Chemopreventive Agent 3,3′-Diindolylmethane Inhibits MDM2 in Colorectal Cancer Cells



Abstract
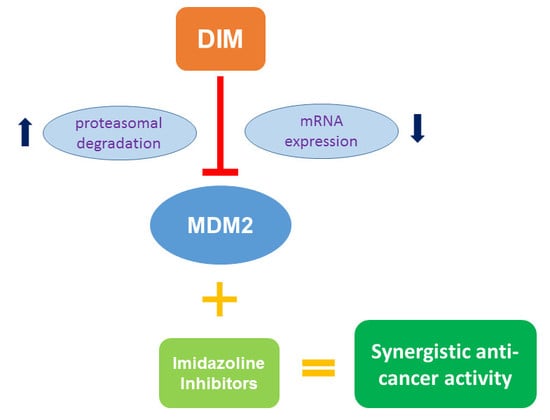
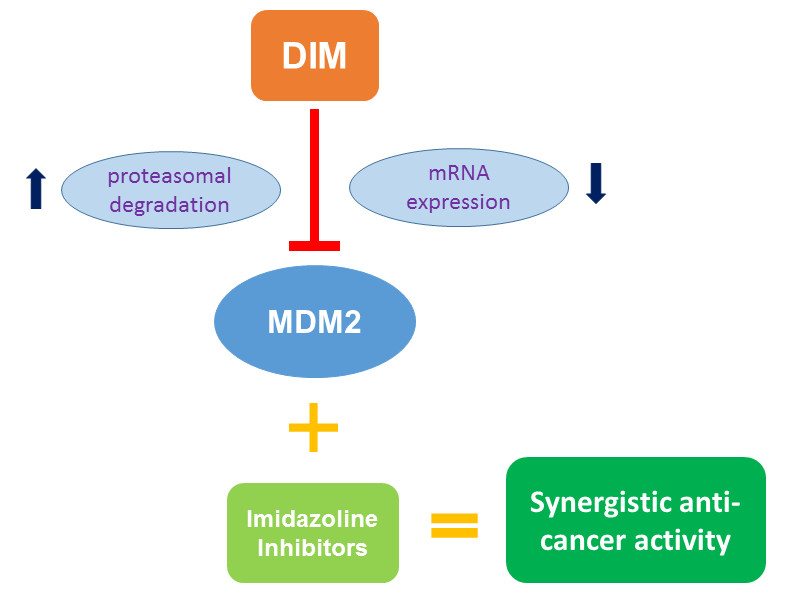
1. Introduction
2. Results
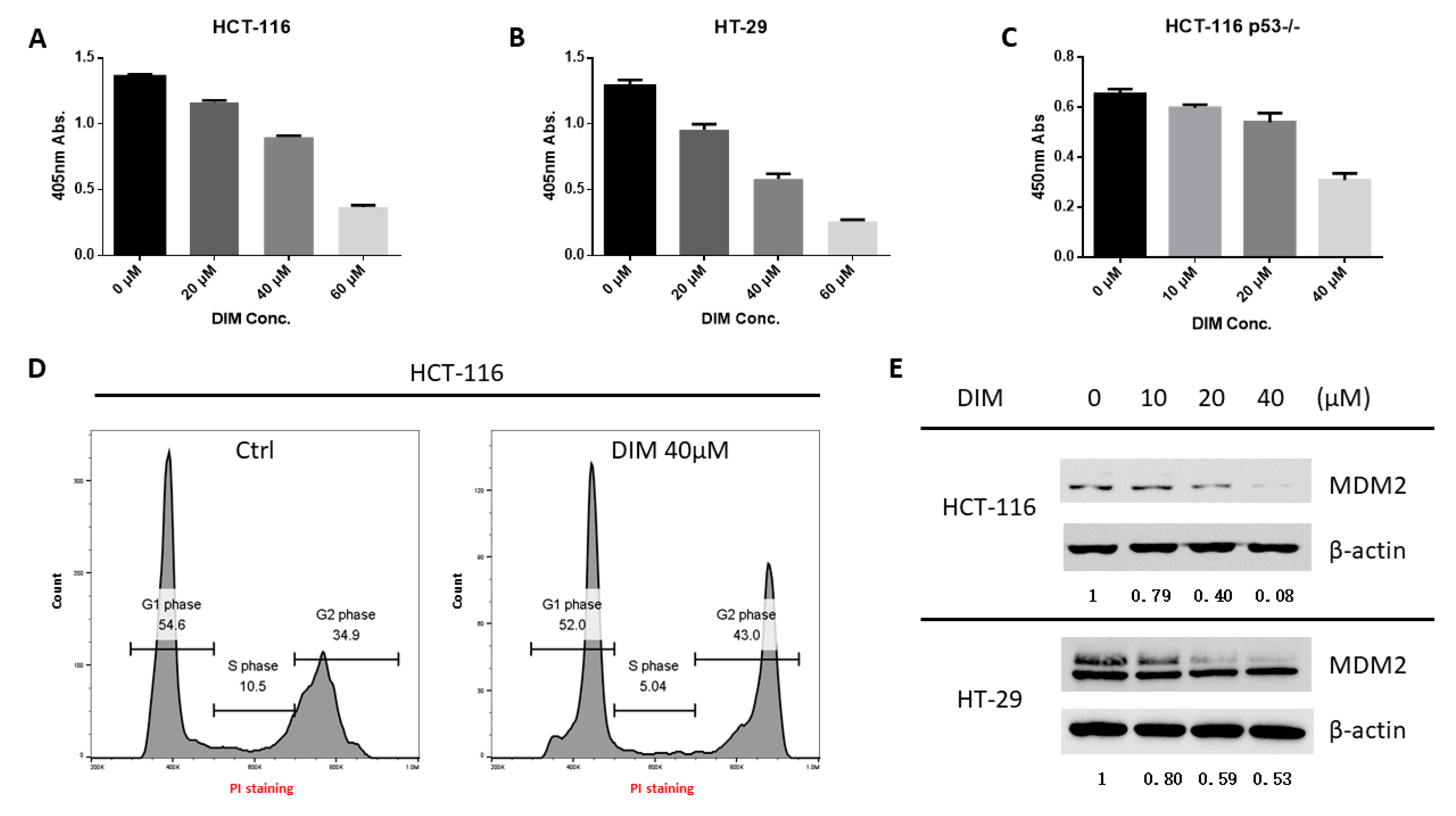
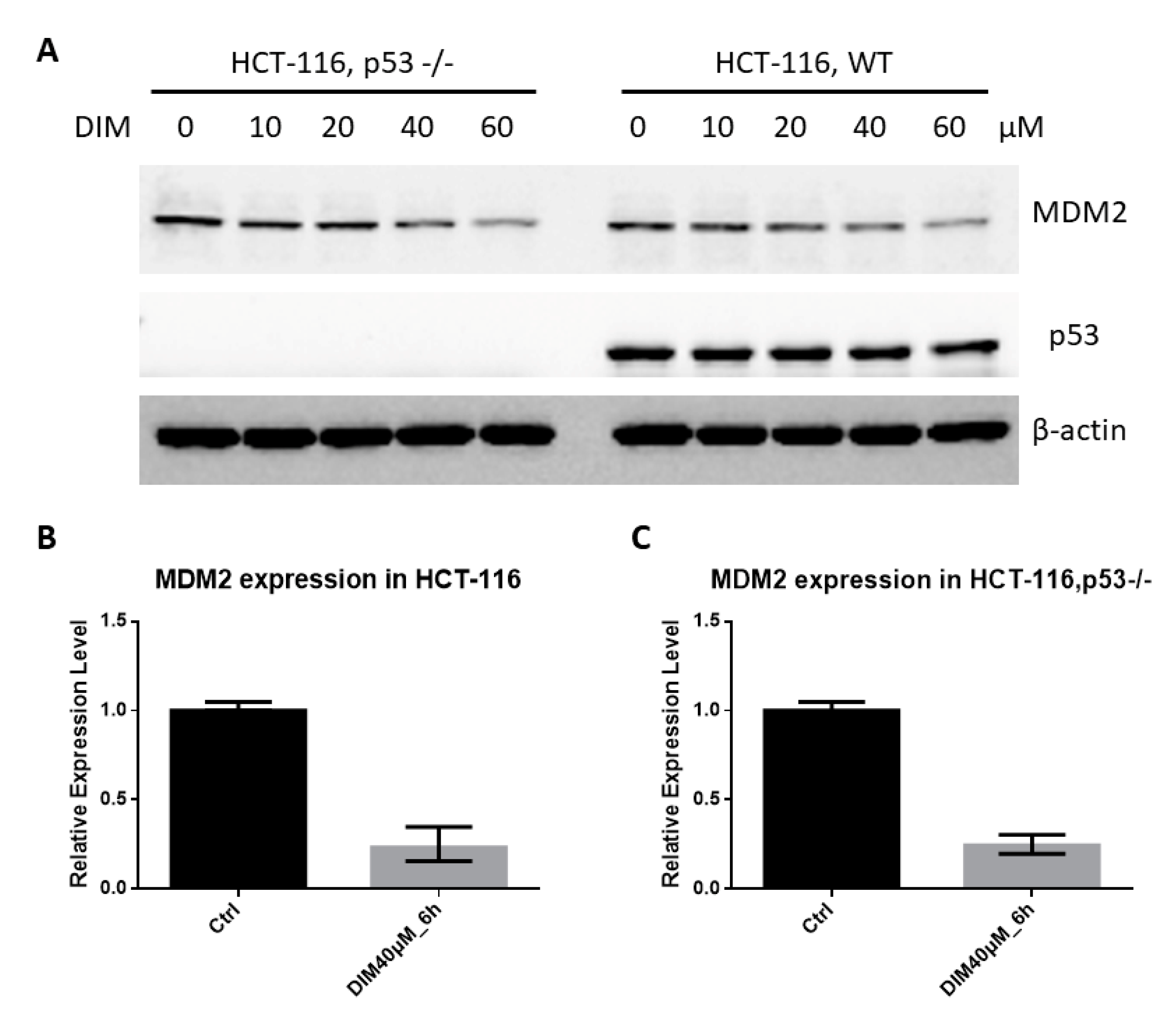
2.1. DIM Inhibits MDM2 Protein in Colorectal Cancer Cells
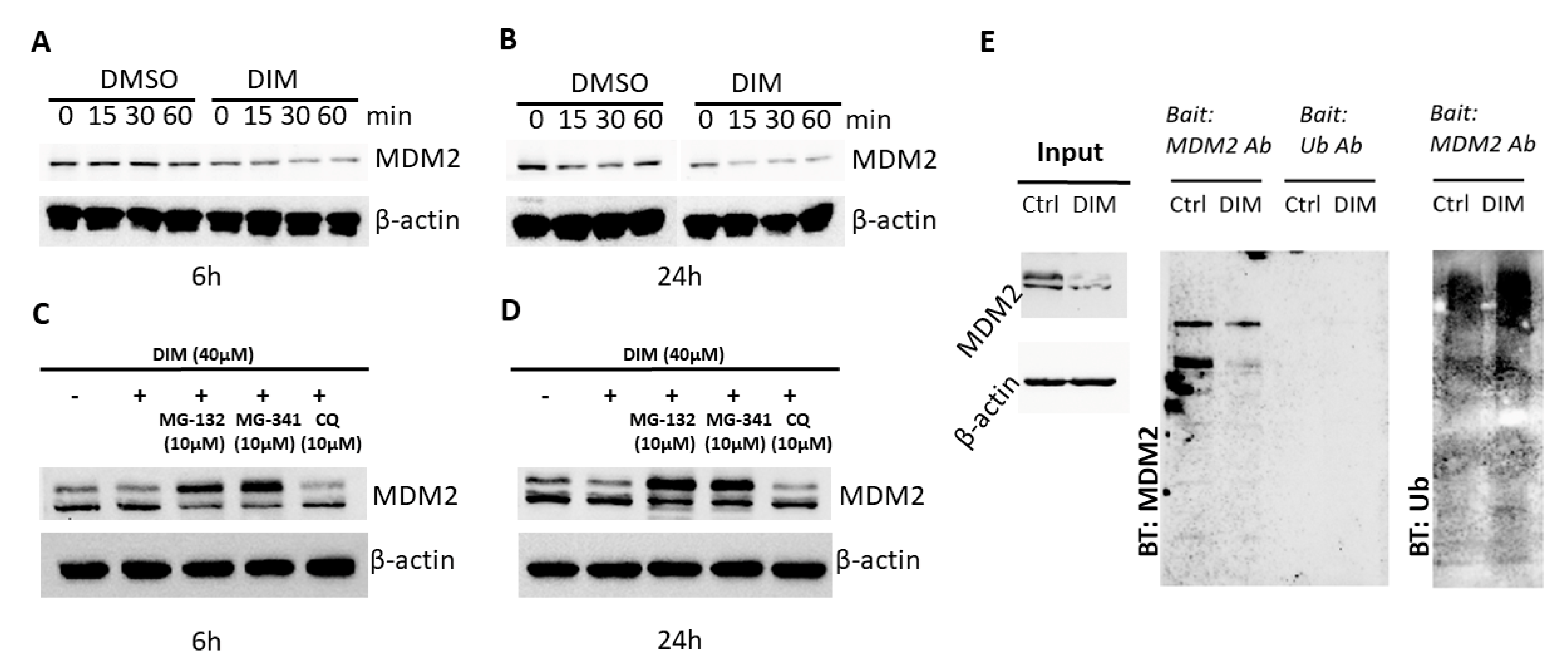
2.2. DIM Induces Proteasome-Mediated MDM2 Degradation
