Aldosterone Induces DNA Damage and Activation of Nrf2 Mainly in Tubuli of Mouse Kidneys



Abstract
1. Introduction
2. Results
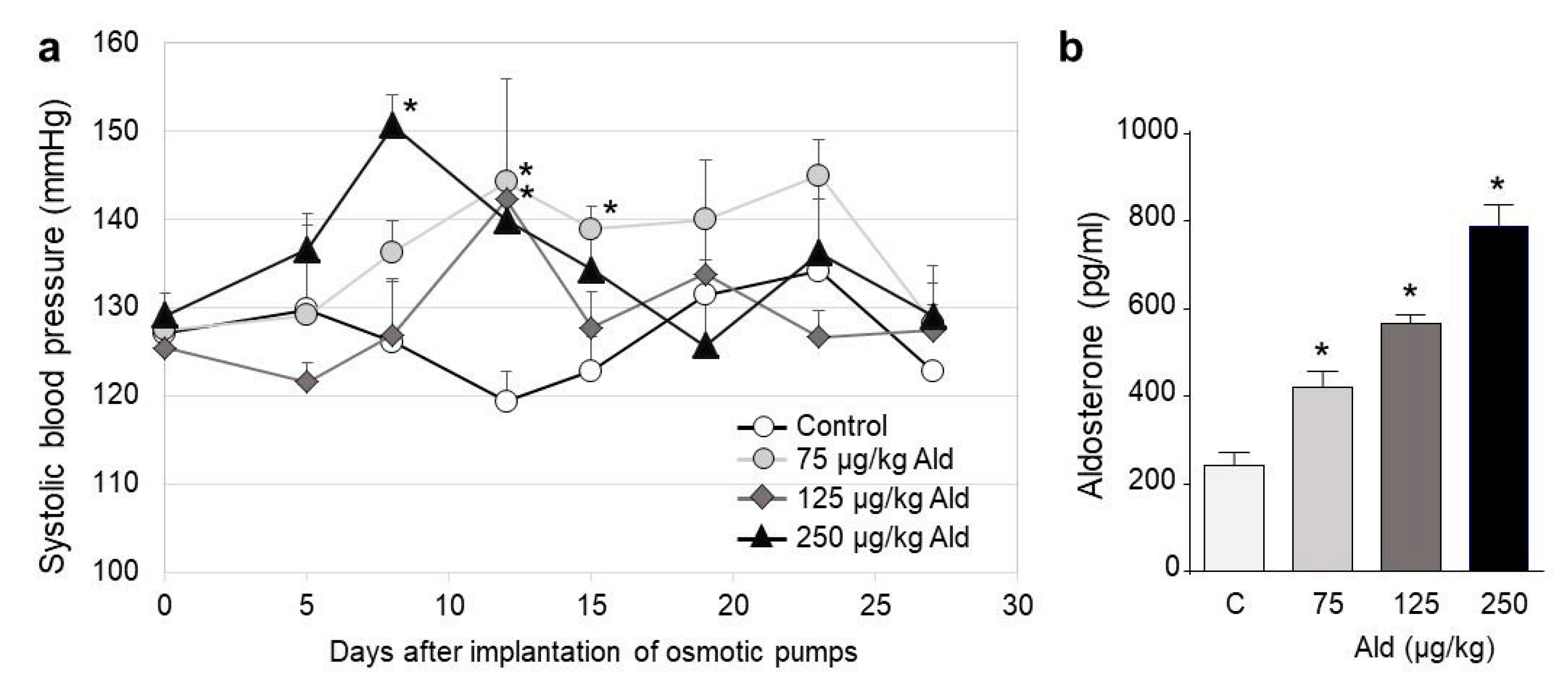
2.1. Blood Pressure Changes and Clinical Characteristics
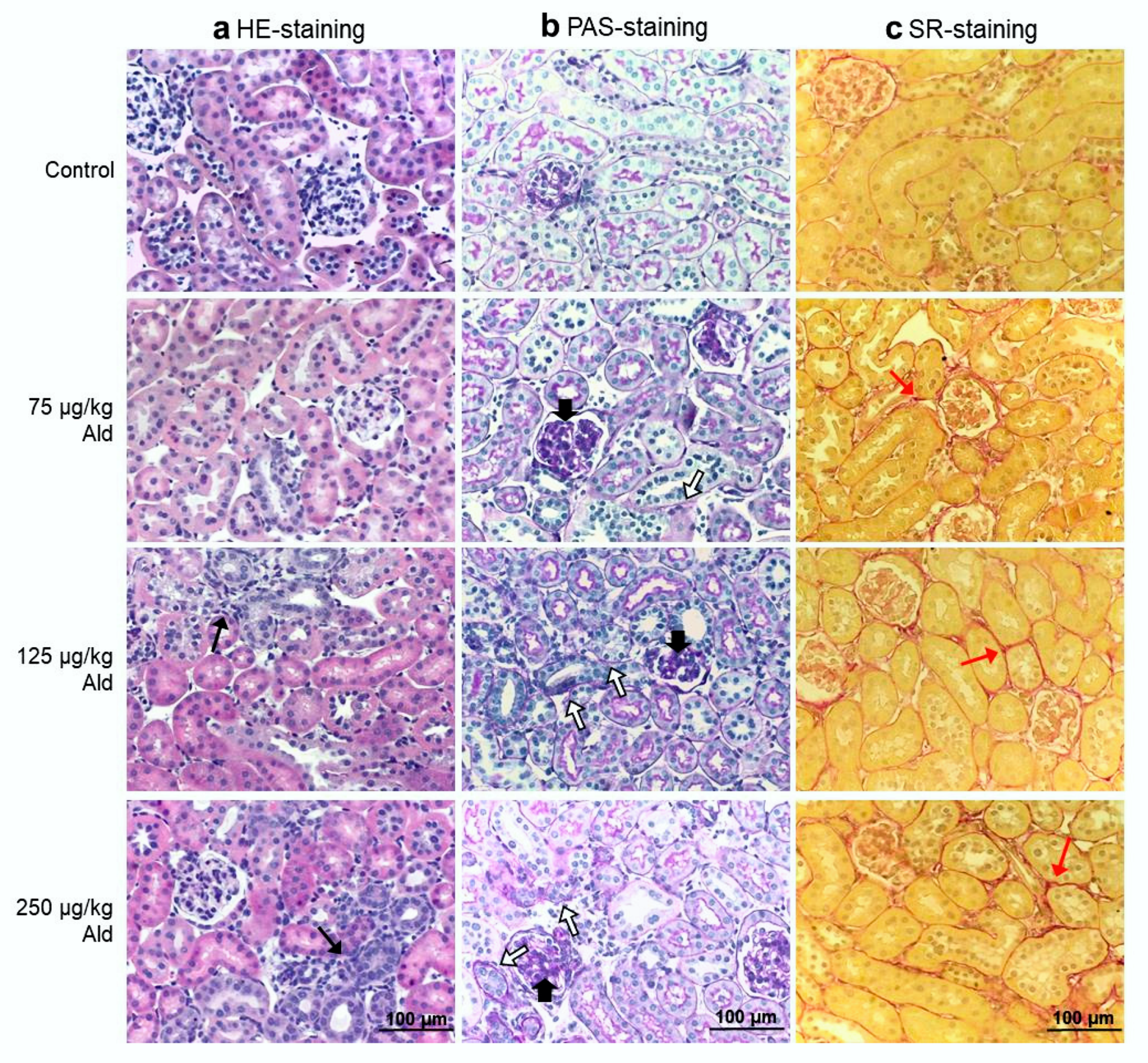
2.2. Renal Function, Histopathological Changes of the Kidney and Systemic Inflammatory Cytokines
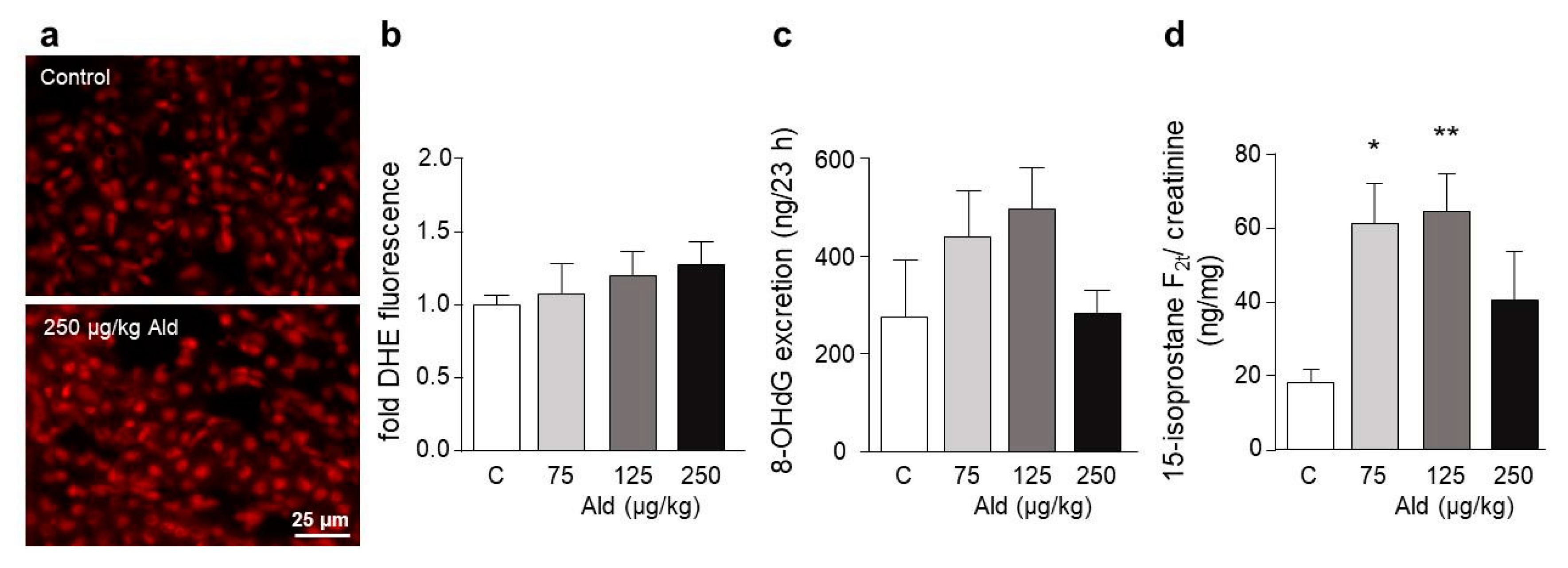
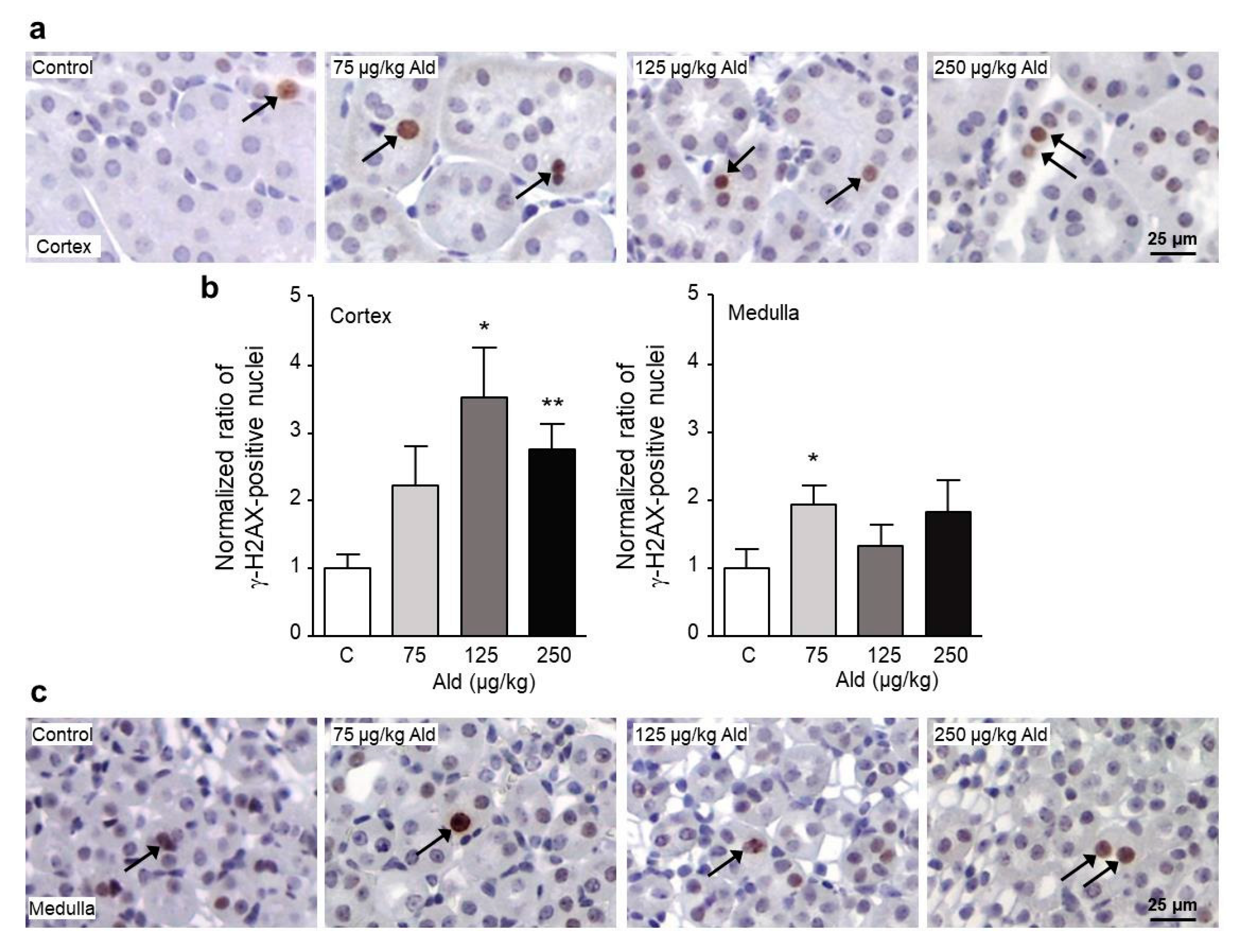
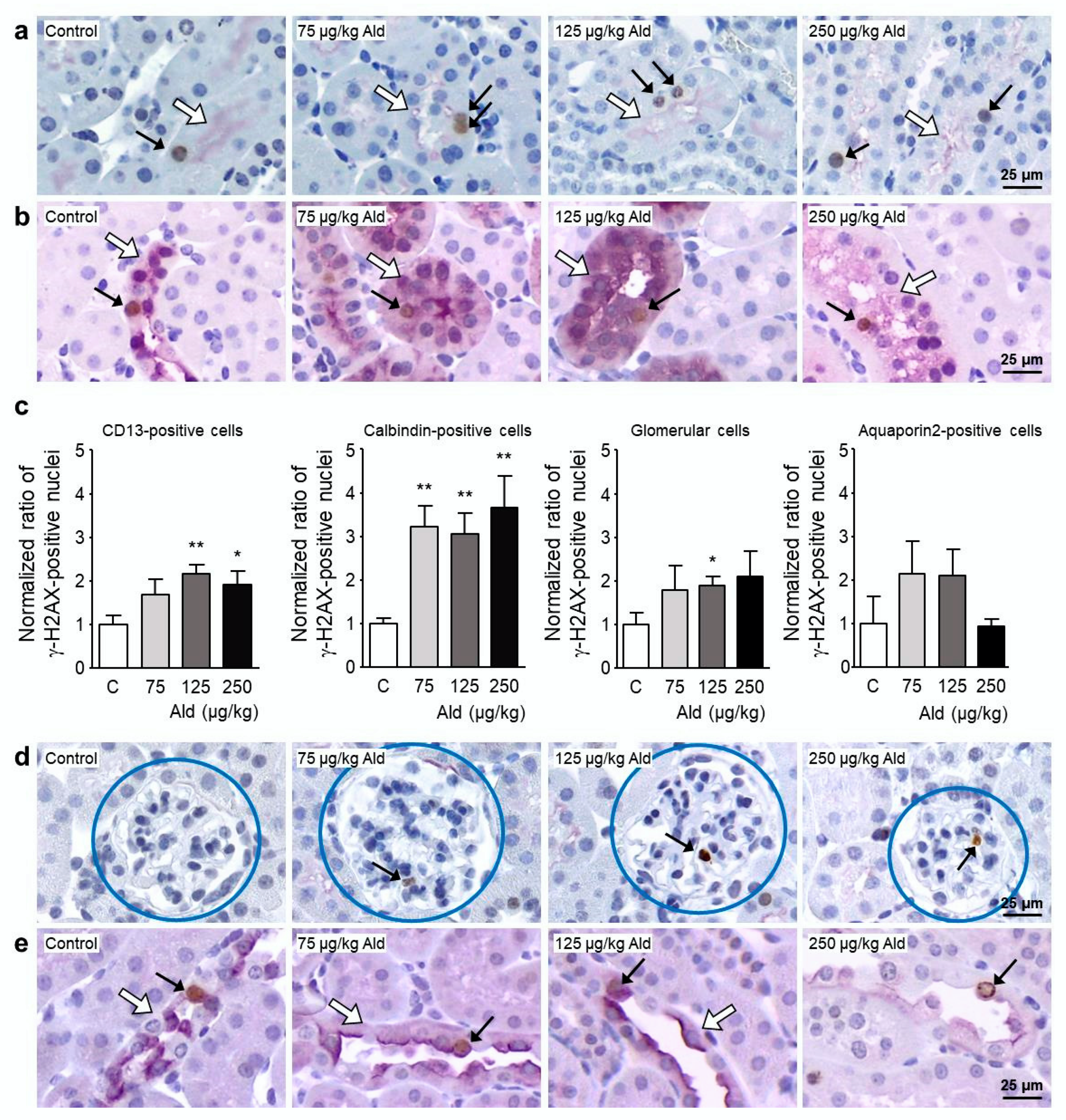
2.3. Oxidative Stress and DNA Damage Induced by Aldosterone in the Kidney
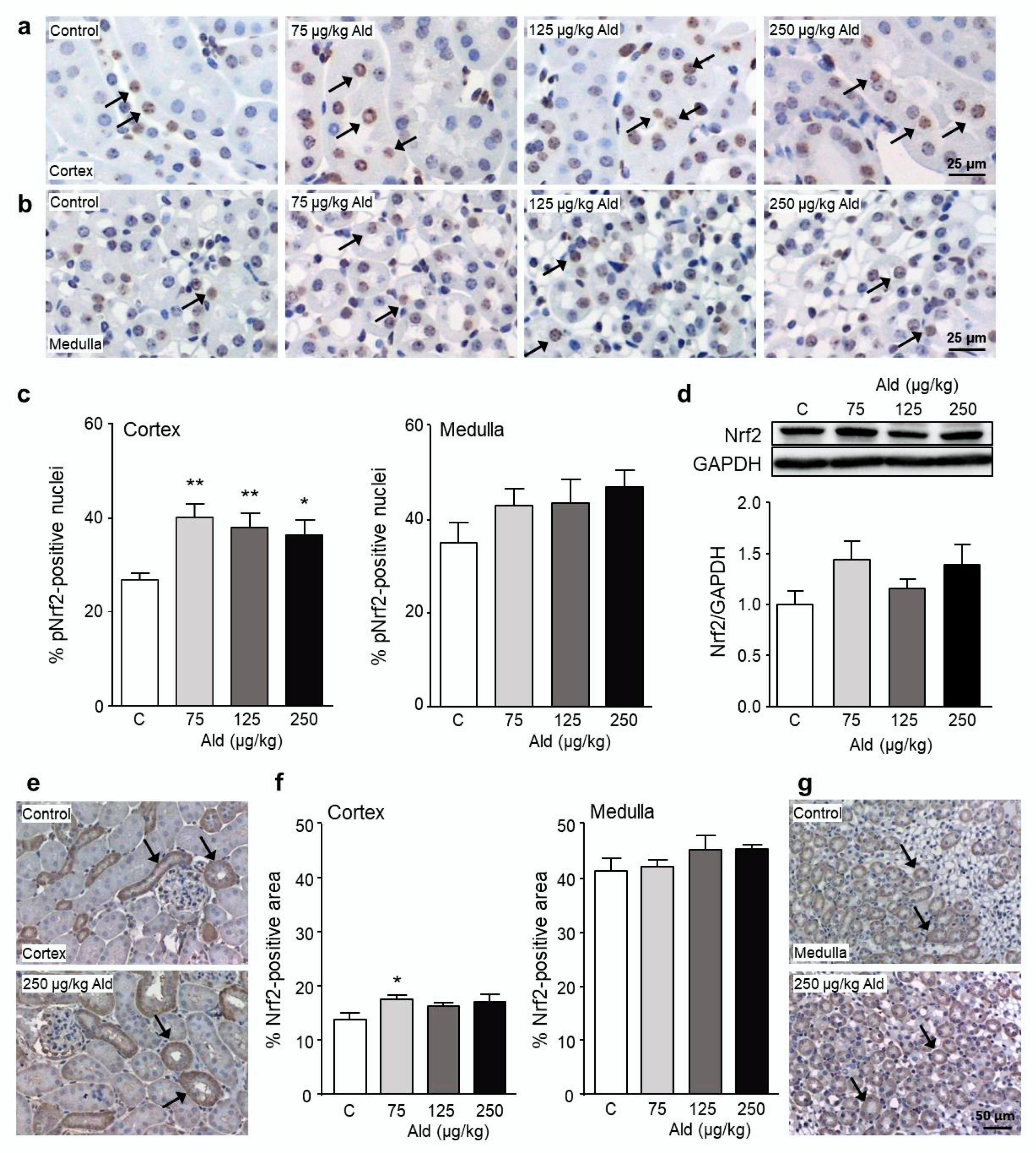
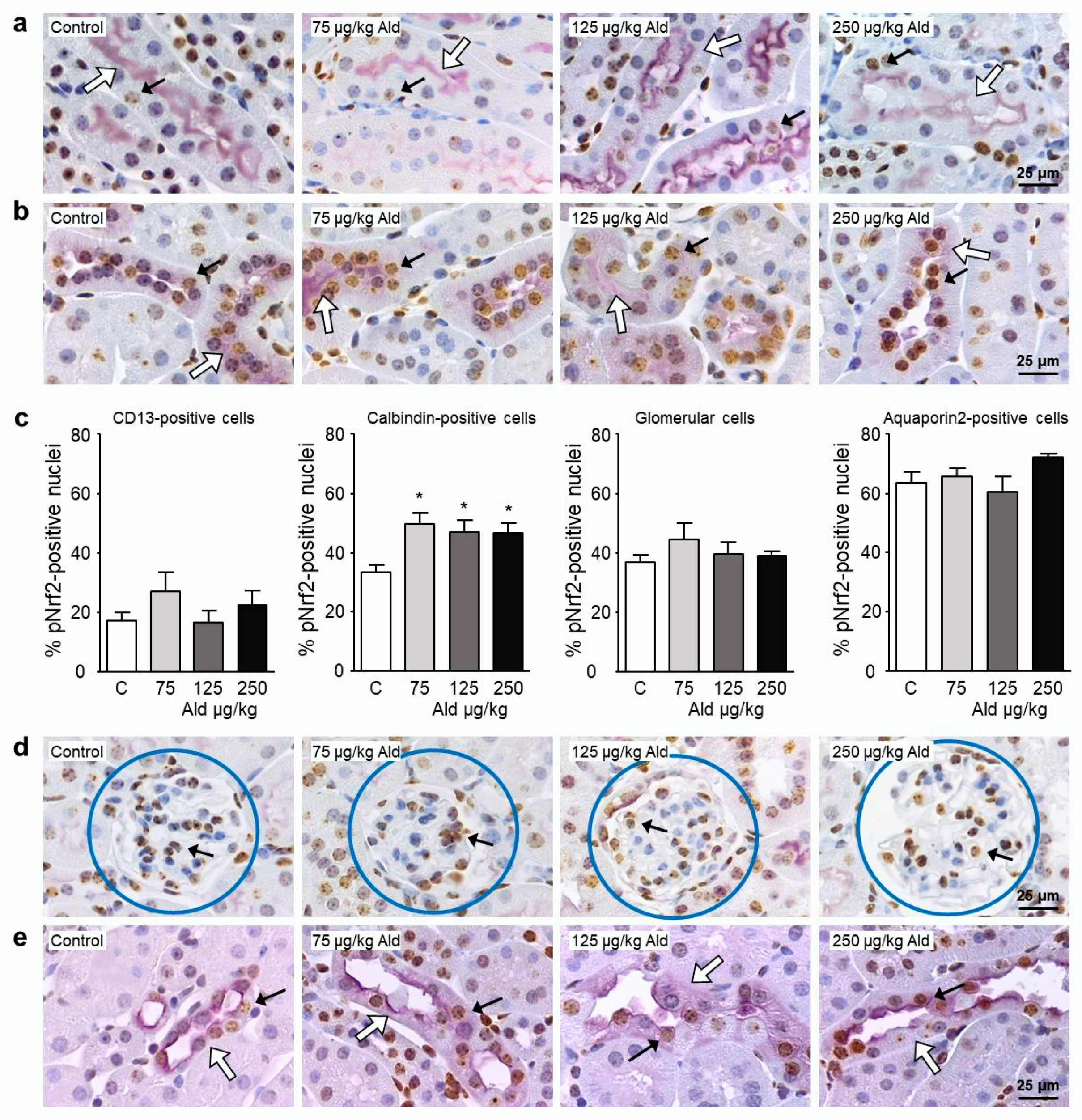
2.4. Abundance and Localization of Nrf2 in the Kidney
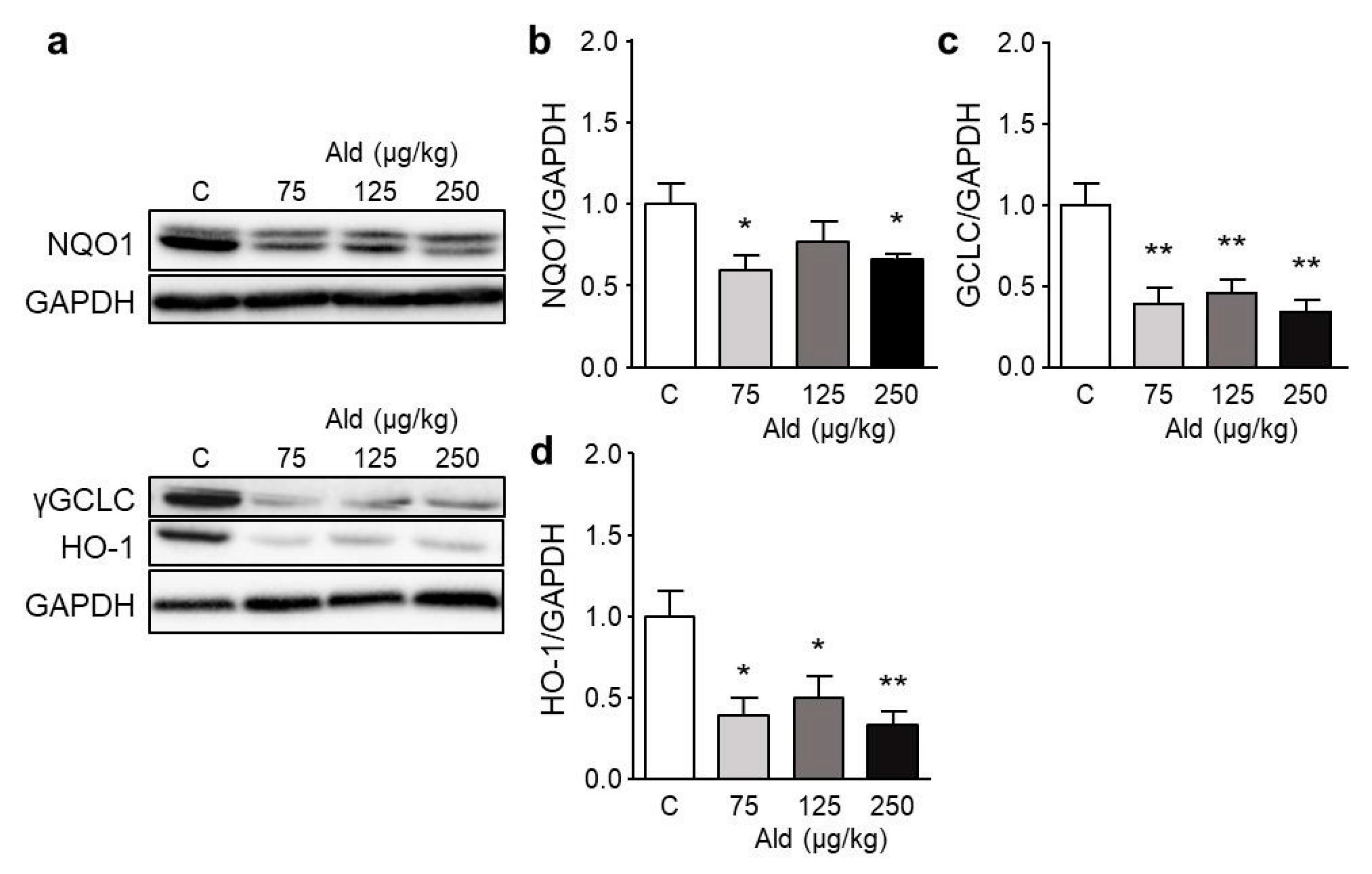
2.5. Effect of Aldosterone on Expression of Selected Genes and on Nrf2 Target Proteins
3. Discussion
4. Materials and Methods
4.1. Animal Treatment
4.2. Quantification of Aldosterone
4.3. Parameters of Renal Function
4.4. Histopathology
4.5. Immunohistochemistry
4.6. Double Staining
4.7. Detection of Reactive Oxygen Species
4.8. Quantification of 8-oxodG in Kidneys and 8-OHdG in Urine
4.9. Quantification of 15-Isoprostane F2t in Urine
4.10. Western Blot
4.11. Quantitative RT-PCR
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 8-OHdG | 8-hydroxy-2′-deoxyguanosine |
| 8-oxodG | 8-oxo-2′-deoxyguanosine |
| αENaC | α-subunit of the epithelial sodium channel |
| γ-H2AX | phosphorylated histone 2AX |
| ACEi | angiotensin converting enzyme inhibitors |
| Ald | aldosterone |
| Apex1 | apurinic/apyrimidinic endonuclease 1 |
| ARB | angiotensin II receptor blockers |
| ARE | antioxidant responsive element |
| ATF3 | activating transcription factor 3 |
