Signaling Through Purinergic Receptor P2Y2 Enhances Macrophage IL-1β Production


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
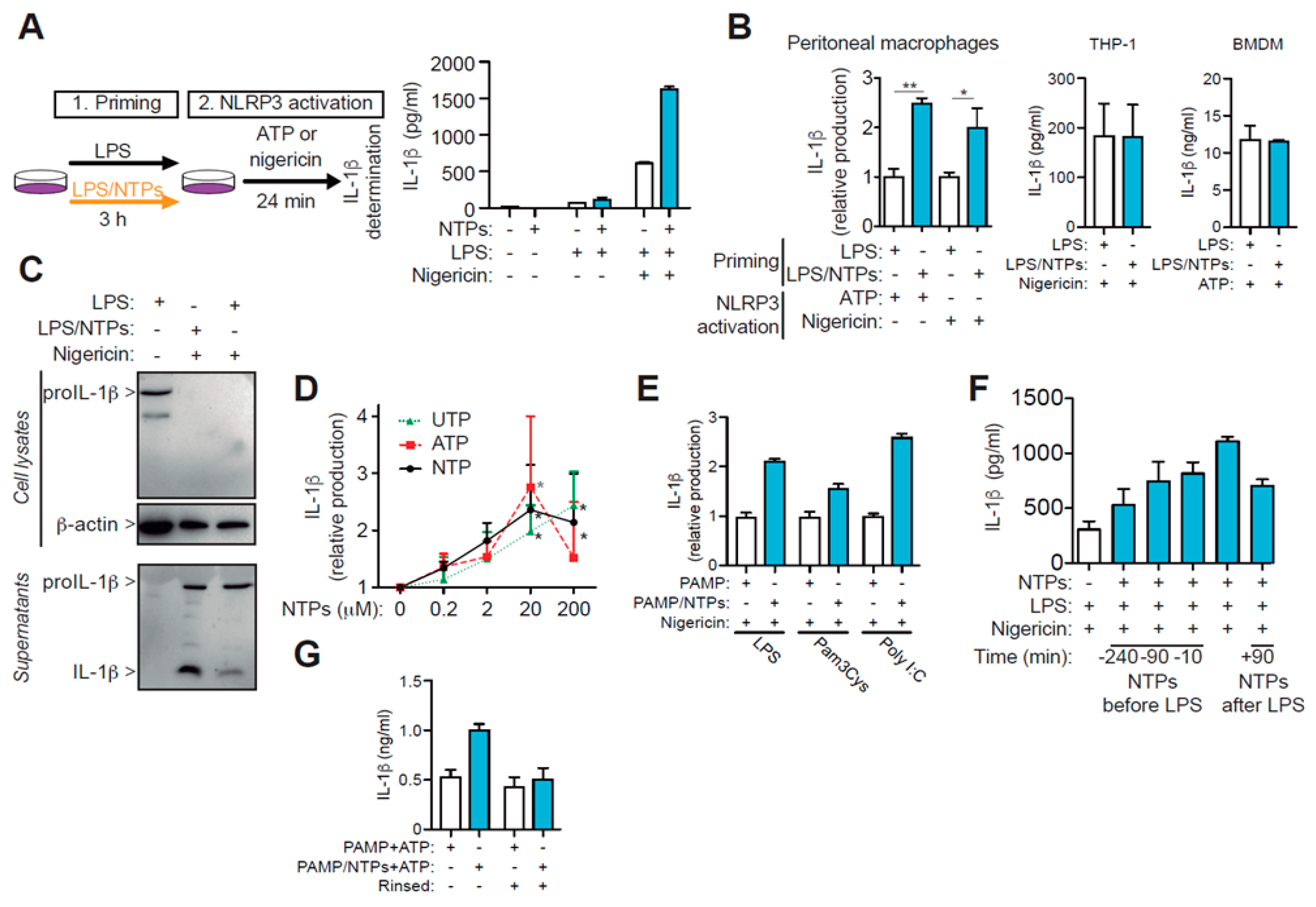
2.1. ATP and UTP Nucleotides Enhance IL-1β Production by Macrophages
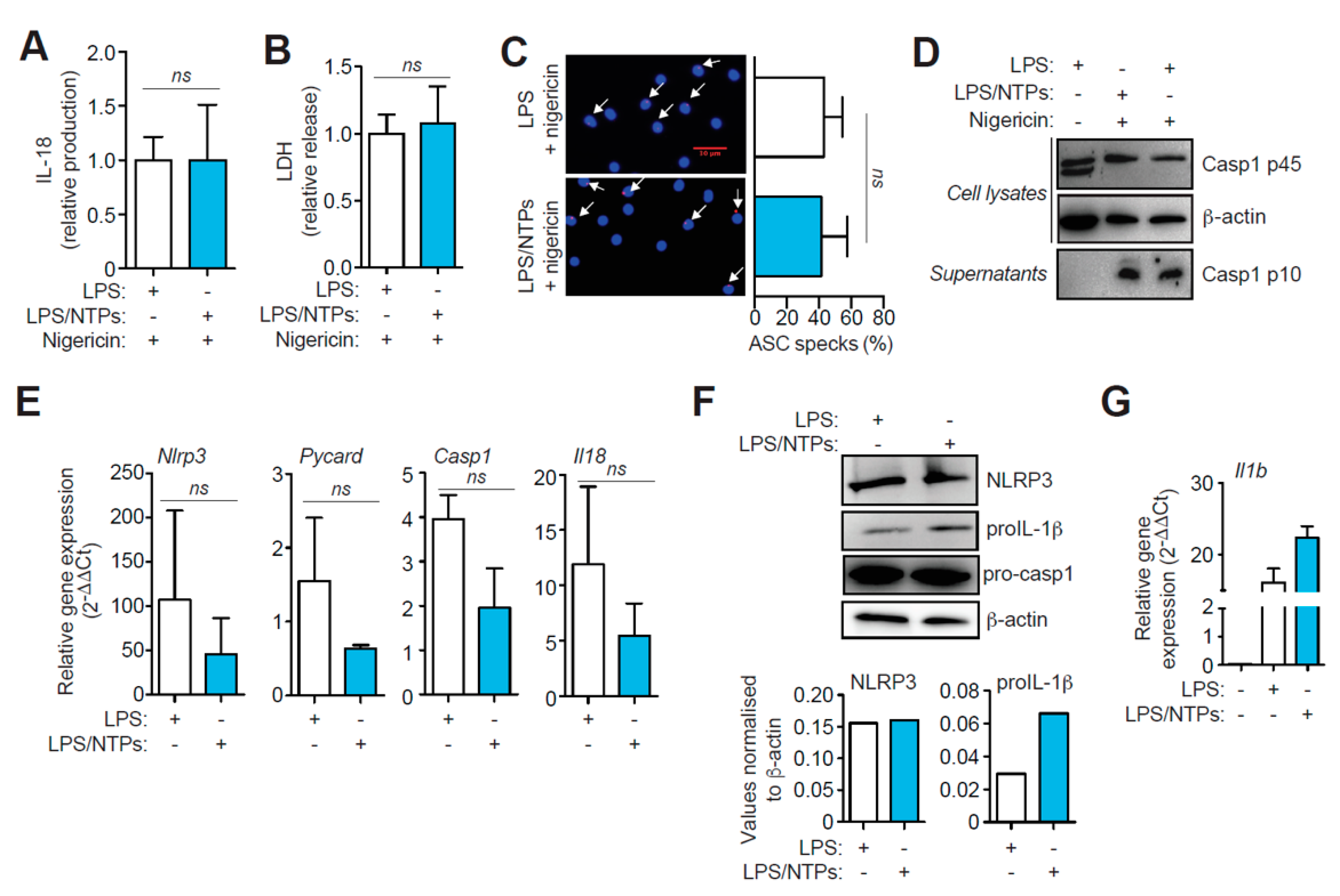
2.2. Nucleotides Increase Il1b Gene Expression, but Do not Affect NLRP3 Priming nor Activation
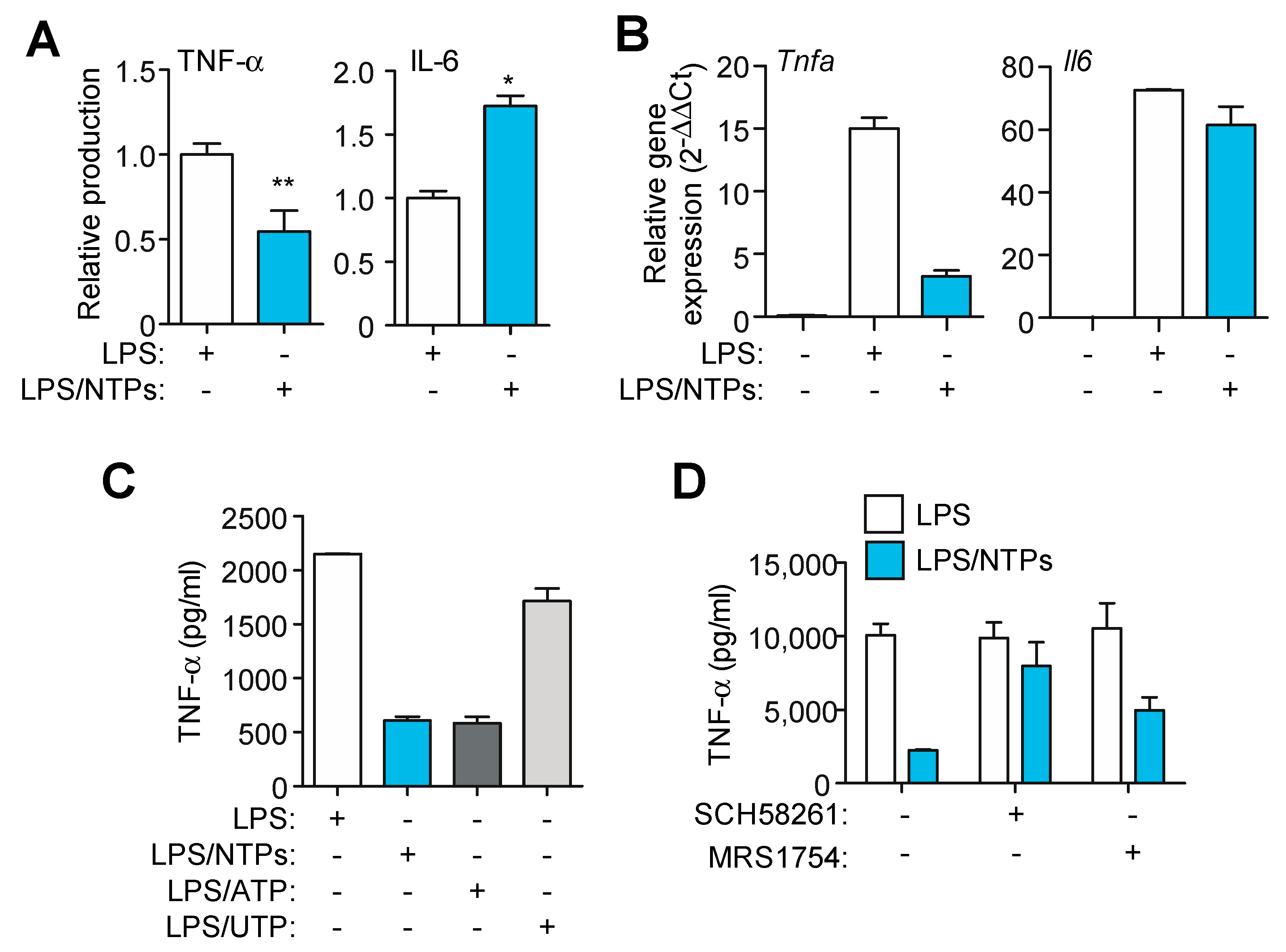
2.3. Nucleotides Induce a Specific Proinflammatory Signature during LPS Priming
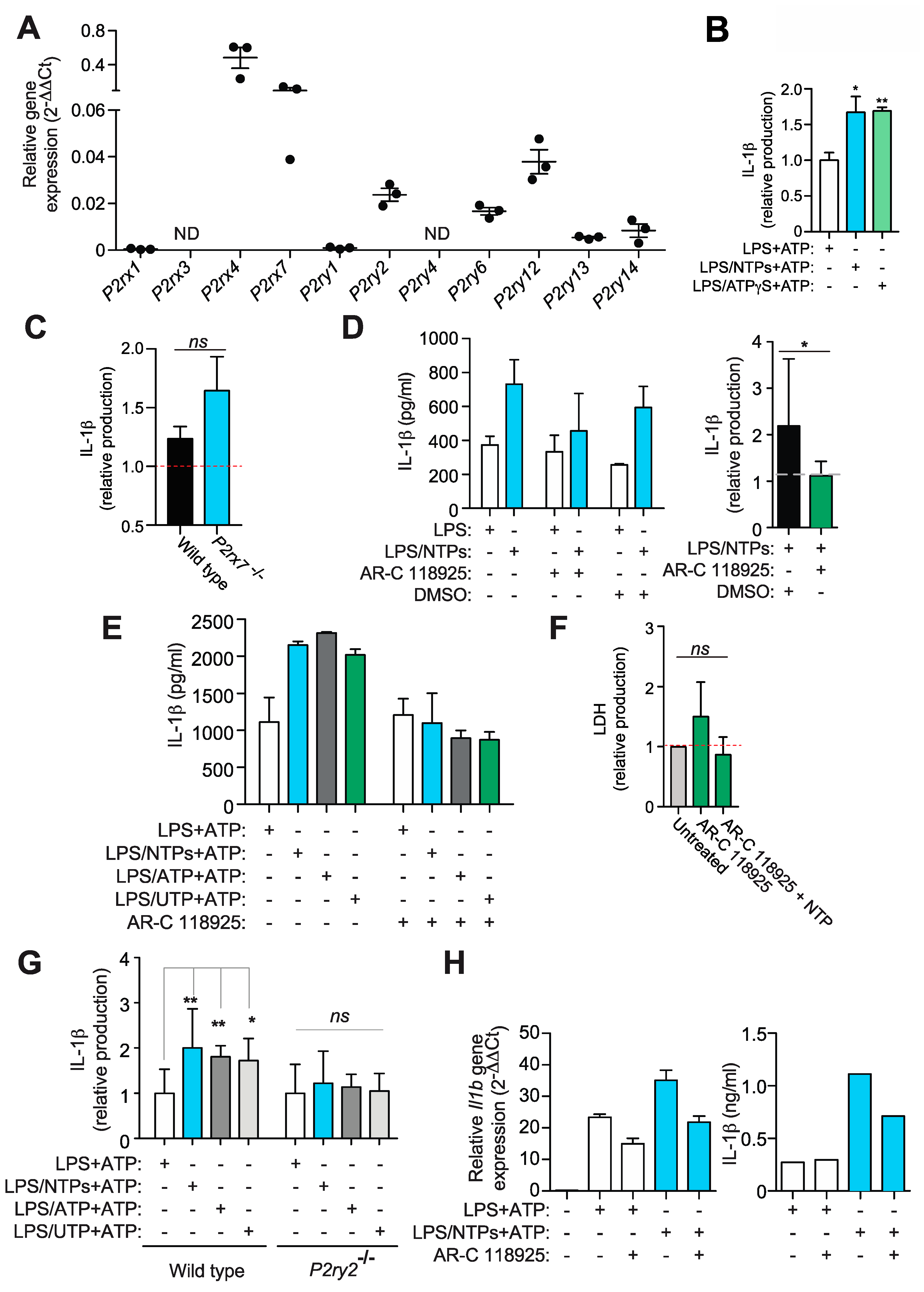
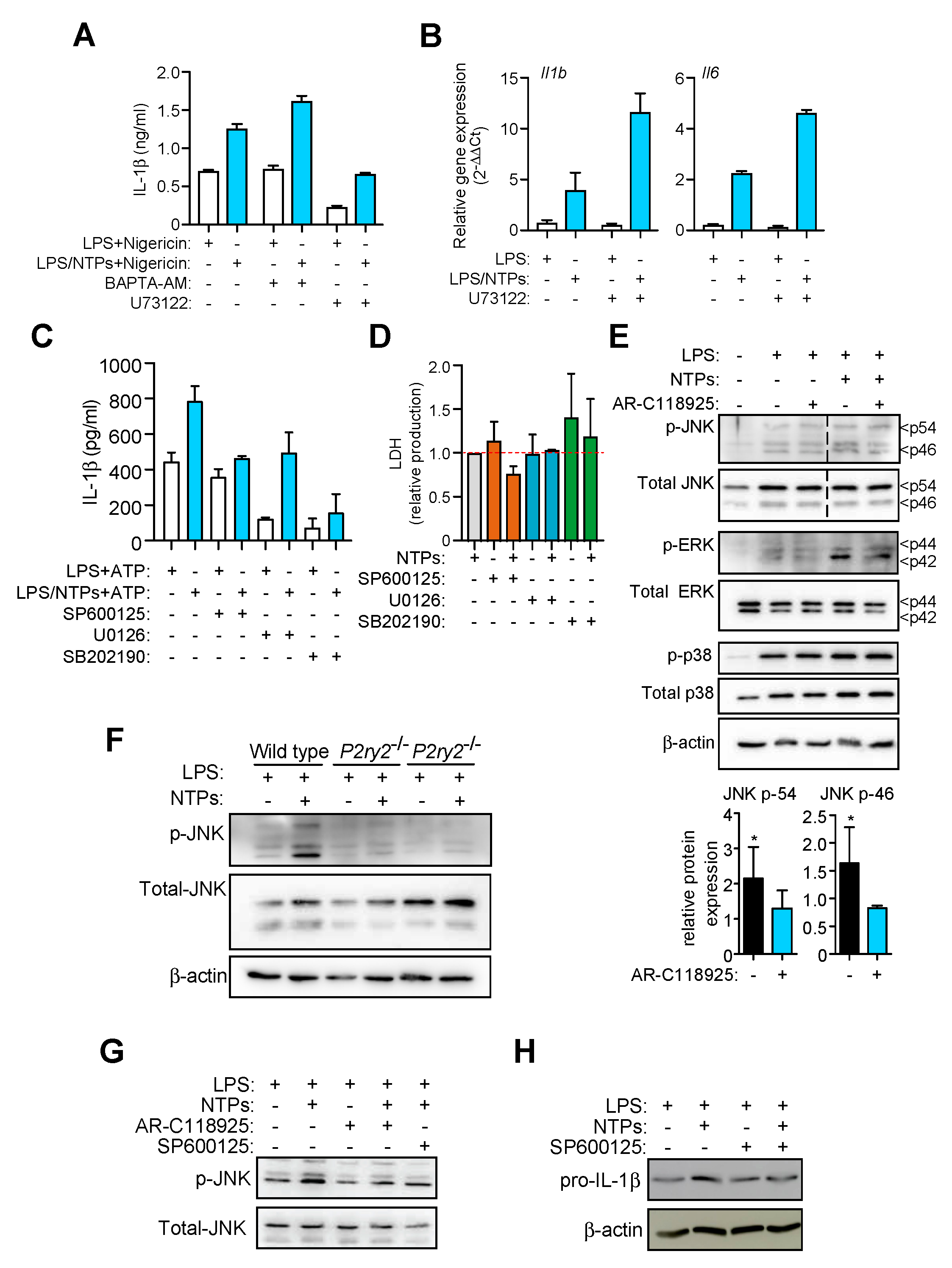
2.4. Nucleotides Activate P2Y2 Purinergic Receptor to Increase IL-1β Production
2.5. P2Y2R-Induced JNK Activation is Responsible for Increased in IL-1β Production
2.6. High Cell Density Disables Nucleotide-Induced IL-1β Production
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Isolation and Culture of Macrophages
4.4. LDH Determination
4.5. ELISA
4.6. Microscopy
4.7. Quantitative Reverse Transcriptase-PCR Analysis
4.8. Western Blot Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP-γS | Adenosine 5′-O-(3-thiotriphosphate) |
| BMDM | Bone marrow derived macrophages |
| eATP | Extracellular adenosine 5’-triphosphate |
| IL | Interleukin |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| NTPs | Nucleotides, in this work a mixture of ATP and UTP |
| P2X7R | Purinergic P2X receptor 7 |
| P2Y2R | Purinergic P2Y2 receptor |
| PAMP | Pathogen associated molecular patterns |
| PLC | Phospholipase C |
| RPMs | Residential peritoneal macrophages |
