Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells


 , , , , and
, , , , and 
Abstract
1. Introduction
2. Results
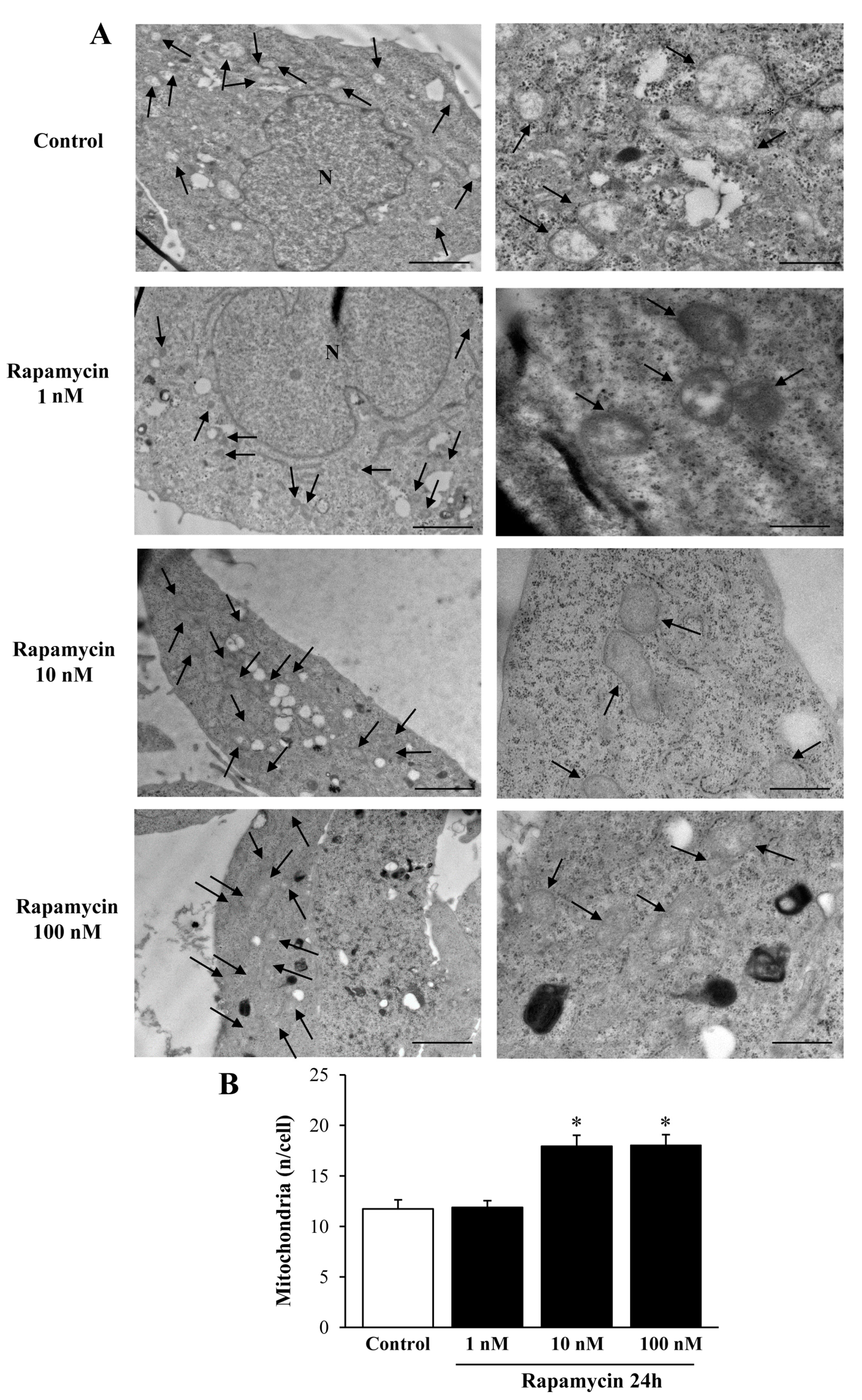
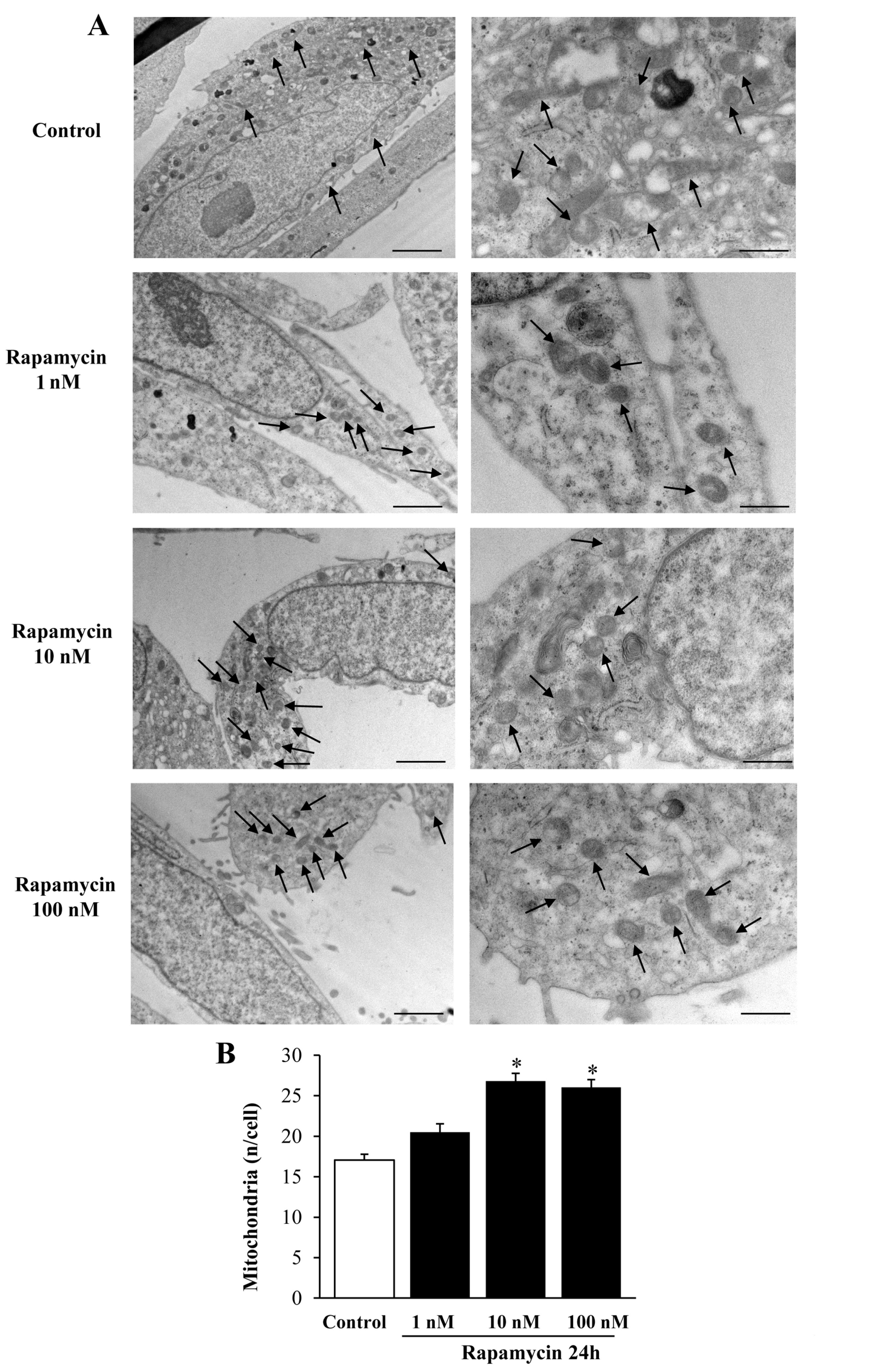