2.3. DIM Inhibition of MDM2 Is P53-Independent
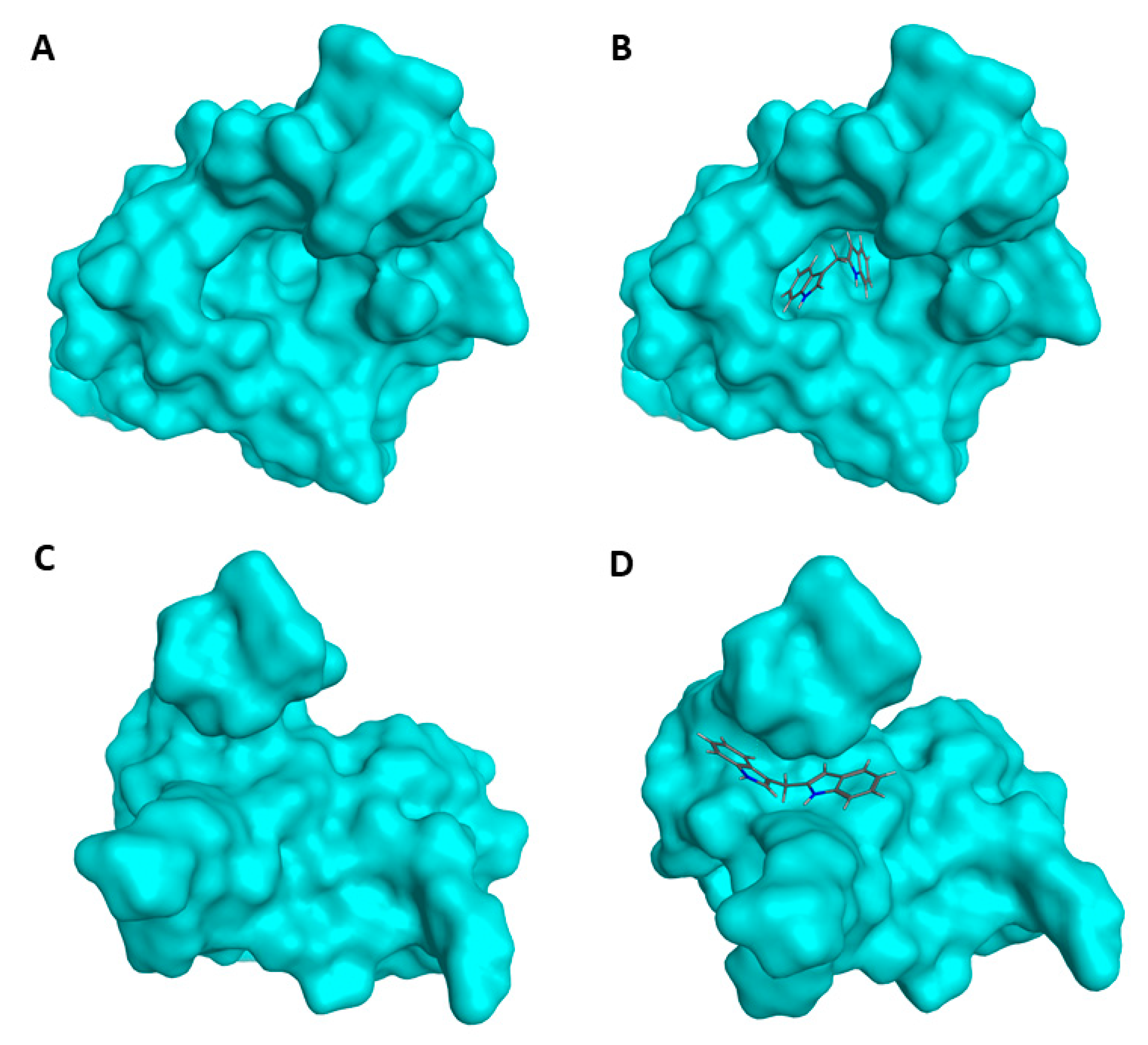
2.4. Prediction of DIM-MDM2 Interaction
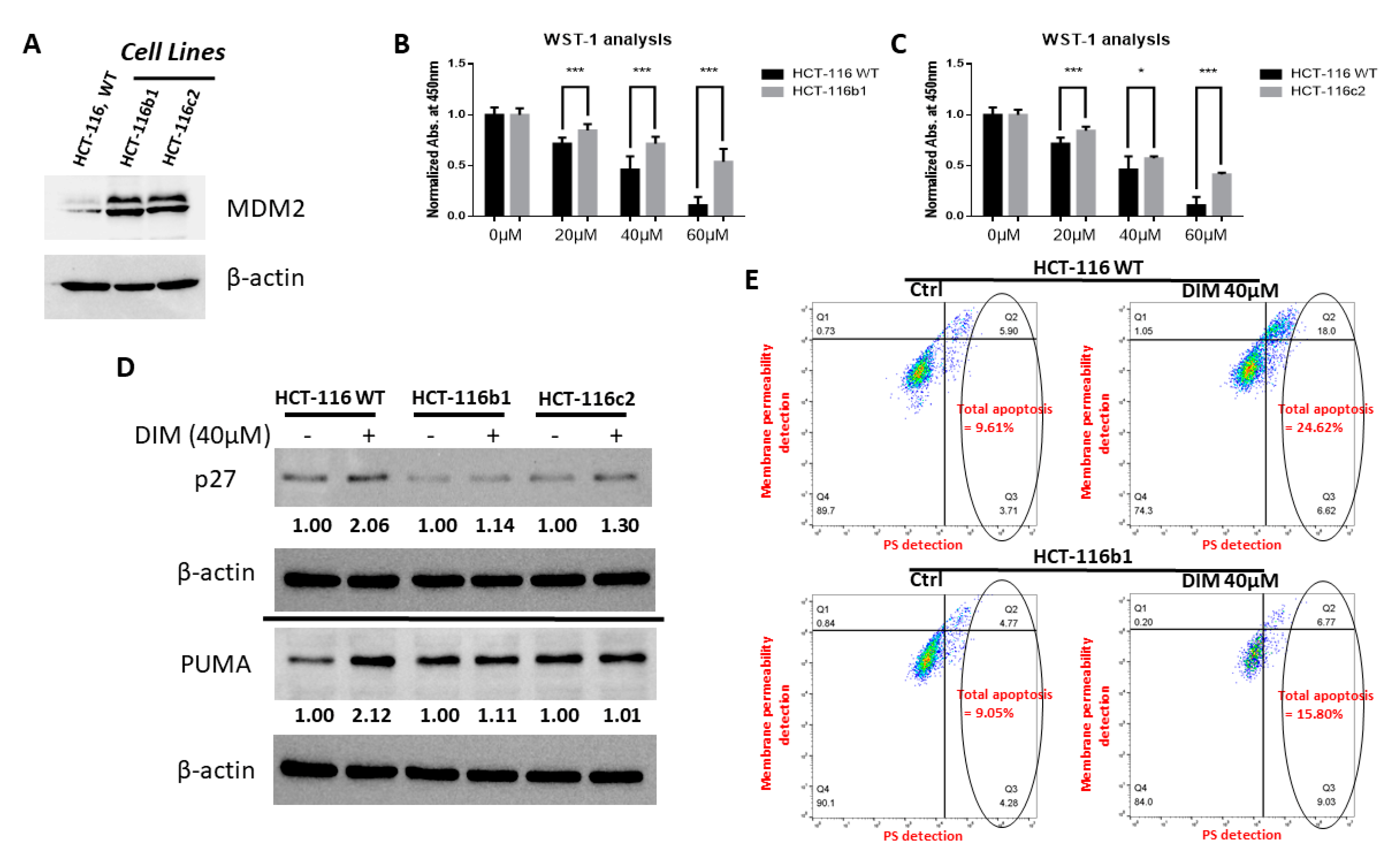
2.5. MDM2 Inhibition Contributes to DIM’s Anti-Cancer Activity
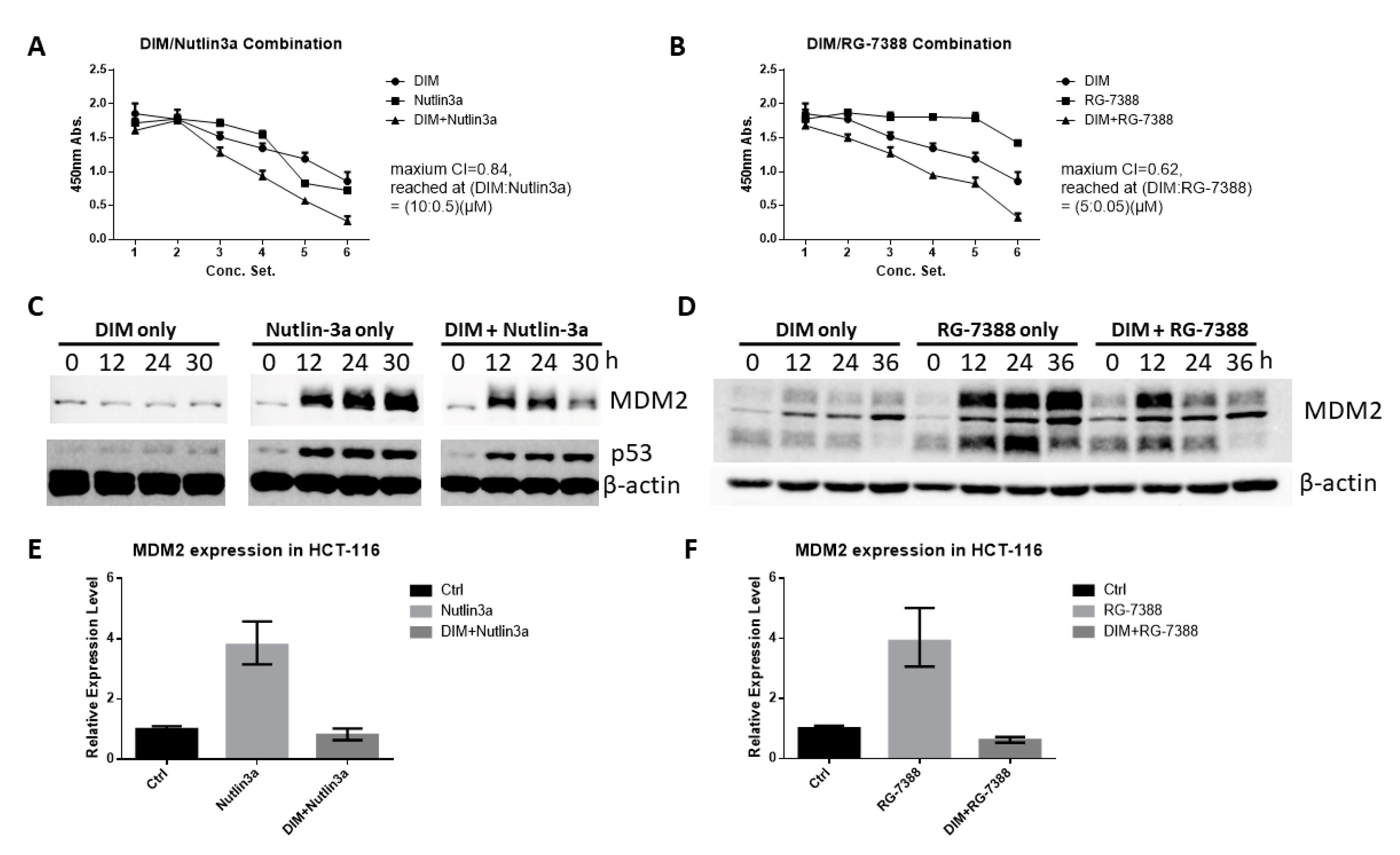
2.6. DIM Enhances the Anti-Cancer Activity of Cis-Imidazoline MDM2 Inhibitors
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Drugs and Chemicals
4.3. WST-1 Assay
4.4. Flow Cytometry