| TLR | Toll-like receptors |
References
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nat. 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Brož, P.; Pelegrin, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2019, 20, 143–157. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef]
- Semino, C.; Carta, S.; Gattorno, M.; Sitia, R.; Rubartelli, A. Progressive waves of IL-1β release by primary human monocytes via sequential activation of vesicular and gasdermin D-mediated secretory pathways. Cell Death Dis. 2018, 9, 1088. [Google Scholar] [CrossRef]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef]
- Martinez-García, J.J.; Martínez-Banaclocha, H.; Angosto-Bazarra, D.; De Torre-Minguela, C.; Baroja-Mazo, A.; Alarcón-Vila, C.; Martinez-Alarcon, L.; Amores-Iniesta, J.; Martín-Sánchez, F.; Ercole, G.A.; et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovitch, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Hamidzadeh, K.; Mosser, D.M. Purinergic Signaling to Terminate TLR Responses in Macrophages. Front. Immunol. 2016, 7, 197. [Google Scholar] [CrossRef]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kubes, P. A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell 2016, 165, 668–678. [Google Scholar] [CrossRef]
- Ghosn, E.E.B.; Cassado, A.A.; Govoni, G.R.; Fukuhara, T.; Yang, Y.; Monack, D.M.; Bortoluci, K.R.; Almeida, S.; Herzenberg, L.A.; Herzenberg, L.A. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc. Natl. Acad. Sci. USA 2010, 107, 2568–2573. [Google Scholar] [CrossRef]
