Role of Human Leukocyte Antigens at the Feto-Maternal Interface in Normal and Pathological Pregnancy: An Update

 ,
, 
Abstract
1. Introduction
2. Methods
3. Mechanisms of Fetal Immune Escape from Maternal Recognition
3.1. Anatomy of Maternal–Fetal Interface
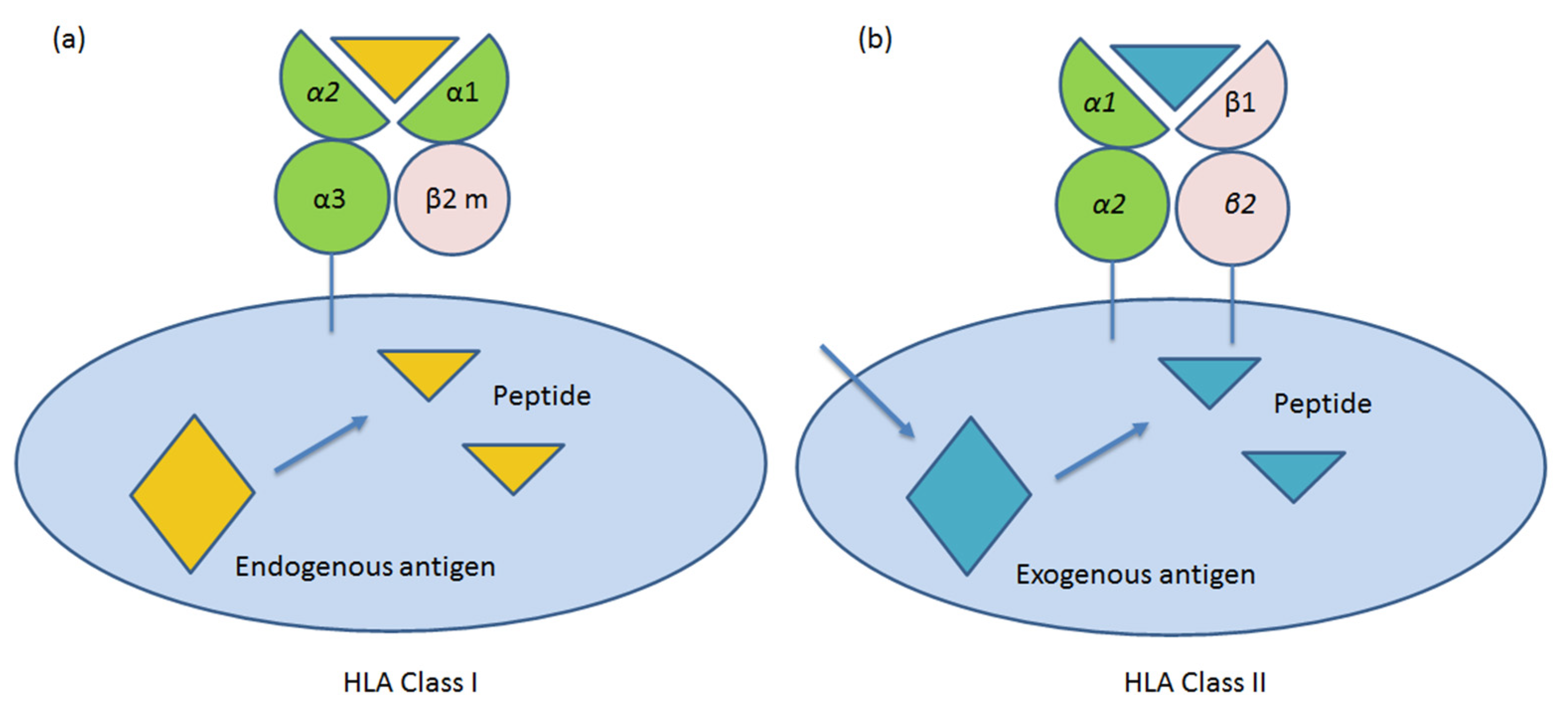
3.2. Peculiar Expression of HLA Class I and II in the Human Placenta
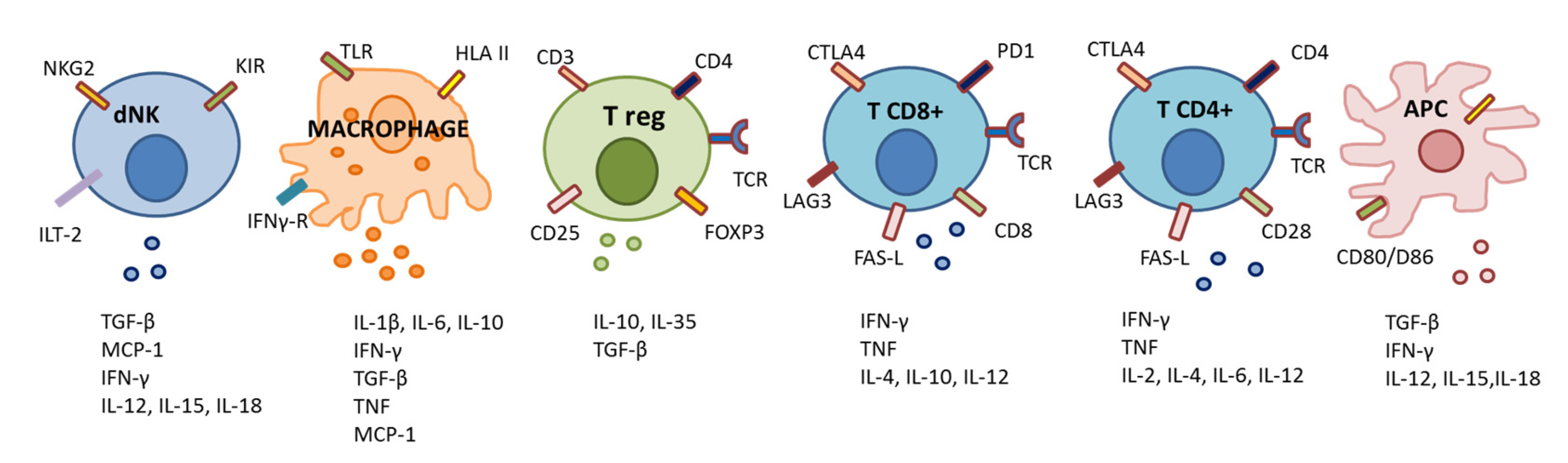
3.3. Other Mechanism of Modulation of Maternal Cell-Mediated Immunity
4. Abnormal Expression of Placental HLA Molecules in Obstetric Syndromes
4.1. Miscarriage
4.2. Pre-Eclampsia
4.3. Preterm Delivery
4.4. Others
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CT | Cytotrophoblast |
| ST | Syncytiotrophoblast |
| EVT | Extravillous trophoblast |
| HLA | Human Leukocyte Antigen |
| MHC | Major Histocompatibility Complex |
| dNK | Decidual natural killer cells |
| EVs | Extracellular vesicles |
| STEVs | Syncytiotrophoblast-derived extracellular vesicles |
| PE | Pre-eclampsia |
| KIRs | Killer cell Ig-like receptors |
References
- Brosens, I.; Puttemans, P.; Benagiano, G. Placental bed research: I. The placental bed: From spiral arteries remodeling to the great obstetrical syndromes. Am. J. Obstet. Gynecol. 2019, 221, 437–456. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sargent, I.L. Immunology of pre-eclampsia. Am. J. Reprod. Immunol. 2010, 63, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Moser, G.; Windsperger, K.; Pollheimer, J.; de Sousa Lopes, S.C.; Huppertz, B. Human trophoblast invasion: New and unexpected routes and functions. Histochem. Cell Biol. 2018, 150, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Tannetta, D.; Collett, G.; Vatish, M.; Redman, C.; Sargent, I. Syncytiotrophoblast extracellular vesicles—Circulating biopsies reflecting placental health. Placenta 2017, 52, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Parham, P.; Norman, P.J.; Abi-Rached, L.; Hilton, H.G.; Guethlein, L.A. Review: Immunogenetics of human placentation. Placenta 2012, 33, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.; Jauniaux, E. What is the placenta? Am. J. Obstet. Gynecol. 2015, 213, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Ander, S.E.; Diamond, M.S.; Coyne, C.B. Immune responses at the maternal-fetal interface. Sci. Immunol. 2019, 4, 6114. [Google Scholar] [CrossRef]
- Parham, P. Molecular definition of the transplantation antigens. FEBS J. 2018, 285, 2728–2745. [Google Scholar] [CrossRef]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef]
- Blees, A.; Januliene, D.; Hofmann, T.; Koller, N.; Schmidt, C.; Trowitzsch, S.; Moeller, A.; Tampé, R. Structure of the human MHC-I peptide-loading complex. Nature 2017, 551, 525–528. [Google Scholar] [CrossRef]