| Atm | ataxia telangiectasia mutated homologue |
| Brca1 | breast cancer 1 |
| CD | cluster of differentiation |
| CKD | chronic kidney disease |
| DHE | dihydroethidium |
| DOCA | desoxycorticosterone acetate |
| EDTA | ethylenediaminetetraacetic acid |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GCLC | γ-glutamate cysteine ligase catalytic subunit |
| GCLM | γ-glutamate-cysteine ligase modifier subunit |
| GPx1 | glutathione peroxidase 1 |
| HE | hematoxylin and eosin stain |
| HO-1 | heme oxygenase 1 |
| IL | interleukin |
| Keap1 | Kelch-like ECH-associated protein 1 |
| Lig | ligase |
| Maf | musculoaponeurotic fibrosarcoma |
| NADPH | reduced nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells |
| NGAL | neutrophil gelatinase-associated lipocalin |
| Nox2 | NADPH oxidase 2 subunit p90 |
| NQO1 | NADPH quinone dehydrogenase 1 |
| Nrf2 | nuclear factor-erythroid-2-related factor 2 |
| Ogg1 | 8-oxoguanine DNA-glucosylase 1 |
| PARP | poly (ADP-ribose) polymerase |
| PAS | periodic acid–Schiff stain |
| PCNA | proliferating cell nuclear antigen |
| pNrf2 | phosphorylated nuclear factor-erythroid-2-related factor 2 |
| pp47phox | phosphorylated NADPH oxidase 2 activator |
| RAAS | renin-angiotensin-aldosterone system |
| ROS | reactive oxygen species |
| SEM | standard error mean |
| SOD1 | superoxide dismutase 1 |
| SR | Sirius red stain |
| TGF-β | transforming growth factor-β |
| TNF-α | tumor necrosis factor-α |
| TrxR1 | thioredoxin reductase 1 |
References
