Non-Classic Disorder of Adrenal Steroidogenesis and Clinical Dilemmas in 21-Hydroxylase Deficiency Combined with Backdoor Androgen Pathway. Mini-Review and Case Report


Abstract
1. Introduction
1.1. Relationship between Adrenal Cortex Structure and Function
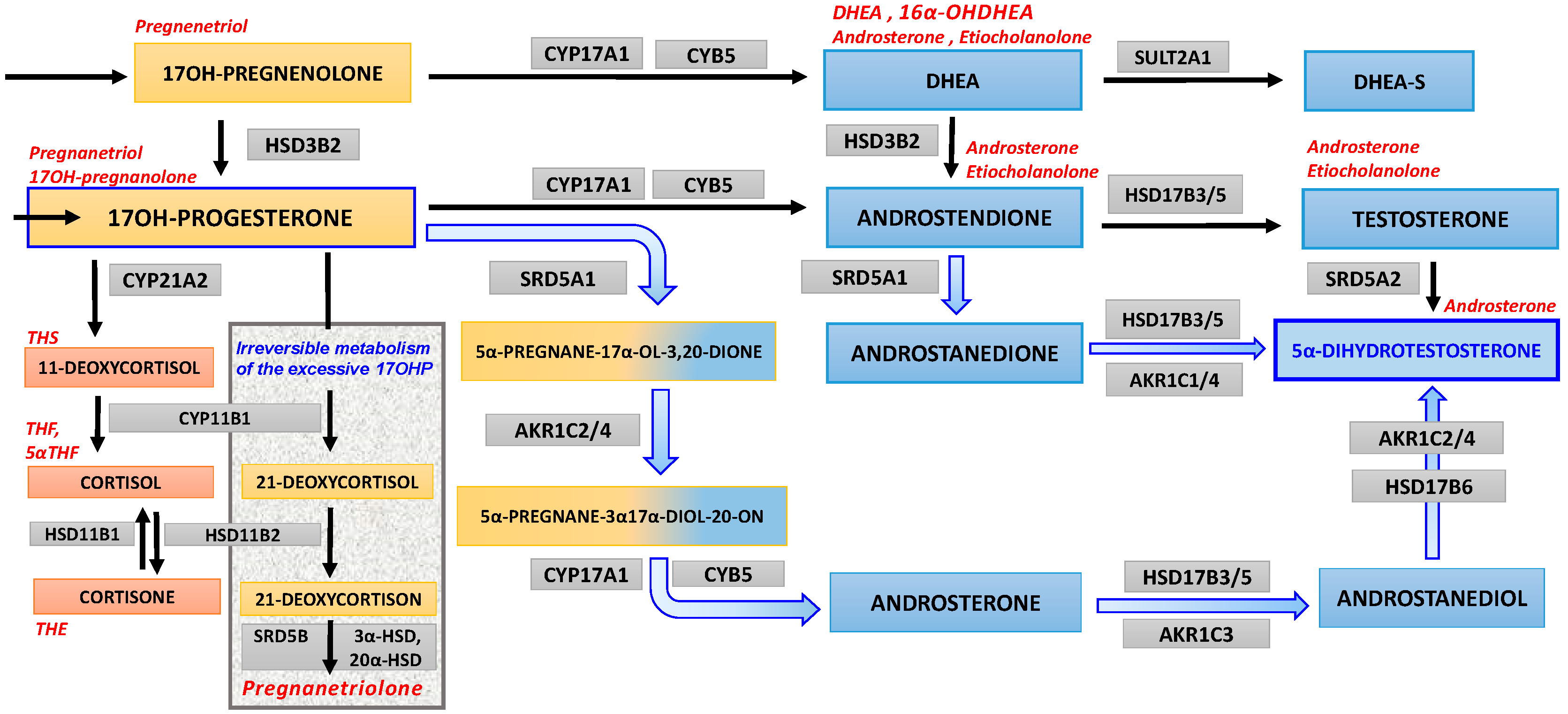
1.2. Congenital Adrenal Hyperplasia and Androgen Biosynthesis
2. Case Report
3. Laboratory Methods
4. Discussion
4.1. Preliminary Suspicion of PCOS
4.2. Early Diagnostics of CAH May Miss Its Non-Classic Forms
4.3. Differential between Non-Classic CAH and PCOS
4.4. Subtle Alterations in Urine Steroid Profile
4.5. 17OHP Excess as Substrate for “Backdoor” Androgen Synthesis
4.6. Information from Steroid Metabolites’ Ratios
4.7. Study Limitation—Lack of 11-Oxygenated-19-Carbon Androgens Evaluation
4.8. Serum Levels vs. Paracrine and Intracrine Impact on Symptoms
4.9. Diagnostic Strategy
4.10. Treatment Options
5. Final Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jacobson, L. Hypothalamic–pituitary–adrenocortical axis regulation. Endocrinol Metab. Clin. N. Am. 2005, 34, 271–292. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R.; Ehrhart-Bornstein, M. Basic and clinical aspects of intraadrenal regulation of steroidogenesis. Z Für. Rheumatol. 2000, 59, II12–II17. [Google Scholar] [CrossRef]
- Einer-Jensen, N.; Carter, A.M. Local transfer of hormones between blood vessels within the adrenal gland may explain the functional interaction between the adrenal cortex and medulla. Med. Hypotheses. 1995, 44, 471–474. [Google Scholar] [CrossRef]
- White, P.C.; Speiser, P.W. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr. Rev. 2000, 21, 245–291. [Google Scholar] [CrossRef] [PubMed]
- Nimkarn, S.; Lin-Su, K.; New, M.I. Steroid 21-hydroxylase deficiency congenital adrenal hyperplasia. Pediatr. Clin. N. Am. 2011, 58, 1281–1300. [Google Scholar] [CrossRef]
- Nimkarn, S.; New, M.I. Steroid 11β-hydroxylase deficiency congenital adrenal hyperplasia. Trends Endocrinol. Metab. 2008, 19, 96–99. [Google Scholar] [CrossRef]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef]
- Migeon, C.J.; Wisniewski, A.B. Congenital adrenal hyperplasia owing to 21-hydroxylase deficiency: Growth, development, and therapeutic considerations. Endocrinol. Metab. Clin. N. Am. 2001, 30, 193–206. [Google Scholar] [CrossRef]
- Charmandari, E.; Chrousos, G.; Merke, D.P. Classic congenital adrenal hyperplasia. In Adrenal Glands: Diagnostic Aspects and Surgical Therapy; Springer: Berlin/Heidelberg, Germany, 2005; p. 107. [Google Scholar]
- Kim, M.S.; Ryabets-Lienhard, A.; Geffner, M.E. Management of congenital adrenal hyperplasia in childhood. Curr. Opin. Endocrinol. Diabetes. Obes. 2012, 19, 483–488. [Google Scholar] [CrossRef]