- Moffett, A.; Chazara, O.; Colucci, F. Maternal allo-recognition of the fetus. Fertil. Steril. 2017, 107, 1269–1272. [Google Scholar] [CrossRef] [PubMed]
- Apps, R.; Murphy, S.P.; Fernando, R.; Gardner, L.; Ahad, T.; Moffett, A. Human leucocyte antigen (HLA) expression of primary trophoblast cells and placental cell lines, determined using single antigen beads to characterize allotype specificities of anti-HLA antibodies. Immunology 2009, 127, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.P.; Tomasi, T.B. Absence of MHC class II antigen expression in trophoblast cells results from a lack of class II transactivator (CIITA) gene expression. Mol. Reprod. Dev. 1998, 51, 1–12. [Google Scholar] [CrossRef]
- Datema, G.; Van Meir, C.A.; Kanhai, H.H.; Van Den Elsen, P.J. Pre-term birth and severe pre-eclampsia are not associated with altered expression of HLA on human trophoblasts. Am. J. Reprod. Immunol. 2003, 49, 193–201. [Google Scholar] [CrossRef]
- Papúchová, H.; Meissner, T.B.; Li, Q.; Strominger, J.L.; Tilburgs, T. The Dual Role of HLA-C in Tolerance and Immunity at the Maternal-Fetal Interface. Front. Immunol. 2019, 10, 2730. [Google Scholar] [CrossRef]
- Olmos-Ortiz, A.; Flores-Espinosa, P.; Mancilla-Herrera, I.; Vega-Sánchez, R.; Díaz, L.; Zaga-Clavellina, V. Innate Immune Cells and Toll-like Receptor-Dependent Responses at the Maternal-Fetal Interface. Int. J. Mol. Sci. 2019, 20, 3654. [Google Scholar] [CrossRef]
- Sharkey, A.M.; Gardner, L.; Hiby, S.; Farrell, L.; Apps, R.; Masters, L.; Goodridge, J.; Lathbury, L.; Stewart, C.A.; Verma, S.; et al. Killer Ig-like receptor expression in uterine NK cells is biased toward recognition of HLA-C and alters with gestational age. J. Immunol. 2008, 181, 39–46. [Google Scholar] [CrossRef]
- Mincheva-Nilsson, L.; Nagaeva, O.; Chen, T.; Stendahl, U.; Antsiferova, J.; Mogren, I.; Hernestål, J.; Baranov, V. Placenta-derived soluble MHC class I chain-related molecules down-regulate NKG2D receptor on peripheral blood mononuclear cells during human pregnancy: A possible novel immune escape mechanism for fetal survival. J. Immunol. 2006, 176, 3585–3592. [Google Scholar] [CrossRef]
- Hedlund, M.; Stenqvist, A.C.; Nagaeva, O.; Kjellberg, L.; Wulff, M.; Baranov, V.; Mincheva-Nilsson, L. Human placenta expresses and secretes NKG2D ligands via exosomes that down-modulate the cognate receptor expression: Evidence for immunosuppressive function. J. Immunol. 2009, 183, 340–351. [Google Scholar] [CrossRef]
- Van der Zwan, A.; Bi, K.; Norwitz, E.R.; Crespo, A.C.; Claas, F.H.J.; Strominger, J.L.; Tilburgs, T. Mixed signature of activation and dysfunction allows human decidual CD8(+) T cells to provide both tolerance and immunity. Proc. Natl. Acad. Sci. USA 2018, 115, 385–390. [Google Scholar] [CrossRef]