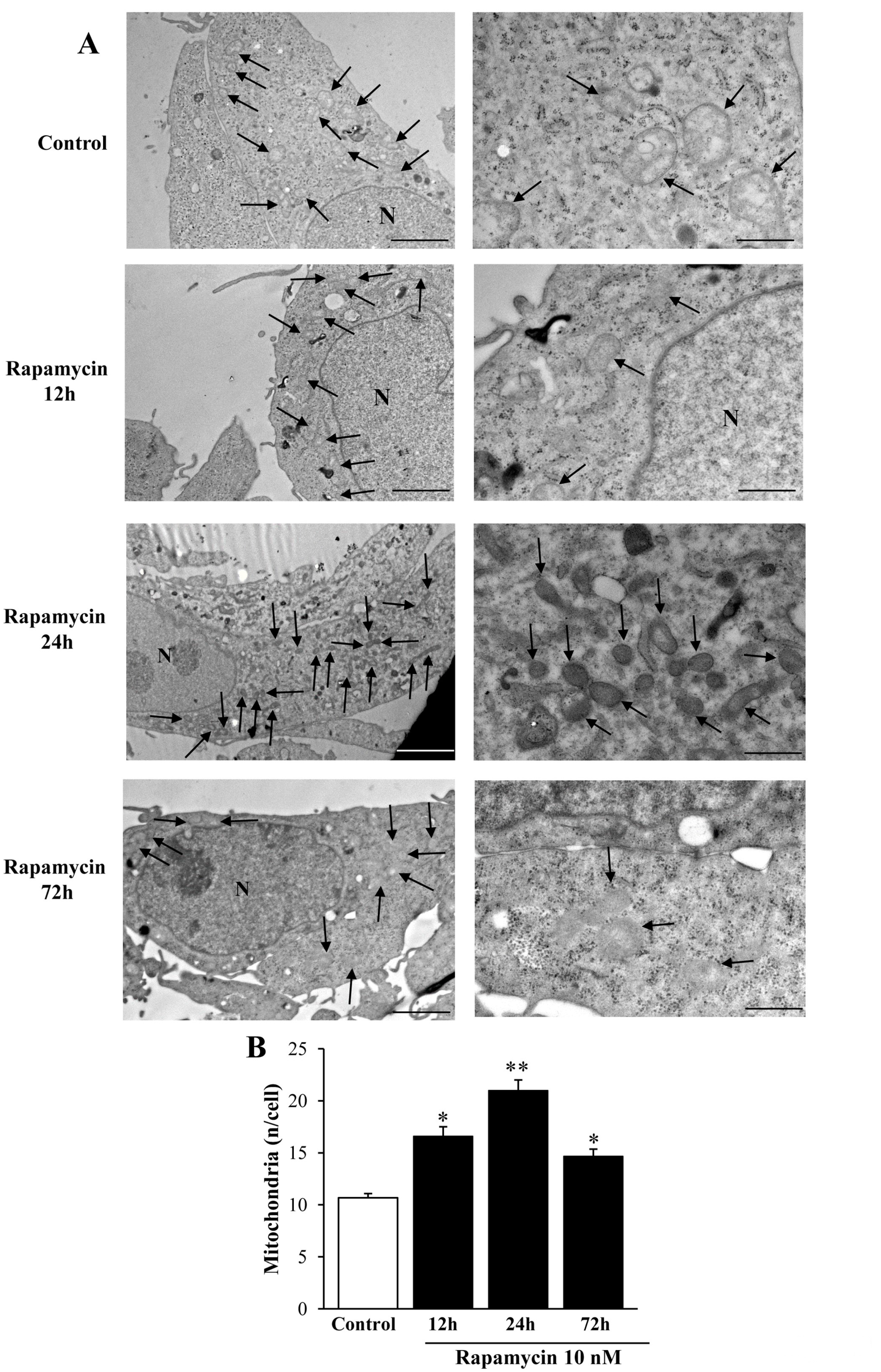
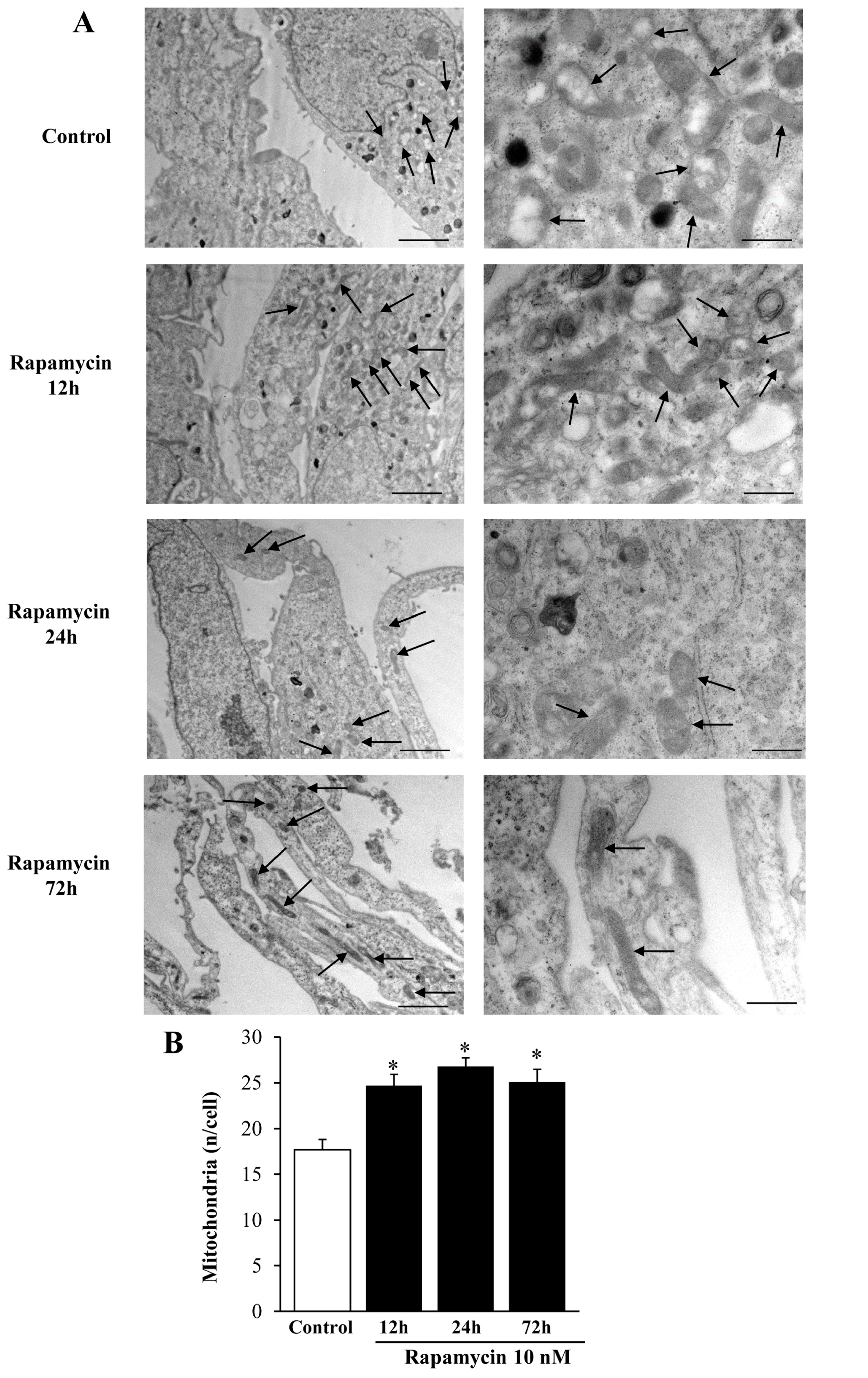
