine | |||
| TGFβ1 | Matrix production Proliferation | Fibronectin and collagen production, DNA synthesis | [28] |
| Thrombin | Migration | MMP inhibition | [29] |
| Matrix Element | Effect | Mechanism | Reference |
|---|---|---|---|
| Glycoprotein | |||
| Fibronectin | Anti-adhesive | Cytoskeletal reorganization | [31,32] |
| Laminin | Anti-adhesive | Inhibit SMC binding to collagen | [31] |
| Elastin | Elastin metabolism | Reduced IEL fragmentation enforces barrier integrity against migration | [33,34] |
| Proteoglycan | |||
| Chondroitin sulfate | Elastin assembly | Release elastin binding protein reduces decreases elastin fiber assembly | [35] |
| Dermatan sulfate | Elastin metabolism | Release elastin binding protein reduces decreases elastin fiber assembly | [35] |
| Glycosaminoglycan | |||
| Hyaluronan | Migration | The influx of water loosens and expands the subendothelial region, SMC binds to hyaluronan through hyaluronan binding protein | [32,36] |
| ECM Mediator | Effect | Mechanism | Reference |
|---|---|---|---|
| Matrix binding receptor | |||
| Integrin | Adhesion Inhibit migration | Increases SMC adhesion to LN | [31] |
| Matrix degrading enzymes | |||
| t-PA | Elastin metabolism | Increases MMP-2 and MMP-9 expression that promotes elastic laminae degradation | [37] |
| LOX | Elastin formation | Catalyze elastin crosslink | [23] |
| Matrix production cytokines | |||
| PGE2 | Hyaluronan deposition | Induces hyaluronan synthase type 2 mRNA | [38] |
| IL-5 | Inhibit proliferation, matrix production | Decreased SMC proliferation and hyaluronan production | [39] |
| TGFβ1 | Adhesion | Increased focal adhesion plaque formation and integrin receptors expression | [40,41] |
| BMP9,10 | Differentiation, matrix production | Bmp9 knockout in mice led to imperfect closure of the DA. Promotes intimal cell differentiation, ECM deposition | [42] |
| Others | |||
| Tropo-elastin | Elastin formation | Decreases elastin binding protein expression | [35] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-Y.; Yeh, J.-L.; Hsu, J.-H. Role of Extracellular Matrix in Pathophysiology of Patent Ductus Arteriosus: Emphasis on Vascular Remodeling. Int. J. Mol. Sci. 2020, 21, 4761. https://doi.org/10.3390/ijms21134761
Lin T-Y, Yeh J-L, Hsu J-H. Role of Extracellular Matrix in Pathophysiology of Patent Ductus Arteriosus: Emphasis on Vascular Remodeling. International Journal of Molecular Sciences. 2020; 21(13):4761. https://doi.org/10.3390/ijms21134761
Chicago/Turabian StyleLin, Ting-Yi, Jwu-Lai Yeh, and Jong-Hau Hsu. 2020. "Role of Extracellular Matrix in Pathophysiology of Patent Ductus Arteriosus: Emphasis on Vascular Remodeling" International Journal of Molecular Sciences 21, no. 13: 4761. https://doi.org/10.3390/ijms21134761
APA StyleLin, T.-Y., Yeh, J.-L., & Hsu, J.-H. (2020). Role of Extracellular Matrix in Pathophysiology of Patent Ductus Arteriosus: Emphasis on Vascular Remodeling. International Journal of Molecular Sciences, 21(13), 4761. https://doi.org/10.3390/ijms21134761