- Gupta, M.K.; Guryev, O.L.; Auchus, R.J. 5alpha-reduced C21 steroids are substrates for human cytochrome P450c17. Arch. Biochem. Biophys. 2003, 418, 151–160. [Google Scholar] [CrossRef]
- Homma, K.; Hasegawa, T.; Nagai, T.; Adachi, M.; Horikawa, R.; Fujiwara, I.; Tajima, T.; Takeda, R.; Fukami, M.; Ogata, T. Urine steroid hormone profile analysis in cytochrome P450 oxidoreductase deficiency: Implication for the backdoor pathway to dihydrotestosterone. J. Clin. Endocrinol. Metab. 2006, 91, 2643–2649. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.; Mallappa, A.; Reisch, N.; Nikolaou, N.; Krone, N.; Hughes, B.A.; O’Neil, D.M.; Whitaker, M.J.; Tomlinson, J.W.; Storbeck, K.H.; et al. Modified-release and conventional glucocorticoids and diurnal androgen excretion in congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2017, 102, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Azziz, R.; Legro, R.; Dewailly, D.; Franks, S.; Tarlatzis, B.; Fauser, B. The Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil. Steril. 2004, 81, 19–25. [Google Scholar]
- Ibáñez, L.; Oberfield, S.E.; Witchel, S.; Auchus, R.J.; Chang, R.J.; Codner, E.; Dabadghao, P.; Darendeliler, F.; Elbarbary, N.S.; Gambineri, A.; et al. An international consortium update: Pathophysiology, diagnosis, and treatment of polycystic ovarian syndrome in adolescence. Horm. Res. Paediatr. 2017, 88, 371–395. [Google Scholar] [CrossRef]
- Gambineri, A.; Fanelli, F.; Prontera, O.; Repaci, A.; Di Dalmazi, G.; Zanotti, L.; Pagotto, U.; Flacco, M.E.; Guidi, J.; Fava, G.A.; et al. Prevalence of hyperandrogenic states in late adolescent and young women: Epidemiological survey on italian high-school students. J. Clin. Endocrinol. Metab. 2013, 98, 1641–1650. [Google Scholar] [CrossRef][Green Version]
- Kurtoğlu, S.; Hatipoğlu, N.; Mazıcıoğlu, M.; Kendirici, M.; Keskin, M.; Kondolot, M. Insulin resistance in obese children and adolescents: HOMA-IR cut-off levels in the prepubertal and pubertal periods. J. Clin. Res. Pediatr. Endocrinol. 2010, 2, 100–106. [Google Scholar] [CrossRef]
- Singh, Y.; Garg, M.K.; Tandon, N.; Marwaha, R.K. A study of insulin resistance by HOMA-IR and its cut-off value to identify metabolic syndrome in urban Indian adolescents. J. Clin. Res. Pediatr. Endocrinol. 2013, 5, 245–251. [Google Scholar]
- Zhou, Z.; Ni, R.; Hong, Y.; Li, Y.; Wang, Y.; Zhao, X.; Yang, D. Defining hyperandrogenaemia according to the free androgen index in Chinese women: A cross-sectional study. Clin. Endocrinol. 2012, 77, 446–452. [Google Scholar] [CrossRef]