2.1. Preliminary Experiments to Assess the Effects of Various Doses and Times of Rapamycin Administration on Mitochondrial Number in Different GBM Cell Lines
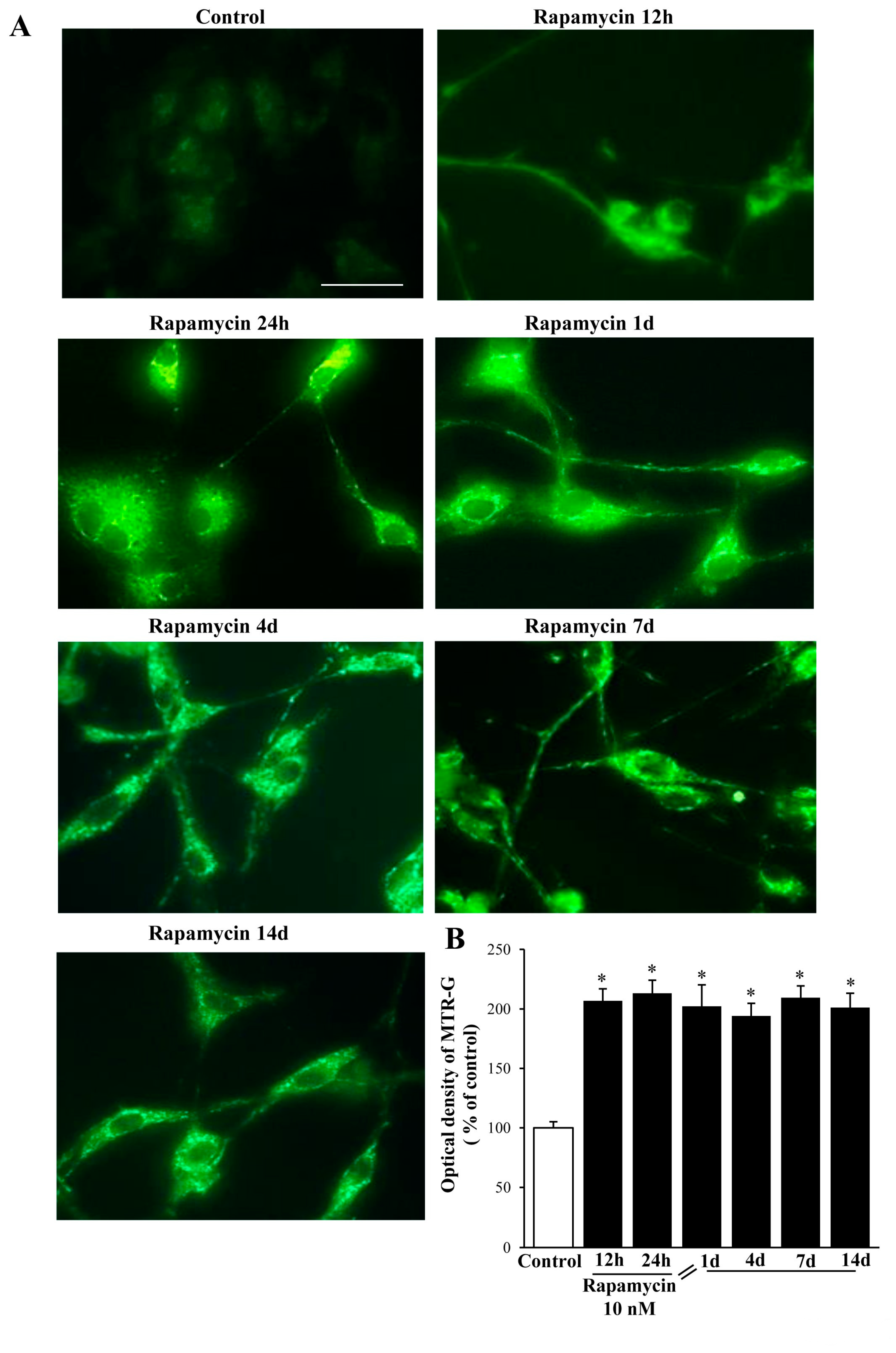