- Tilburgs, T.; Roelen, D.L.; van der Mast, B.J.; van Schip, J.J.; Kleijburg, C.; de Groot-Swings, G.M.; Kanhai, H.H.; Claas, F.H.; Scherjon, S.A. Differential distribution of CD4+CD25bright and CD8+CD28− T-cells in decidua and maternal blood during human pregnancy. Placenta 2006, 27, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Tilburgs, T.; Crespo, Â.C.; van der Zwan, A.; Rybalov, B.; Raj, T.; Stranger, B.; Gardner, L.; Moffett, A.; Strominger, J.L. Human HLAG+ extravillous trophoblasts: Immune-activating cells that interact with decidual leukocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 7219–7224. [Google Scholar] [CrossRef] [PubMed]
- Svensson-Arvelund, J.; Mehta, R.B.; Lindau, R.; Mirrasekhian, E.; Rodriguez-Martinez, H.; Berg, G.; Lash, G.E.; Jenmalm, M.C.; Ernerudh, J. The human fetal placenta promotes tolerance against the semiallogeneic fetus by inducing regulatory T cells and homeostatic M2 macrophages. J. Immunol. 2015, 194, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.; James, E. Tumour and placenta establishment: The importance of antigen processing and presentation. Placenta 2017, 56, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Persson, G.; Jørgensen, N.; Nilsson, L.L.; Andersen, L.H.J.; Hviid, T.V.F. A role for both HLA-F and HLA-G in reproduction and during pregnancy? Hum. Immunol. 2020, 81, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Le Page, M.E.; Goodridge, J.P.; John, E.; Christiansen, F.T.; Witt, C.S. Killer Ig-like receptor 2DL4 does not mediate NK cell IFN-γ responses to soluble HLA-G preparations. J. Immunol. 2014, 192, 732–740. [Google Scholar] [CrossRef]
- Pistoia, V.; Morandi, F.; Wang, X.; Ferrone, S. Soluble HLAG: Are they clinically relevant? In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2007; Volume 17, pp. 469–479. [Google Scholar] [CrossRef]
- Fournel, S.; Aguerre-Girr, M.; Huc, X.; Lenfant, F.; Alam, A.; Toubert, A.; Bensussan, A.; Le Bouteiller, P. Cutting edge: Soluble HLA-G1 triggers CD95/CD95 ligand-mediated apoptosis in activated CD8+ cells by interacting with CD8. J. Immunol. 2000, 164, 6100–6104. [Google Scholar] [CrossRef]
- Naji, A.; Menier, C.; Morandi, F.; Agaugué, S.; Maki, G.; Ferretti, E.; Bruel, S.; Pistoia, V.; Carosella, E.D.; Rouas-Freiss, N. Binding of HLA-G to ITIM-bearing Ig-like transcript 2 receptor suppresses B cell Responses. J. Immunol. 2014, 192, 1536–1546. [Google Scholar] [CrossRef]
- Morandi, F.; Rizzo, R.; Fainardi, E.; Rouas-Freiss, N.; Pistoia, V. Recent Advances in Our Understanding of HLA-G Biology: Lessons from a Wide Spectrum of Human Diseases. J. Immunol. Res. 2016, 4326495. [Google Scholar] [CrossRef]
- Morandi, F.; Rouas-Freiss, N.; Pistoia, V. The emerging role of soluble HLA-G in the control of chemotaxis. Cytokine Growth Factor Rev. 2014, 25, 327–335. [Google Scholar] [CrossRef]