- Ginalska-Malinowska, M. Classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency—The next disease included in the neonatal screening program in Poland. Dev. Period. Med. 2018, 22, 197–200. [Google Scholar]
- Alonso-Fernández, J.R. Pregnanetriolone in paper-borne urine for neonatal screening for 21-hydroxylase deficiency: The place of urine in neonatal screening. Mol. Genet. Metab. Rep. 2016, 8, 99–102. [Google Scholar] [CrossRef]
- Azziz, R.; Sanchez, L.A.; Knochenhauer, E.S.; Moran, C.; Lazenby, J.; Stephens, K.C.; Taylor, K.; Boots, L.R. Androgen excess in women: Experience with over 1000 consecutive patients. J. Clin. Endocrinol. Metab. 2004, 89, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.M.; Barth, J.H. Clinical biochemistry of dihydrotestosterone. Ann. Clin. Biochem. 2013, 50, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Swerdloff, R.S.; Dudley, R.E.; Page, S.T.; Wang, C.; Salameh, W.A. Dihydrotestosterone: Biochemistry, physiology, and clinical implications of elevated blood levels. Endocr. Rev. 2017, 38, 220–254. [Google Scholar] [CrossRef] [PubMed]
- Dhayat, N.A.; Marti, N.; Kollmann, Z.; Troendle, A.; Bally, L.; Escher, G.; Grössl, M.; Ackermann, D.; Ponte, B.; Pruijm, M.; et al. Members of the SKIPOGH Study Group. Urinary steroid profiling in women hints at a diagnostic signature of the polycystic ovary syndrome: A pilot study considering neglected steroid metabolites. PLoS ONE 2018, 13, e0203903. [Google Scholar] [CrossRef]
- Kamrath, C.; Hochberg, Z.; Hartmann, M.F.; Remer, T.; Wudy, S.A. Increased activation of the alternative “backdoor” pathway in patients with 21-hydroxylase deficiency: Evidence from urinary steroid hormone analysis. J. Clin. Endocrinol. Metab. 2012, 97, 367–375. [Google Scholar] [CrossRef]
- Skordis, N.; Shammas, C.; Phedonos, A.A.P.; Kyriakou, A.; Toumba, M.; Neocleous, V.; Phylactou, L.A. Genetic defects of the CYP21A2 gene in girls with premature adrenarche. J. Endocrinol. Investig. 2015, 38, 535–539. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef]
- Cox, R.I.; Finkelstein, M. Pregnane-3α,17α,20α-Triol and Pregnane-3α,17α,20α-Triol-11-one excretion by patients with adrenocortical dysfunction. J. Clin. Investig. 1957, 36, 1726–1735. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Antignac, J.P.; Le Bizec, B.; Morvan, M.-L.; Svechnikov, K.; Söder, O.; Savchuk, L.; Monteiro, A.; Soffientini, U.; Johnston, Z.C.; et al. Alternative (backdoor) androgen production and masculinization in the human fetus. PLoS Biol. 2019, 17, e3000002. [Google Scholar]
- Auchus, R.J. The backdoor pathway to dihydrotestosterone. Trends Endocrinol. Metab. TEM 2004, 15, 432–438. [Google Scholar] [CrossRef]