2.2. Rapamycin Produces a Two-Fold Increase in MitoTracker-Green (MTR-G)
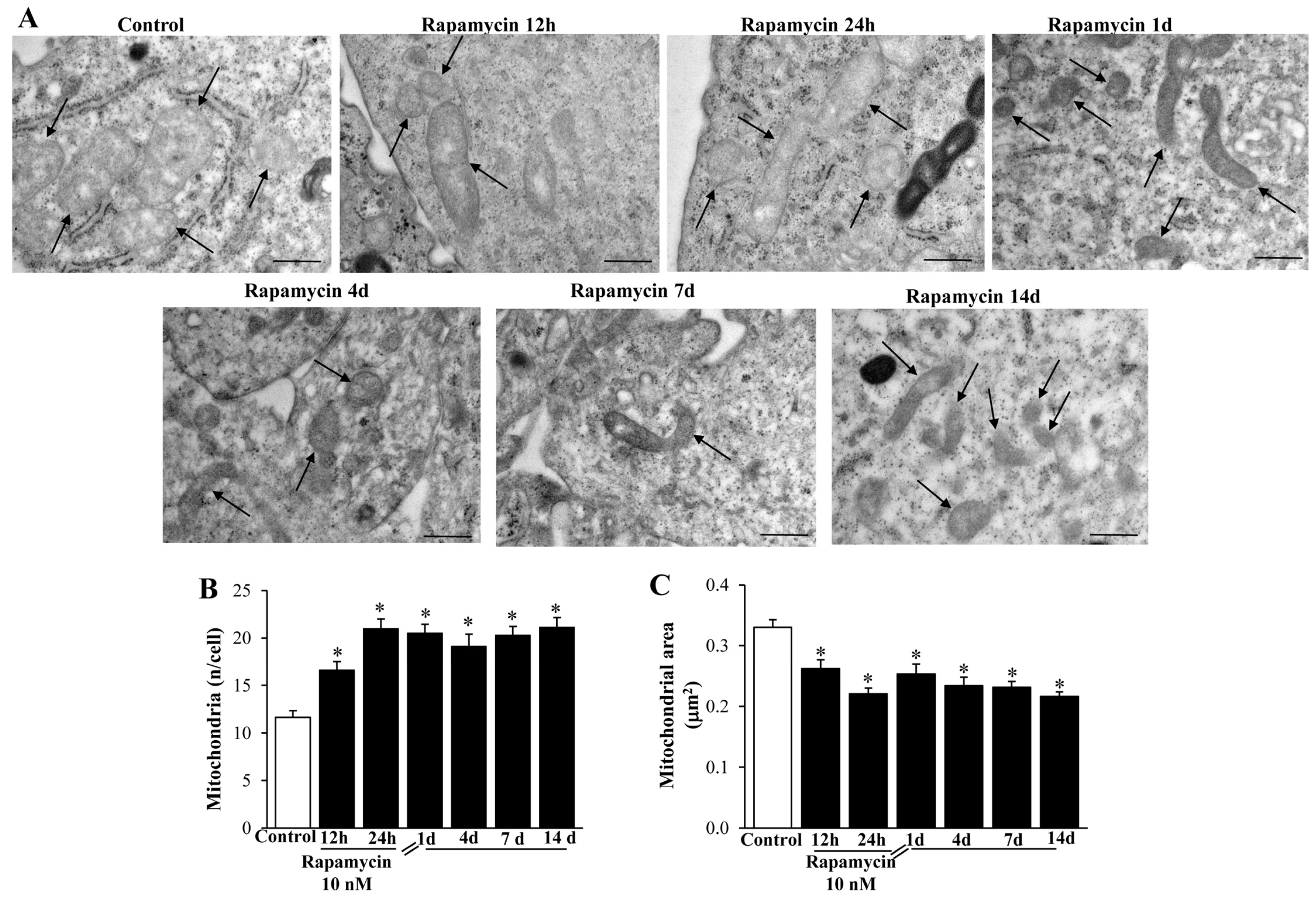