- Shavit, L.; Lifschitz, M.D.; Epstein, M. Aldosterone blockade and the mineralocorticoid receptor in the management of chronic kidney disease: Current concepts and emerging treatment paradigms. Kidney Int. 2012, 81, 955–968. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Girerd, S.; Jaisser, F. Mineralocorticoid receptor antagonists and kidney diseases: Pathophysiological basis. Kidney Int. 2019, 96, 302–319. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Esposito, E.; Attley, J.; Cuzzocrea, S. Targeting inflammation: New therapeutic approaches in chronic kidney disease (CKD). Pharmacol. Res. 2014, 81, 91–102. [Google Scholar] [CrossRef]
- Kuchta, A.; Pacanis, A.; Kortas-Stempak, B.; Cwiklinska, A.; Zietkiewicz, M.; Renke, M.; Rutkowski, B. Estimation of oxidative stress markers in chronic kidney disease. Kidney Blood Press. Res. 2011, 34, 12–19. [Google Scholar] [CrossRef]
- Dincer, Y.; Sekercioglu, N.; Pekpak, M.; Gunes, K.N.; Akcay, T. Assessment of DNA oxidation and antioxidant activity in hypertensive patients with chronic kidney disease. Ren. Fail. 2008, 30, 1006–1011. [Google Scholar] [CrossRef]
- Pellegrino, D.; La Russa, D.; Marrone, A. Oxidative Imbalance and Kidney Damage: New Study Perspectives from Animal Models to Hospitalized Patients. Antioxidants 2019, 8, 594. [Google Scholar] [CrossRef]
- Sindhu, R.K.; Ehdaie, A.; Farmand, F.; Dhaliwal, K.K.; Nguyen, T.; Zhan, C.D.; Roberts, C.K.; Vaziri, N.D. Expression of catalase and glutathione peroxidase in renal insufficiency. Biochim. Biophys. Acta 2005, 1743, 86–92. [Google Scholar] [CrossRef]
- Sahni, N.; Gupta, K.L.; Rana, S.V.; Prasad, R.; Bhalla, A.K. Intake of antioxidants and their status in chronic kidney disease patients. J. Ren. Nutr. 2012, 22, 389–399. [Google Scholar] [CrossRef]