4.5. Blocking of Protein Synthesis and Degradation
4.6. Western Blotting
4.7. Co-Immunoprecipitation (Co-IP)
4.8. Real-Time PCR
4.9. Molecular Modeling
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DIM | 3,3′- Diindolylmethane |
| MDM2 | Mouse double minute 2 homolog |
| MDMX | Mouse double minute X |
| I3C | Indol-3-cabinol |
| CRC | Colorectal cancer |
| mRNA | Messenger RNA |
| PCR | Polymerase chain reaction |
| TKI | Tyrosine kinase inhibitors |
| EGFR | Epidermal growth factor receptor |
| RTK | Receptor tyrosine kinase |
| CHX | Cycloheximide |
| PI | Propidium iodide |
| PS | Phosphatidylserine |
| CQ | Chloroquine |
| MOE | Molecular Operating Environment |
| RING | Really interesting new gene |
| RRP | Recurrent respiratory papillomatosis |
References
- Mattiuzzi, C.; Sanchis-Gomar, F.; Lippi, G. Concise update on colorectal cancer epidemiology. Ann. Transl. Med. 2019, 7, 609. [Google Scholar] [CrossRef] [PubMed]
- Giusti, R.M.; Shastri, K.A.; Cohen, M.H.; Keegan, P.; Pazdur, R. FDA drug approval summary: Panitumumab (Vectibix). Oncologist 2007, 12, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Ettrich, T.J.; Seufferlein, T. Regorafenib. In Small Molecules in Oncology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 185–196. [Google Scholar]
- Graham, J.; Muhsin, M.; Kirkpatrick, P. Cetuximab. Nat. Rev. Drug Discov. 2004, 3, 549–550. [Google Scholar] [PubMed]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Saiki, A.Y.; Caenepeel, S. The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb. Perspect. Med. 2016, 6, a026336. [Google Scholar] [CrossRef]
- National Cancer Institute. Clinical Trials Information: MDM2 Inhibitor. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/r?d=C162996&loc=0&pn=2&rl=2 (accessed on 27 January 2020).