- Stokes, L.; Surprenant, A. Purinergic P2Y2 receptors induce increased MCP-1/CCL2 synthesis and release from rat alveolar and peritoneal macrophages. J. Immunol. 2007, 179, 6016–6023. [Google Scholar] [CrossRef] [PubMed]
- Ivar, K.V.; Harden, T.K. Molecular Pharmacology, Physiology, and Structure of the P2Y Receptors. Adv. Pharmacol. 2011, 61, 373–415. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Dixon, C.J.; White, P.J.; Hall, J.F.; Kingston, S.; Boarder, M.R. Regulation of Human Hepatocytes by P2Y Receptors: Control of Glycogen Phosphorylase, Ca2+, and Mitogen-Activated Protein Kinases. J. Pharmacol. Exp. Ther. 2005, 313, 1305–1313. [Google Scholar] [CrossRef]
- Hao, Y.; Liang, J.F.; Chow, A.W.; Cheung, W.-T.; Ko, W.-H. P2Y6 Receptor-Mediated Proinflammatory Signaling in Human Bronchial Epithelia. PLoS ONE 2014, 9, e106235. [Google Scholar] [CrossRef]
- Taylor, P.R.; Martínez-Pomares, L.; Stacey, M.; Lin, H.-H.; Brown, G.D.; Gordon, S. MACROPHAGE RECEPTORS AND IMMUNE RECOGNITION. Annu. Rev. Immunol. 2005, 23, 901–944. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Ouyang, X.; Ghani, A.; Malik, A.; Wilder, T.; Colegio, O.; Flavell, R.A.; Cronstein, B.; Mehal, W. Adenosine is required for sustained inflammasome activation via the A₂A receptor and the HIF-1α pathway. Nat. Commun. 2013, 4, 2909. [Google Scholar] [CrossRef] [PubMed]