- Tepel, M.; van der Giet, M.; Statz, M.; Jankowski, J.; Zidek, W. The antioxidant acetylcysteine reduces cardiovascular events in patients with end-stage renal failure: A randomized, controlled trial. Circulation 2003, 107, 992–995. [Google Scholar] [CrossRef]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef]
- Pedruzzi, L.M.; Cardozo, L.F.; Daleprane, J.B.; Stockler-Pinto, M.B.; Monteiro, E.B.; Leite, M., Jr.; Vaziri, N.D.; Mafra, D. Systemic inflammation and oxidative stress in hemodialysis patients are associated with down-regulation of Nrf2. J. Nephrol. 2015, 28, 495–501. [Google Scholar] [CrossRef]
- Jerotic, D.; Matic, M.; Suvakov, S.; Vucicevic, K.; Damjanovic, T.; Savic-Radojevic, A.; Pljesa-Ercegovac, M.; Coric, V.; Stefanovic, A.; Ivanisevic, J.; et al. Association of Nrf2, SOD2 and GPX1 Polymorphisms with Biomarkers of Oxidative Distress and Survival in End-Stage Renal Disease Patients. Toxins 2019, 11, 431. [Google Scholar] [CrossRef]
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Wang, Y.; Senitko, M.; Meyer, C.; Wigley, W.C.; Ferguson, D.A.; Grossman, E.; Chen, J.; Zhou, X.J.; Hartono, J.; et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARgamma, and HO-1. Am. J. Physiol. Ren. Physiol. 2011, 300, F1180–F1192. [Google Scholar] [CrossRef]
- Pergola, P.E.; Krauth, M.; Huff, J.W.; Ferguson, D.A.; Ruiz, S.; Meyer, C.J.; Warnock, D.G. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am. J. Nephrol. 2011, 33, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Agarwal, R.; Audhya, P.; Bakris, G.L.; Chin, M.; Krauth, M.; Lambers Heerspink, H.J.; Meyer, C.J.; McMurray, J.J.; et al. Rationale and trial design of Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes: The Occurrence of Renal Events (BEACON). Am. J. Nephrol. 2013, 37, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Reisman, S.A.; Bakris, G.L.; O’Grady, M.; Linde, P.G.; McCullough, P.A.; Packham, D.; Vaziri, N.D.; Ward, K.W.; Warnock, D.G.; et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 2014, 39, 499–508. [Google Scholar] [CrossRef]