- Baudhuin, J.; Migraine, J.; Faivre, V.; Loumagne, L.; Lukaszewicz, A.C.; Payen, D.; Favier, B. Exocytosis acts as a modulator of the ILT4-mediated inhibition of neutrophil functions. Proc. Natl. Acad. Sci. USA 2013, 110, 17957–17962. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, V.M.; Straszewski-Chavez, S.L.; Guller, S.; Mor, G. First trimester trophoblast cells secrete Fas ligand which induces immune cell apoptosis. Mol. Hum. Reprod. 2004, 10, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Piao, H.L.; Wang, S.C.; Tao, Y.; Fu, Q.; Du, M.R.; Li, D.J. CXCL12/CXCR4 signal involved in the regulation of trophoblasts on peripheral NK cells leading to Th2 bias at the maternal-fetal interface. Eur Rev. Med. Pharmacol Sci. 2015, 19, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hao, S.; Chen, X.; Zhao, H.; Du, L.; Ren, H.; Wang, C.; Mao, H. Human placental trophoblast cells contribute to maternal-fetal tolerance through expressing IL-35 and mediating iTR35 conversion. Nat. Commun. 2019, 10, 4601. [Google Scholar] [CrossRef]
- Saito, S.; Sakai, M.; Sasaki, Y.; Nakashima, A.; Shiozaki, A. Inadequate tolerance induction may induce pre-eclampsia. J. Reprod. Immunol. 2007, 76, 30–39. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Placental oxidative stress: From miscarriage to preeclampsia. J. Soc. Gynecol. Investig. 2004, 11, 342–352. [Google Scholar] [CrossRef]
- Ford, H.B.; Schust, D.J. Recurrent pregnancy loss: Etiology, diagnosis, and therapy. Rev. Obstet. Gynecol. 2009, 2, 76–83. [Google Scholar]
- Wang, X.; Li, B.; Wang, J.; Lei, J.; Liu, C.; Ma, Y.; Zhao, H. Evidence That miR-133a Causes Recurrent Spontaneous Abortion by Reducing HLA-G Expression. Reprod. Biomed. Online 2012, 25, 415–424. [Google Scholar] [CrossRef]
- Craenmehr, M.H.C.; Nederlof, I.; Cao, M.; Drabbels, J.J.M.; Spruyt-Gerritse, M.J.; Anholts, J.D.H.; Kapsenberg, H.M.; Stegehuis, J.A.; van der Keur, C.; Fasse, E.; et al. Increased HLA-G Expression in Term Placenta of Women with a History of Recurrent Miscarriage Despite Their Genetic Predisposition to Decreased HLA-G Levels. Int. J. Mol. Sci. 2019, 20, 625. [Google Scholar] [CrossRef]
- Seshadri, S.; Sunkara, S.K. Natural killer cells in female infertility and recurrent miscarriage: A systematic review and meta-analysis. Hum. Reprod. Update 2014, 20, 429–438. [Google Scholar] [CrossRef]