- Sipka, S.; Kovács, I.; Szántó, S.; Szegedi, G.; Brugós, L.; Bruckner, G.; József, S.A. Adenosine inhibits the release of interleukin-1beta in activated human peripheral mononuclear cells. Cytokine 2005, 31, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Uratsuji, H.; Tada, Y.; Kawashima, T.; Kamata, M.; Hau, C.S.; Asano, Y.; Sugaya, M.; Kadono, T.; Asahina, A.; Sato, S.; et al. P2Y6 Receptor Signaling Pathway Mediates Inflammatory Responses Induced by Monosodium Urate Crystals. J. Immunol. 2011, 188, 436–444. [Google Scholar] [CrossRef]
- Gombault, A.; Baron, L.; Couillin, I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 2013, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Surprenant, A.; North, R.A. Signaling at Purinergic P2X Receptors. Annu. Rev. Physiol. 2009, 71, 333–359. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Ben, D.D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.S.; Wang, J.; Bhagwat, S.; Munger, J.; Janssen, W.J.; Wright, T.W.; Elliott, M.R. CD73 regulates anti-inflammatory signaling between apoptotic cells and endotoxin-conditioned tissue macrophages. Cell Death Differ. 2017, 24, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Vounotrypidis, P.; Kouklakis, G.; Anagnostopoulos, K.; Zezos, P.; Polychronidis, A.; Maltezos, E.; Efremidou, E.; Pitiakoudis, M.; Lyratzopoulos, N. Interleukin-1 associations in inflammatory bowel disease and the enteropathic seronegative spondylarthritis. Autoimmun. Highlights 2013, 4, 87–94. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef]
- Wen, H.; Ting, J.P.-Y.; O’Neill, L.A.J. A role for the NLRP3 inflammasome in metabolic diseases—Did Warburg miss inflammation? Nat. Immunol. 2012, 13, 352–357. [Google Scholar] [CrossRef] [PubMed]