- Queisser, N.; Amann, K.; Hey, V.; Habib, S.L.; Schupp, N. Blood pressure has only minor influence on aldosterone-induced oxidative stress and DNA damage in vivo. Free Radic. Biol. Med. 2013, 54, 17–25. [Google Scholar] [CrossRef]
- Queisser, N.; Oteiza, P.I.; Link, S.; Hey, V.; Stopper, H.; Schupp, N. Aldosterone activates transcription factor Nrf2 in kidney cells both in vitro and in vivo. Antioxid. Redox Signal. 2014, 21, 2126–2142. [Google Scholar] [CrossRef] [PubMed]
- Schupp, N.; Kolkhof, P.; Queisser, N.; Gärtner, S.; Schmid, U.; Kretschmer, A.; Hartmann, E.; Oli, R.G.; Schäfer, S.; Stopper, H. Mineralocorticoid receptor-mediated DNA damage in kidneys of DOCA-salt hypertensive rats. FASEB J. 2011, 25, 968–978. [Google Scholar] [CrossRef]
- Keidar, S.; Kaplan, M.; Pavlotzky, E.; Coleman, R.; Hayek, T.; Hamoud, S.; Aviram, M. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: A possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation 2004, 109, 2213–2220. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Miana, M.; Reboul, P.; Cachofeiro, V.; Martinez-Martinez, E.; de Boer, R.A.; Poirier, F.; Lacolley, P.; Zannad, F.; Rossignol, P.; et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Waeckel, L.; Potier, L.; Chollet, C.; Taveau, C.; Bruneval, P.; Roussel, R.; Alhenc-Gelas, F.; Bouby, N. Antihypertensive role of tissue kallikrein in hyperaldosteronism in the mouse. Endocrinology 2012, 153, 3886–3896. [Google Scholar] [CrossRef]