- Meuleman, T.; van Beelen, E.; Kaaja, R.J.; van Lith, J.M.; Claas, F.H.; Bloemenkamp, K.W. HLA-C antibodies in women with recurrent miscarriage suggests that antibody mediated rejection is one of the mechanisms leading to recurrent miscarriage. J. Reprod. Immunol. 2016, 116, 28–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Inada, K.; Shima, T.; Ito, M.; Ushijima, A.; Saito, S. Helios-positive functional regulatory T cells are decreased in decidua of miscarriage cases with normal fetal chromosomal content. J. Reprod. Immunol. 2015, 107, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.I.; Jost, M.; Spratte, J.; Schaier, M.; Mahnke, K.; Meuer, S.; Zeier, M.; Steinborn, A. Differentiation of ICOS+ and ICOS− recent thymic emigrant regulatory T cells (RTE T regs) during normal pregnancy, pre-eclampsia and HELLP syndrome. Clin. Exp. Immunol. 2016, 183, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Tian, M.; Hu, X.; Wang, L.; Ji, J.; Liao, A. The altered PD-1/PD-L1 pathway delivers the “one-two punch” effects to promote the Treg/Th17 imbalance in pre-eclampsia. Cell. Mol. Immunol. 2018, 15, 710–723. [Google Scholar] [CrossRef]
- Athanassakis, I.; Aifantis, Y.; Makrygiannakis, A.; Koumantakis, E.; Vassiliadis, S. Placental tissue from human miscarriages expresses class II HLA-DR antigens. Am. J. Reprod. Immunol. 1995, 34, 281–287. [Google Scholar] [CrossRef]
- Labarrere, C.A.; Faulk, W.P. Class II reactivity of human villous trophoblast in chronic inflammation of unestablished etiology. Transplantation 1990, 50, 812–816. [Google Scholar] [CrossRef]
- Abalos, E.; Cuesta, C.; Carroli, G.; Qureshi, Z.; Widmer, M.; Vogel, J.P.; Souza, J.P. Who Multicountry Survey on Maternal and Newborn Health Research Network. Preeclampsia, eclampsia and adverse maternal and perinatal outcomes: A secondary analysis of the World Health Organization multicountry survey on maternal and newborn health. BJOG 2014, 121, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hiby, S.E.; Walker, J.J.; O’Shaughnessy, K.M.; Redman, C.W.; Carrington, M.; Trowsdale, J.; Moffet, A. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J. Exp. Med. 2004, 200, 957–965. [Google Scholar] [CrossRef]
- Larsen, T.G.; Hackmon, R.; Geraghty, D.E.; Hviid, T.V.F. Fetal human leukocyte antigen-C and maternal killer-cell immunoglobulin-like receptors in cases of severe preeclampsia. Placenta 2019, 75, 27–33. [Google Scholar] [CrossRef]
- Tersigni, C.; Redman, C.W.; Dragovic, R.; Tannetta, D.; Scambia, G.; Di Simone, N.; Sargent, I.; Vatish, M. HLA-DR is aberrantly expressed at feto-maternal interface in pre-eclampsia. J. Reprod. Immunol. 2018, 129, 48–52. [Google Scholar] [CrossRef]
- Labarrere, C.A.; Faulk, W.P. Intercellular adhesion molecule-1 (ICAM-1) and HLADR antigens are expressed on endovascular cytotrophoblasts in abnormal pregnancies. Am. J. Reprod. Immunol. 1995, 33, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Van den Elsen, P.J.; Gobin, S.J.; van der Stoep, N.; Datema, G.; Viëtor, H.E. Transcriptional control of MHC genes in fetal trophoblast cells. J. Reprod. Immunol. 2001, 52, 129–145. [Google Scholar] [CrossRef]