- A Sim, J.; Park, C.-K.; Oh, S.B.; Evans, R.J.; A North, R. P2X1and P2X4receptor currents in mouse macrophages. Br. J. Pharmacol. 2007, 152, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, H.; Fujiwaki, T.; Tsukimoto, M.; Kawano, A.; Harada, H.; Kojima, S. P2X4 receptor regulates P2X7 receptor-dependent IL-1β and IL-18 release in mouse bone marrow-derived dendritic cells. Biochem. Biophys. Res. Commun. 2013, 432, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-C.; Choi, C.H.; Najwane, S.-S.; Larry, J.; Kalina, R.A.; Hanen, S.; Özlem, Y.; David, M.O. P2X4 Assembles with P2X7 and Pannexin-1 in Gingival Epithelial Cells and Modulates ATP-induced Reactive Oxygen Species Production and Inflammasome Activation. PLoS ONE 2013, 8, e70210. [Google Scholar] [CrossRef]
- Stachon, P.; Geis, S.; Peikert, A.; Heidenreich, A.; Michel, N.A.; Ünal, F.; Hoppe, N.; Dufner, B.; Schulte, L.; Marchini, T.; et al. Extracellular ATP Induces Vascular Inflammation and Atherosclerosis via Purinergic Receptor Y2in MiceHighlights. Arter. Thromb. Vasc. Boil. 2016, 36, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Adamson, S.E.; Montgomery, G.; Seaman, S.A.; Peirce-Cottler, S.M.; Leitinger, N. Myeloid P2Y2 receptor promotes acute inflammation but is dispensable for chronic high-fat diet-induced metabolic dysfunction. Purinergic Signal. 2017, 14, 19–26. [Google Scholar] [CrossRef]
- Jin, H.; Ko, Y.S.; Kim, H.J. P2Y2R-mediated inflammasome activation is involved in tumor progression in breast cancer cells and in radiotherapy-resistant breast cancer. Int. J. Oncol. 2018, 53, 1953–1966. [Google Scholar] [CrossRef]