- Ding, Q.; Zhang, Z.; Liu, J.-J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C. Discovery of RG7388, a potent and selective p53–MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef]
- Vassilev, L.T. Small-molecule antagonists of p53-MDM2 binding: Research tools and potential therapeutics. Cell Cycle 2004, 3, 417–419. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Guo, B. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010, 70, 646–654. [Google Scholar] [CrossRef]
- Thomson, C.A.; Ho, E.; Strom, M.B. Chemopreventive properties of 3,3′-diindolylmethane in breast cancer: Evidence from experimental and human studies. Nutr. Rev. 2016, 74, 432–443. [Google Scholar] [CrossRef]
- Gamet-Payrastre, L.; Lumeau, S.; Gasc, N.; Cassar, G.; Rollin, P.; Tulliez, J. Selective cytostatic and cytotoxic effects of glucosinolates hydrolysis products on human colon cancer cells in vitro. Anticancer Drugs 1998, 9, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Park, S.Y.; Shin, H.K.; Kwon, D.Y.; Surh, Y.J.; Park, J.H. Activation of caspase-8 contributes to 3,3′-Diindolylmethane-induced apoptosis in colon cancer cells. J. Nutr. 2007, 137, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Park, J.H.Y. Induction of G1 and G2/M cell cycle arrests by the dietary compound 3, 3’-diindolylmethane in HT-29 human colon cancer cells. BMC Gastroenterol. 2009, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.; Dashwood, W.-M.; Li, L.; Yin, T.; Ulusan, A.M.; Shatzer, K.; Gao, S.; Ruan, K.-H.; Hu, M. Acute changes in colonic PGE 2 levels as a biomarker of efficacy after treatment of the Pirc (F344/NTac-Apc am1137) rat with celecoxib. Inflamm. Res. 2020, 69, 131–137. [Google Scholar] [CrossRef]
- Yun, C.; Dashwood, W.-M.; Kwong, L.N.; Gao, S.; Yin, T.; Ling, Q.; Singh, R.; Dashwood, R.H.; Hu, M. Accurate quantification of PGE2 in the polyposis in rat colon (Pirc) model by surrogate analyte-based UPLC–MS/MS. J. Pharm. Biomed. Anal. 2018, 148, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Juven, T.; Barak, Y.; Zauberman, A.; George, D.; Oren, M. Wild type p53 can mediate sequence-specific transactivation of an internal promoter within the mdm2 gene. Oncogene 1993, 8, 3411–3416. [Google Scholar] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, H.; Hu, W. The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim. Et Biophys. Sin. 2014, 46, 180–189. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Bohlman, S.; Manfredi, J.J. p53-independent effects of Mdm2. Sub-Cell. Biochem. 2014, 85, 235–246. [Google Scholar] [CrossRef]
- Ganguli, G.; Wasylyk, B. p53-independent functions of MDM2. Mol. Cancer Res. MCR 2003, 1, 1027–1035. [Google Scholar]
- Chou, T.; Martin, N. CompuSyn Software for Drug Combinations and for General Dose-Effect Analysis, and User’s Guide; Combosyn Inc.: Paramus, NJ, USA, 2007. [Google Scholar]
- Rosen, C.A.; Bryson, P.C. Indole-3-carbinol for recurrent respiratory papillomatosis: Long-term results. J. Voice 2004, 18, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Anderton, M.J.; Manson, M.M.; Verschoyle, R.D.; Gescher, A.; Lamb, J.H.; Farmer, P.B.; Steward, W.P.; Williams, M.L. Pharmacokinetics and tissue disposition of indole-3-carbinol and its acid condensation products after oral administration to mice. Clin. Cancer Res. 2004, 10, 5233–5241. [Google Scholar] [CrossRef] [PubMed]
- Arneson, D.; Hurwitz, A.; McMahon, L.; Robaugh, D. Presence of 3, 3′-Diindolylmethane in human plasma after oral administration of Indole-3-carbinol. Proc. Am. Assoc. Cancer Res. 1999, 40, 2833. [Google Scholar]
- Thomson, C.A.; Chow, H.H.S.; Wertheim, B.C.; Roe, D.J.; Stopeck, A.; Maskarinec, G.; Altbach, M.; Chalasani, P.; Huang, C.; Strom, M.B.; et al. A randomized, placebo-controlled trial of diindolylmethane for breast cancer biomarker modulation in patients taking tamoxifen. Breast Cancer Res. Treat. 2017, 165, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Dalessandri, K.M.; Firestone, G.L.; Fitch, M.D.; Bradlow, H.L.; Bjeldanes, L.F. Pilot study: Effect of 3,3′-diindolylmethane supplements on urinary hormone metabolites in postmenopausal women with a history of early-stage breast cancer. Nutr. Cancer 2004, 50, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Tou, J.C.; Hong, C.; Kim, H.A.; Riby, J.E.; Firestone, G.L.; Bjeldanes, L.F. 3,3′-Diindolylmethane inhibits angiogenesis and the growth of transplantable human breast carcinoma in athymic mice. Carcinogenesis 2005, 26, 771–778. [Google Scholar] [CrossRef]