- Christakoudi, S.; Cowan, D.A.; Christakudis, G.; Taylor, N.F. 21-hydroxylase deficiency in the neonate—Trends in steroid anabolism and catabolism during the first weeks of life. J. Steroid. Biochem. Mol. Biol. 2013, 138, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Dhayat, N.A.; Dick, B.; Frey, B.M.; d’Uscio, C.H.; Vogt, B.; Flück, C.E. Androgen biosynthesis during minipuberty favors the backdoor pathway over the classic pathway: Insights into enzyme activities and steroid fluxes in healthy infants during the first year of life from the urinary steroid metabolome. J. Steroid. Biochem. Mol. Biol. 2017, 165, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Fiandalo, M.V.; John Wilton, J.; Mohler, J.L. Roles for the backdoor pathway of androgen metabolism in prostate cancer response to castration and drug treatment. Int. J. Biol. Sci. 2014, 10, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Krone, N.; Hughes, B.A.; Lavery, G.G.; Stewart, P.M.; Arlt, W.; Shackleton, C.H.L. Gas chromatography/mass spectrometry (GC/MS) remains a pre-eminent discovery tool in clinical steroid investigations even in the era of fast liquid chromatography tandem mass spectrometry (LC/MS/MS). J. Steroid. Biochem. Mol. Biol. 2010, 121, 496–504. [Google Scholar] [CrossRef]
- Turcu, A.F.; Nanba, A.T.; Chomic, R.; Upadhyay, S.K.; Giordano, T.J.; Shields, J.J.; Merke, D.P.; Rainey, W.E.; Auchus, R.J. Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency. Eur. J. Endocrinol. 2016, 174, 601–609. [Google Scholar] [CrossRef]
- O’Reilly, M.W.; Kempegowda, P.; Jenkinson, C.; Taylor, A.E.; Quanson, J.L.; Storbeck, K.H.; Arlt, W. 11-Oxygenated C19 steroids are the predominant androgens in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 840–848. [Google Scholar] [CrossRef]
- Rege, J.; Turcu, A.F.; Kasa-Vubu, J.Z.; Lerario, A.M.; Auchus, G.C.; Auchus, R.J.; Smith, J.M.; White, P.C.; Rainey, W.E. 11-ketotestosterone is the dominant circulating bioactive androgen during normal and premature adrenarche. J. Clin. Endocrinol. Metab. 2018, 103, 4589–4598. [Google Scholar] [CrossRef]
- Turcu, A.F.; Auchus, R.J. Clinical significance of 11-oxygenated androgens. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 252–259. [Google Scholar] [CrossRef]
- Turcu, A.F.; Rege, J.; Auchus, R.J.; Rainey, W.E. 11-Oxygenated androgens in health and disease. Nat. Rev. Endocrinol. 2020, 16, 284–296. [Google Scholar] [CrossRef]
- Azziz, R.; Black, V.; Hines, G.A.; Fox, L.M.; Boots, L.R. Adrenal androgen excess in the polycystic ovary syndrome: Sensitivity and responsivity of the hypothalamic-pituitary-adrenal axis. J. Clin. Endocrinol. Metab. 1998, 83, 2317–2323. [Google Scholar] [CrossRef]