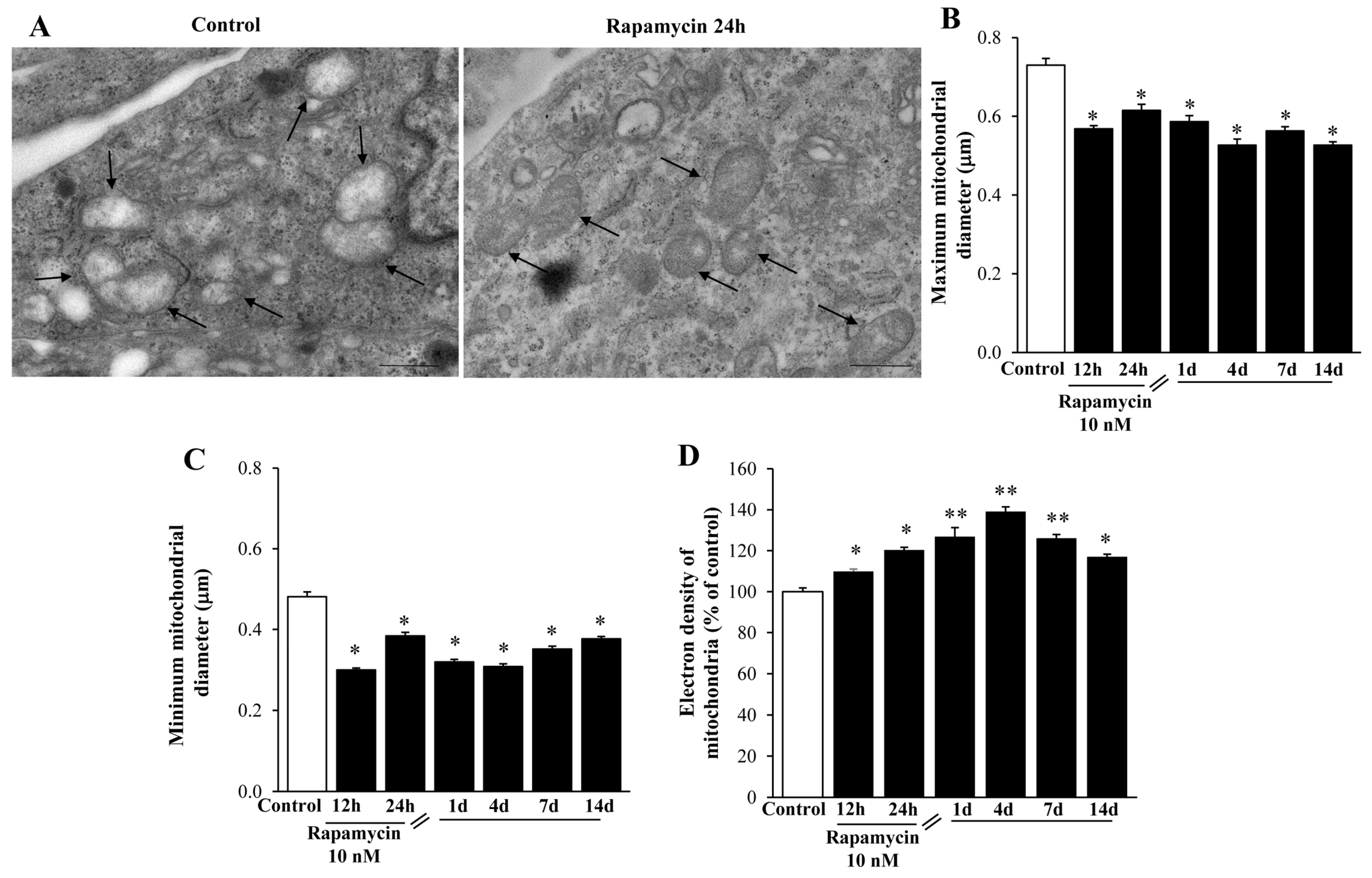
2.3. Rapamycin Modifies Mitochondrial Number, Size, and