- Bhatnagar, N.; Li, X.; Chen, Y.; Zhou, X.; Garrett, S.H.; Guo, B. 3,3′-diindolylmethane enhances the efficacy of butyrate in colon cancer prevention through down-regulation of survivin. Cancer Prev. Res. 2009, 2, 581–589. [Google Scholar] [CrossRef]
- Sepkovic, D.W.; Stein, J.; Carlisle, A.D.; Ksieski, H.B.; Auborn, K.; Bradlow, H.L. Diindolylmethane inhibits cervical dysplasia, alters estrogen metabolism, and enhances immune response in the K14-HPV16 transgenic mouse model. Cancer Epidemiol. Biomark. 2009, 18, 2957–2964. [Google Scholar] [CrossRef]
- Aronchik, I.; Kundu, A.; Quirit, J.G.; Firestone, G.L. The antiproliferative response of indole-3-carbinol in human melanoma cells is triggered by an interaction with NEDD4-1 and disruption of wild-type PTEN degradation. Mol. Cancer Res. 2014, 12, 1621–1634. [Google Scholar] [CrossRef]
- Lee, Y.-R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.-Y.; Fu, T.-M.; Chen, H.; Ishikawa, T.; Chiang, S.-Y.; Katon, J. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Konopleva, M.; McQueen, T.; O’Brien, S.; Plunkett, W.; Andreeff, M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood 2006, 108, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Zhang, X.; Srivenugopal, K.S.; Wang, M.H.; Wang, W.; Zhang, R. Targeting MDM2-p53 interaction for cancer therapy: Are we there yet? Curr. Med. Chem. 2014, 21, 553–574. [Google Scholar] [CrossRef]
- Arena, G.; Riscal, R.; Linares, L.K.; Le Cam, L. MDM2 controls gene expression independently of p53 in both normal and cancer cells. Cell Death Differ. 2018, 25, 1533–1535. [Google Scholar] [CrossRef]
- Erba, H.P.; Becker, P.S.; Shami, P.J.; Grunwald, M.R.; Flesher, D.L.; Zhu, M.; Rasmussen, E.; Henary, H.A.; Anderson, A.A.; Wang, E.S. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019, 3, 1939–1949. [Google Scholar] [CrossRef]
- Khurana, A.; Shafer, D.A. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: Perspectives on the therapeutic potential of idasanutlin (RG7388). Oncotargets Ther. 2019, 12, 2903–2910. [Google Scholar] [CrossRef]
- Honda, R.; Yasuda, H. Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef]
- Gu, J.J.; Kawai, H.; Nie, L.G.; Kitao, H.; Wiederschain, D.; Jochemsen, A.G.; Parant, J.; Lozano, G.; Yuan, Z.M. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J. Biol. Chem. 2002, 277, 19251–19254. [Google Scholar] [CrossRef]
- Okino, S.T.; Pookot, D.; Basak, S.; Dahiya, R. Toxic and chemopreventive ligands preferentially activate distinct aryl hydrocarbon receptor pathways: Implications for cancer prevention. Cancer Prev. Res. 2009, 2, 251–256. [Google Scholar] [CrossRef]
- Ohtake, F.; Fujii-Kuriyama, Y.; Kato, S. AhR acts as an E3 ubiquitin ligase to modulate steroid receptor functions. Biochem. Pharmacol. 2009, 77, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2018.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Leong, H.; Riby, J.E.; Firestone, G.L.; Bjeldanes, L.F. Potent ligand-independent estrogen receptor activation by 3,3′-diindolylmethane is mediated by cross talk between the protein kinase A and mitogen-activated protein kinase signaling pathways. Mol Endocrinol. 2004, 18, 291–302. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration Setting # | 1 | 2 | 3 | 4 | 5 | 6 | Unit |
|---|---|---|---|---|---|---|---|
| DIM | 0 | 5 | 10 | 20 | 30 | 40 | μM |
| Nutlin-3a | 0 | 0.1 | 0.5 | 1 | 5 | 10 | μM |
| RG-7388 | 0 | 0.05 | 0.1 | 0.5 | 1 | 5 | μM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, X.; Liu, J.; Cho, K.B.; Kedika, S.; Guo, B. Chemopreventive Agent 3,3′-Diindolylmethane Inhibits MDM2 in Colorectal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 4642. https://doi.org/10.3390/ijms21134642
Gao X, Liu J, Cho KB, Kedika S, Guo B. Chemopreventive Agent 3,3′-Diindolylmethane Inhibits MDM2 in Colorectal Cancer Cells. International Journal of Molecular Sciences. 2020; 21(13):4642. https://doi.org/10.3390/ijms21134642
Chicago/Turabian StyleGao, Xiang, Jingwen Liu, Kwang Bog Cho, Samanthreddy Kedika, and Bin Guo. 2020. "Chemopreventive Agent 3,3′-Diindolylmethane Inhibits MDM2 in Colorectal Cancer Cells" International Journal of Molecular Sciences 21, no. 13: 4642. https://doi.org/10.3390/ijms21134642
APA StyleGao, X., Liu, J., Cho, K. B., Kedika, S., & Guo, B. (2020). Chemopreventive Agent 3,3′-Diindolylmethane Inhibits MDM2 in Colorectal Cancer Cells. International Journal of Molecular Sciences, 21(13), 4642. https://doi.org/10.3390/ijms21134642