- Kato, Y.; Mori, K.; Kasahara, M.; Osaki, K.; Ishii, A.; Mori, K.P.; Toda, N.; Ohno, S.; Kuwabara, T.; Tokudome, T.; et al. Natriuretic peptide receptor guanylyl cyclase-A pathway counteracts glomerular injury evoked by aldosterone through p38 mitogen-activated protein kinase inhibition. Sci. Rep. 2017, 7, 46624. [Google Scholar] [CrossRef]
- Butler, M.J.; Ramnath, R.; Kadoya, H.; Desposito, D.; Riquier-Brison, A.; Ferguson, J.K.; Onions, K.L.; Ogier, A.S.; ElHegni, H.; Coward, R.J.; et al. Aldosterone induces albuminuria via matrix metalloproteinase-dependent damage of the endothelial glycocalyx. Kidney Int. 2019, 95, 94–107. [Google Scholar] [CrossRef]
- Brand, S.; Amann, K.; Schupp, N. Angiotensin II-induced hypertension dose-dependently leads to oxidative stress and DNA damage in mouse kidneys and hearts. J. Hypertens. 2013, 31, 333–344. [Google Scholar] [CrossRef]
- Hartmann, C.; Schulz, I.; Epe, B.; Schupp, N. Angiotensin II-induced hypertension increases the mutant frequency in rat kidney. Arch. Toxicol. 2019, 93, 2045–2055. [Google Scholar] [CrossRef]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed]
- Numazawa, S.; Ishikawa, M.; Yoshida, A.; Tanaka, S.; Yoshida, T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am. J. Physiol. Cell Physiol. 2003, 285, C334–C342. [Google Scholar] [CrossRef]
- Lim, J.S.; Ahn, K.Y. Potassium Depletion Upregulates Expression of Nrf2 Transcription Factor in Rat Kidney. Kidney Res. Clin. Pract. 2011, 30, 239–245. [Google Scholar]
- Lyu, Z.; Mao, Z.; Li, Q.; Xia, Y.; Liu, Y.; He, Q.; Wang, Y.; Zhao, H.; Lu, Z.; Zhou, Q. PPARgamma maintains the metabolic heterogeneity and homeostasis of renal tubules. EBioMedicine 2018, 38, 178–190. [Google Scholar] [CrossRef]