- Labrie, F.; Luu-The, V.; Labrie, C.; Bélanger, A.; Simard, J.; Lin, S.X.; Pelletier, G. Endocrine and intracrine sources of androgens in women: Inhibition of breast cancer and other roles of androgens and their precursor dehydroepiandrosterone. Endocr. Rev. 2003, 24, 152–182. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, L.; Arlt, W.; Storbeck, K.H. Intracrine androgen biosynthesis, metabolism and action revisited. Mol. Cell Endocrinol. 2018, 465, 4–26. [Google Scholar] [CrossRef] [PubMed]
- Livadas, S.; Dracopoulou, M.; Dastamani, A.; Sertedaki, A.; Maniati-Christidi, M.; Magiakou, A.M.; Kanaka-Gantenbein, C.; Chrousos, G.P.; Dacou-Voutetakis, C. The spectrum of clinical, hormonal and molecular findings in 280 individuals with nonclassical congenital adrenal hyperplasia caused by mutations of the CYP21A2 gene. Clin. Endocrinol. 2015, 82, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Livadas, S.; Bothou, C. Management of the female with non-classical congenital adrenal hyperplasia (NCCAH): A patient-oriented approach. Front. Endocrinol. 2019, 10, 366. [Google Scholar] [CrossRef] [PubMed]
- Kurtoğlu, S.; Hatipoğlu, N. Non-classical congenital adrenal hyperplasia in childhood. J. Clin. Res. Pediatr. Endocrinol. 2017, 9, 1–7. [Google Scholar] [CrossRef]
- Accetta, S.G.; Di Domênico, K.; Ritter, C.G.; Ritter, A.T.; Capp, E.; Spritzer, P.M. Anthropometric and endocrine features in girls with isolated premature pubarche or non-classical congenital adrenal hyperplasia. J. Pediatr. Endocrinol. Metab. 2004, 17, 767–773. [Google Scholar] [CrossRef]
- Lucas-Herald, A.K.; Rodie, M.; Lucaccioni, L.; Shapiro, D.; McNeilly, J.; Shaikh, M.G.; Ahmed, S.F. The pitfalls associated with urinary steroid metabolite ratios in children undergoing investigations for suspected disorders of steroid synthesis. Int. J. Pediatr. Endocrinol. 2015, 2015, 10. [Google Scholar] [CrossRef][Green Version]
- Ambroziak, U.; Kępczyńska-Nyk, A.; Kuryłowicz, A.; Małunowicz, E.M.; Wójcicka, A.; Miśkiewicz, P.; Macech, M. The diagnosis of nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency, based on serum basal or post-ACTH stimulation 17-hydroxyprogesterone, can lead to false-positive diagnosis. Clin. Endocrinol. 2016, 84, 23–29. [Google Scholar] [CrossRef]
- Török, D.; Halász, Z.; Garami, M.; Homoki, J.; Fekete, G.; Sólyom, J. Limited value of serum steroid measurements in identification of mild form of 21-hydroxylase deficiency. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2003, 111, 27–32. [Google Scholar] [CrossRef]
- Krone, N.; Arlt, W. Genetics of congenital adrenal hyperplasia. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 181–192. [Google Scholar] [CrossRef]