- Del Rey, A.; Renigunta, V.; Dalpke, A.H.; Leipziger, J.; Matos, J.E.; Robaye, B.; Zuzarte, M.; Kavelaars, A.; Hanley, P.J. Knock-out Mice Reveal the Contributions of P2Y and P2X Receptors to Nucleotide-induced Ca2+ Signaling in Macrophages. J. Biol. Chem. 2006, 281, 35147–35155. [Google Scholar] [CrossRef]
- Isfort, K.; Ebert, F.; Bornhorst, J.; Sargin, S.; Kardakaris, R.; Pasparakis, M.; Bähler, M.; Schwerdtle, T.; Schwab, A.; Hanley, P.J. Real-time Imaging Reveals That P2Y2 and P2Y12 Receptor Agonists Are Not Chemoattractants and Macrophage Chemotaxis to Complement C5a Is Phosphatidylinositol 3-Kinase (PI3K)- and p38 Mitogen-activated Protein Kinase (MAPK)-independent. J. Biol. Chem. 2011, 286, 44776–44787. [Google Scholar] [CrossRef]
- Ding, L.; Ma, W.; Littmann, T.; Camp, R.; Shen, J. The P2Y2 Nucleotide Receptor Mediates Tissue Factor Expression in Human Coronary Artery Endothelial Cells. J. Biol. Chem. 2011, 286, 27027–27038. [Google Scholar] [CrossRef]
- Lee, A.H.; Ledderose, C.; Li, X.; Slubowski, C.J.; Sueyoshi, K.; Staudenmaier, L.; Bao, Y.; Zhang, J.; Junger, W.G. Adenosine Triphosphate Release is Required for Toll-Like Receptor-Induced Monocyte/Macrophage Activation, Inflammasome Signaling, Interleukin-1β Production, and the Host Immune Response to Infection. Crit. Care Med. 2018, 46, e1183–e1189. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, K.; Ledderose, C.; Shen, Y.; Lee, A.H.; Shapiro, N.I.; Junger, W.G. Lipopolysaccharide suppresses T cells by generating extracellular ATP that impairs their mitochondrial function via P2Y11 receptors. J. Biol. Chem. 2019, 294, 6283–6293. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Gimenez, L.; Renaudin, F.; Jalabert, M.; Gras, P.; Gosset, M.; Rey, C.; Sarda, S.; Collet, C.; Cohen-Solal, M.; Combes, C.; et al. Inflammatory Potential of Four Different Phases of Calcium Pyrophosphate Relies on NF-κB Activation and MAPK Pathways. Front. Immunol. 2018, 9, 2248. [Google Scholar] [CrossRef] [PubMed]
- Riera-Borrull, M.; Cuevas, V.D.; Alonso, B.; Vega, M.A.; Joven, J.; Izquierdo, E.; Corbí, A.L. Palmitate Conditions Macrophages for Enhanced Responses toward Inflammatory Stimuli via JNK Activation. J. Immunol. 2017, 199, 3858–3869. [Google Scholar] [CrossRef] [PubMed]
- Riteau, N.; Baron, L.; Villeret, B.; Guillou, N.; Savigny, F.; Ryffel, B.; Rassendren, F.; Le Bert, M.; Gombault, A.; Couillin, I. ATP release and purinergic signaling: A common pathway for particle-mediated inflammasome activation. Cell Death Dis. 2012, 3, e403. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, Z.-S.; Xue, W.; Bai, Z.-F.; Wang, Q.-Y.; Dai, J.; Liu, X.; Huang, Y.-J.; Cai, H.; Zhan, X.-Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197.e6. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK Expression by Macrophages Promotes Obesity-Induced Insulin Resistance and Inflammation. Science 2012, 339, 218–222. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Labasi, J.M.; Petrushova, N.; Donovan, C.; McCurdy, S.; Lira, P.; Payette, M.M.; Brissette, W.; Wicks, J.R.; Audoly, L.; Gabel, C.A. Absence of the P2X7 receptor alters leukocyte function and attenuates an inflammatory response. J. Immunol. 2002, 168, 6436–6445. [Google Scholar] [CrossRef]