- Shepard, M.; Dhulipala, P.; Kabaria, S.; Abraham, N.G.; Lianos, E.A. Heme oxygenase-1 localization in the rat nephron. Nephron 2002, 92, 660–664. [Google Scholar] [CrossRef]
- Brand, S.; Amann, K.; Mandel, P.; Zimnol, A.; Schupp, N. Oxidative DNA damage in kidneys and heart of hypertensive mice is prevented by blocking angiotensin II and aldosterone receptors. PLoS ONE 2014, 9, e115715. [Google Scholar] [CrossRef]
- Ali, B.H.; Al-Salam, S.; Al Suleimani, Y.; Al Kalbani, J.; Al Bahlani, S.; Ashique, M.; Manoj, P.; Al Dhahli, B.; Al Abri, N.; Naser, H.T.; et al. Curcumin Ameliorates Kidney Function and Oxidative Stress in Experimental Chronic Kidney Disease. Basic Clin. Pharmacol. Toxicol. 2018, 122, 65–73. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568–572. [Google Scholar] [CrossRef]
- Kang, S.J.; You, A.; Kwak, M.K. Suppression of Nrf2 signaling by angiotensin II in murine renal epithelial cells. Arch. Pharmacal Res. 2011, 34, 829–836. [Google Scholar] [CrossRef]
- Westhoff, J.H.; Hilgers, K.F.; Steinbach, M.P.; Hartner, A.; Klanke, B.; Amann, K.; Melk, A. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension 2008, 52, 123–129. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control | 75 µg/kg Ald | 125 µg/kg Ald | 250 µg/kg Ald |
|---|---|---|---|---|
| Body weight (g) | 27.3 ± 0.4 | 27.7 ± 0.3 | 27.1 ± 0.9 | 26.0 ± 0.8 |
| Kidney/body weight (‰) | 6.3 ± 0.2 | 8.7 ± 0.4 *** | 9.6 ± 0.3 *** | 9.7 ± 0.2 *** |
| Heart/body weight (‰) | 4.9 ± 0.1 | 5.8 ± 0.5 | 5.7 ± 0.2 * | 5.5 ± 0.1 ** |
| Drinking volume (mL/23 h) | 7.6 ± 1.7 | 16.8 ± 2.1 ** | 19.7 ± 5.9 | 12.7 ± 5.2 |
| Diuresis (mL/23 h) | 3.5 ± 2.3 | 9.9 ± 2.4 | 12.0 ± 5.2 | 9.3 ± 3.9 |
| Creatinine clearance (mL/h) | 2.4 ± 0.5 | 4.0 ± 0.8 | 4.6 ± 1.0 | 3.0 ± 0.4 |