- Geirsson, A.; Paliwal, I.; Lynch, R.J.; Bothwell, A.L.; Hammond, G.L. Class II transactivator promoter activity is suppressed through regulation by a trophoblast noncoding RNA. Transplantation 2003, 76, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.P.; Choi, J.C.; Holtz, R. Regulation of major histocompatibility complex class II gene expression in trophoblast cells. Reprod. Biol. Endocrinol. 2004, 2, 52. [Google Scholar] [CrossRef][Green Version]
- Geppert, T.D.; Lipsky, P.E. Antigen presentation by interferon-gamma-treated endothelial cells and fibroblasts: Differential ability to function as antigen presenting cells despite comparable. J. Immunol. 1985, 135, 3750–3762. [Google Scholar]
- Stout, M.J.; Cao, B.; Landeau, M.; French, J.; Macones, G.A.; Mysorekar, I.U. Increased human leukocyte antigen-G expression at the maternal-fetal interface is associated with preterm birth. J. Matern. Fetal Neonatal Med. 2015, 28, 454–459. [Google Scholar] [CrossRef]
- Jauniaux, E.; Burton, G.J. Pathophysiology of placenta accreta spectrum disorders: A review of current findings. Clin. Obstet. Gynecol. 2018, 61, 743–754. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| HLA-A, -B | HLA-C | HLA-G | HLA-E | HLA-DP | HLA-DQ | HLA-DR | |
|---|---|---|---|---|---|---|---|
| Fetal tissues 1 | + | + | − | + | + | + | + |
| ST 2 | − | − | − | − | − | − | − |
| VT | − | − | − | − | − | − | − |
| EVT | − | + | + | + | − | − | − |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tersigni, C.; Meli, F.; Neri, C.; Iacoangeli, A.; Franco, R.; Lanzone, A.; Scambia, G.; Di Simone, N. Role of Human Leukocyte Antigens at the Feto-Maternal Interface in Normal and Pathological Pregnancy: An Update. Int. J. Mol. Sci. 2020, 21, 4756. https://doi.org/10.3390/ijms21134756
Tersigni C, Meli F, Neri C, Iacoangeli A, Franco R, Lanzone A, Scambia G, Di Simone N. Role of Human Leukocyte Antigens at the Feto-Maternal Interface in Normal and Pathological Pregnancy: An Update. International Journal of Molecular Sciences. 2020; 21(13):4756. https://doi.org/10.3390/ijms21134756
Chicago/Turabian StyleTersigni, Chiara, Federica Meli, Caterina Neri, Azzurra Iacoangeli, Rita Franco, Antonio Lanzone, Giovanni Scambia, and Nicoletta Di Simone. 2020. "Role of Human Leukocyte Antigens at the Feto-Maternal Interface in Normal and Pathological Pregnancy: An Update" International Journal of Molecular Sciences 21, no. 13: 4756. https://doi.org/10.3390/ijms21134756
APA StyleTersigni, C., Meli, F., Neri, C., Iacoangeli, A., Franco, R., Lanzone, A., Scambia, G., & Di Simone, N. (2020). Role of Human Leukocyte Antigens at the Feto-Maternal Interface in Normal and Pathological Pregnancy: An Update. International Journal of Molecular Sciences, 21(13), 4756. https://doi.org/10.3390/ijms21134756