- Compan, V.; Martín-Sánchez, F.; Baroja-Mazo, A.; Lopez-Castejon, G.; Gomez, A.I.; Verkhratsky, A.; Brough, D.; Pelegrin, P. Apoptosis-associated speck-like protein containing a CARD forms specks but does not activate caspase-1 in the absence of NLRP3 during macrophage swelling. J. Immunol. 2014, 194, 1261–1273. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Rosa, G.; Gómez, A.I.; Baños, M.C.; Pelegrín, P. Signaling Through Purinergic Receptor P2Y2 Enhances Macrophage IL-1β Production. Int. J. Mol. Sci. 2020, 21, 4686. https://doi.org/10.3390/ijms21134686
de la Rosa G, Gómez AI, Baños MC, Pelegrín P. Signaling Through Purinergic Receptor P2Y2 Enhances Macrophage IL-1β Production. International Journal of Molecular Sciences. 2020; 21(13):4686. https://doi.org/10.3390/ijms21134686
Chicago/Turabian Stylede la Rosa, Gonzalo, Ana I. Gómez, María C. Baños, and Pablo Pelegrín. 2020. "Signaling Through Purinergic Receptor P2Y2 Enhances Macrophage IL-1β Production" International Journal of Molecular Sciences 21, no. 13: 4686. https://doi.org/10.3390/ijms21134686
APA Stylede la Rosa, G., Gómez, A. I., Baños, M. C., & Pelegrín, P. (2020). Signaling Through Purinergic Receptor P2Y2 Enhances Macrophage IL-1β Production. International Journal of Molecular Sciences, 21(13), 4686. https://doi.org/10.3390/ijms21134686