| Albumin/creatinine (µg/mg) | 111 ± 15 | 1776 ± 678 * | 2520 ± 1563 * | 2716 ± 913 * |
| NGAL/creatinine (ng/mg) | 138 ± 30 | 541 ± 70 ** | 817 ± 156 * | 1087 ± 221 ** |
| Parameter | Control | 75 µg/kg Ald | 125 µg/kg Ald | 250 µg/kg Ald | |
|---|---|---|---|---|---|
| Inflamed area (%) | 0.5 ± 0.1 | 0.4 ± 0.1 | 1.61 ± 0.5* | 1.9 ± 0.6 * | |
| Atrophic basal membranes (%) | 4.7 ± 1.8 | 20.2 ± 6.0 * | 23.9 ± 7.5 ** | 27.3 ± 5.0 ** | |
| Collagen deposition (%) | 1.9 ± 0.7 | 7.5 ± 1.5 ** | 10.9 ± 1.7 ** | 11.7 ± 1.7 *** | |
| Glomerular damage | GSI | 0.58 ± 0.10 | 1.18 ± 0.12 ** | 1.32 ± 0.08 *** | 0.97 ± 0.11 * |
| MSI | 0.57 ± 0.11 | 0.84 ± 0.05 | 0.83 ± 0.02 | 0.93 ± 0.09 ** | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balhorn, R.; Hartmann, C.; Schupp, N. Aldosterone Induces DNA Damage and Activation of Nrf2 Mainly in Tubuli of Mouse Kidneys. Int. J. Mol. Sci. 2020, 21, 4679. https://doi.org/10.3390/ijms21134679
Balhorn R, Hartmann C, Schupp N. Aldosterone Induces DNA Damage and Activation of Nrf2 Mainly in Tubuli of Mouse Kidneys. International Journal of Molecular Sciences. 2020; 21(13):4679. https://doi.org/10.3390/ijms21134679
Chicago/Turabian StyleBalhorn, Ronja, Christina Hartmann, and Nicole Schupp. 2020. "Aldosterone Induces DNA Damage and Activation of Nrf2 Mainly in Tubuli of Mouse Kidneys" International Journal of Molecular Sciences 21, no. 13: 4679. https://doi.org/10.3390/ijms21134679
APA StyleBalhorn, R., Hartmann, C., & Schupp, N. (2020). Aldosterone Induces DNA Damage and Activation of Nrf2 Mainly in Tubuli of Mouse Kidneys. International Journal of Molecular Sciences, 21(13), 4679. https://doi.org/10.3390/ijms21134679