- Pignatelli, D.; Carvalho, B.L.; Palmeiro, A.; Barros, A.; Guerreiro, S.G.; Macut, D. The complexities in genotyping of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Front. Endocrinol. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pall, M.; Azziz, R.; Beires, J.; Pignatelli, D. The phenotype of hirsute women: A comparison of polycystic ovary syndrome and 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Fertil. Steril. 2010, 94, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, M.; Debono, M.; Huatan, H.; Merke, D.; Arlt, W.; Ross, R.J. An oral multiparticulate, modified-release, hydrocortisone replacement therapy that provides physiological cortisol exposure. Clin. Endocrinol. 2014, 80, 554–561. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Hormones | Value | Reference Values |
|---|---|---|
| LH 1, (mIU/mL) | 9.80 | 1.80–11.78 |
| FSH 2, (mIU/mL) | 6.30 | 3.00–8.10 |
| E2 3, (pg/mL) | 38.00 | 21.00–251.00 |
| ACTH 4, (pg/mL) | 84.90 | 10.00–60.00 |
| T 5, (nmol/L) | 2.26 | 0.38–2.74 |
| DHEA-S 6, (μmol/L) | 11.39 | 3.17–14.39 |
| Time (min) | Glucose (mg/dL) | Insulin (μU/mL) | C-Peptide (pmol/mL) |
|---|---|---|---|
| 0′ | 84 | 7.60 | 0.59 |
| 30′ | 160 | 45.50 | - |
| 60′ | 146 | 53.80 | 3.57 |
| 90′ | 111 | 45.70 | - |
| 120′ | 107 | 38.50 | 3.02 |
| 150′ | 111 | 32.70 | - |
| Time (min) | F 1 (ng/mL) | 17OHP 2 (ng/mL) | ANDR 3 (ng/mL) |
|---|---|---|---|
| 0′ | 212.00 | 1.66 | 3.00 |
| 60′ | 272.00 | 2.29 | 3.10 |
| 120′ | 261.00 | 2.38 | 3.59 |
| Steroid Metabolites | Results (μg/24 h) | Reference Range Values |
|---|---|---|
| AN (androsterone) | 2102.5 | (710–3140) |
| ET (etiocholanolone) | 1994.7 | (380–2590) |
| 11-OAN/ET (11-ketoandrosterone/etiocholanolone) | 103.5 | |
| 11-OHAN (11-hydroxy-androsterone) | 271.6 | (180–912) |
| 11-OHET (11-hydroxy-etiocholanolone) | 55.0 | (54–750) |
| ET/AN | 0.9 | |
| DHEA (dehydroepiandrosterone) | 438.2 | (73–559) |
| 5-AND (5-androstenediol) | 131.5 | (16–180) |
| 16α-OHDHEA (16alpha-hydroxy-DHEA) | 610.2 | (50–490) |
| An-3-ol (5-androstenetriol) | 357.5 | (130–610) |
| 5-PT (5-pregnenetriol) | 175.9 | (80–390) |
| 16-OHPN (16alpha-hydroxy-pregnenolone) | 0.0 | |
| 17-OHPN (5beta) (17-hydroxy-pregnanolone) | 124.9 | (25–208) |
| 17-OHPN (5alpha) (17-hydroxy-pregnanolone) | 6.1 | (0.5–9) |
| PT (pregnanetriol) | 385.0 | (185–885) |
| PTN (pregnanetriolone) | 142.5 | (10–50) |
| PD (pregnanediol) | 227.2 | <900 |
| E1 (estrone) | 3.7 | (0–52) |
| E2 (estradiol) | 1.4 | (0–15) |
| E3 (estriol) | 4.0 | (1–30) |
| THS (tetrahydro-11-deoxycortisol) | 36.2 | (20–72) |
| THDOC (tetrahydro-11-deoxycorticosterone) | 4.9 | (<16) |
| THA (tetrahydro-11-dehydrocorticosterone) | 83.2 | (50–260) |
| allo-THA (allo-tetrahydro-11-dehydrocorticosterone) | 24.6 | |
| THB (tetrahydro-corticosterone) | 66.2 | (20–256) |
| allo-THB (allo-tetrahydro-corticosterone) | 79.0 | (78–543) |
| THAldo (tetrahydro-aldosterone) | 25.6 | (7–51) |
| THE (tetrahydro-cortisone) | 1995.6 | (585–3960) |
| THF (tetrahydro-cortisol) | 771.4 | (315–2060) |
| allo-THF (allo-tetrahydro-cortisol) | 425.0 | (420–2660) |
| THF/allo-THF | 2.2 | |
| THF+allo-THF/THF | 0.6 | (0.55–1.2) |
| a-CTN (alpha-cortolone) | 1065.8 | (360–1420) |
| b-CTN (beta-cortolone) | 273.8 | (75–925) |
| a-CT (alpha-cortol) | 154.7 | (30–800) |
| b-CT (beta-cortol) | 110.7 | (60–300) |
| E (cortisone) | 70.3 | (17–115) |
| F (cortisol) | 46.8 | (9–52) |
| F/E (cortisol/cortisone) | 0.7 | (0.35–0.75) |
| 6b-OHF (6beta-hydrocortisol) | 0.0 | |
| 20a-DHF (20alfa/beta-dihydrocortisol) | 0.0 |
| ACTH 1 (pg/mL) | DHEA-S 2 (μmol/L) | ANDR 3 (ng/mL) | T4 (nmol/L) | DHT 5 (pg/mL) | F 6 (ng/mL) | |
|---|---|---|---|---|---|---|
| baseline reference | 10.00–60.00 | 3.17–14.39 | 0.14–2.92 | 0.38–2.74 | 24.00–368.00 | 37.00–194.00 |
| values baseline | 37.20 | 16.40 | 2.59 | 3.84 | 578.00 | 203.00 |
| post-test | 0.90 | 4.52 | 1.78 | 2.16 | 337.00 | <10.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumińska, M.; Bogusz-Górna, K.; Wegner, D.; Fichna, M. Non-Classic Disorder of Adrenal Steroidogenesis and Clinical Dilemmas in 21-Hydroxylase Deficiency Combined with Backdoor Androgen Pathway. Mini-Review and Case Report. Int. J. Mol. Sci. 2020, 21, 4622. https://doi.org/10.3390/ijms21134622
Sumińska M, Bogusz-Górna K, Wegner D, Fichna M. Non-Classic Disorder of Adrenal Steroidogenesis and Clinical Dilemmas in 21-Hydroxylase Deficiency Combined with Backdoor Androgen Pathway. Mini-Review and Case Report. International Journal of Molecular Sciences. 2020; 21(13):4622. https://doi.org/10.3390/ijms21134622
Chicago/Turabian StyleSumińska, Marta, Klaudia Bogusz-Górna, Dominika Wegner, and Marta Fichna. 2020. "Non-Classic Disorder of Adrenal Steroidogenesis and Clinical Dilemmas in 21-Hydroxylase Deficiency Combined with Backdoor Androgen Pathway. Mini-Review and Case Report" International Journal of Molecular Sciences 21, no. 13: 4622. https://doi.org/10.3390/ijms21134622
APA StyleSumińska, M., Bogusz-Górna, K., Wegner, D., & Fichna, M. (2020). Non-Classic Disorder of Adrenal Steroidogenesis and Clinical Dilemmas in 21-Hydroxylase Deficiency Combined with Backdoor Androgen Pathway. Mini-Review and Case Report. International Journal of Molecular Sciences, 21(13), 4622. https://doi.org/10.3390/ijms21134622