Phenotype
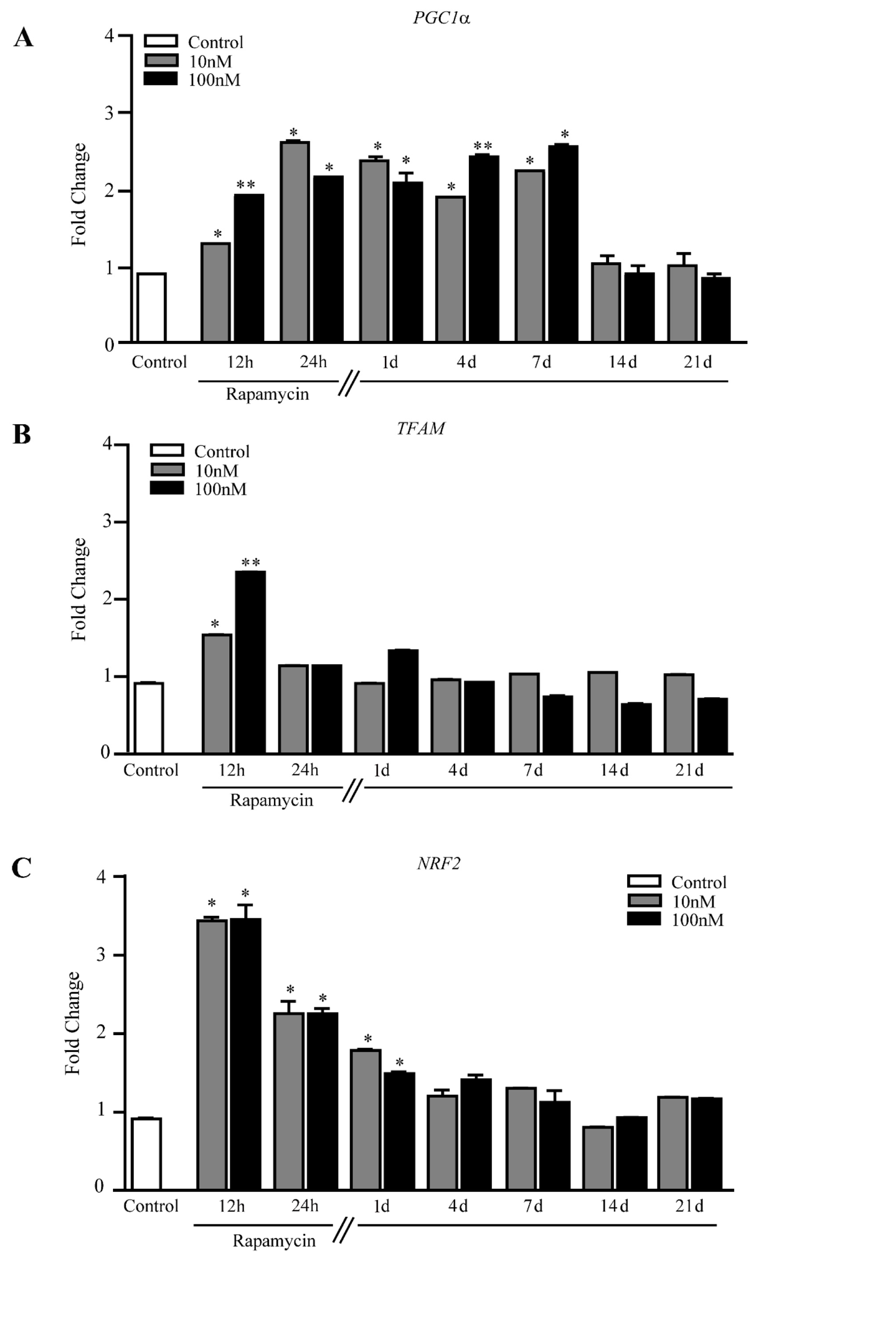
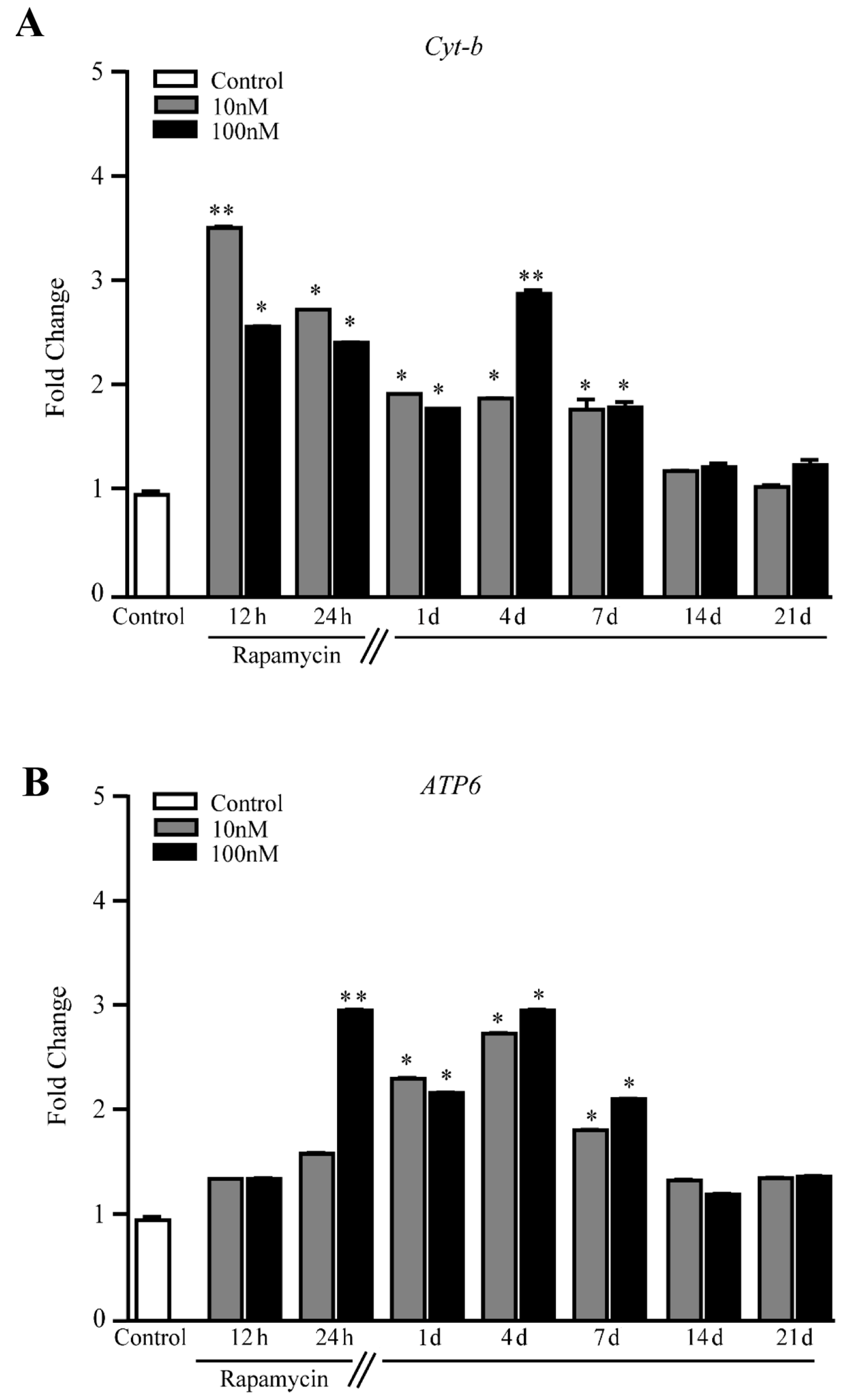
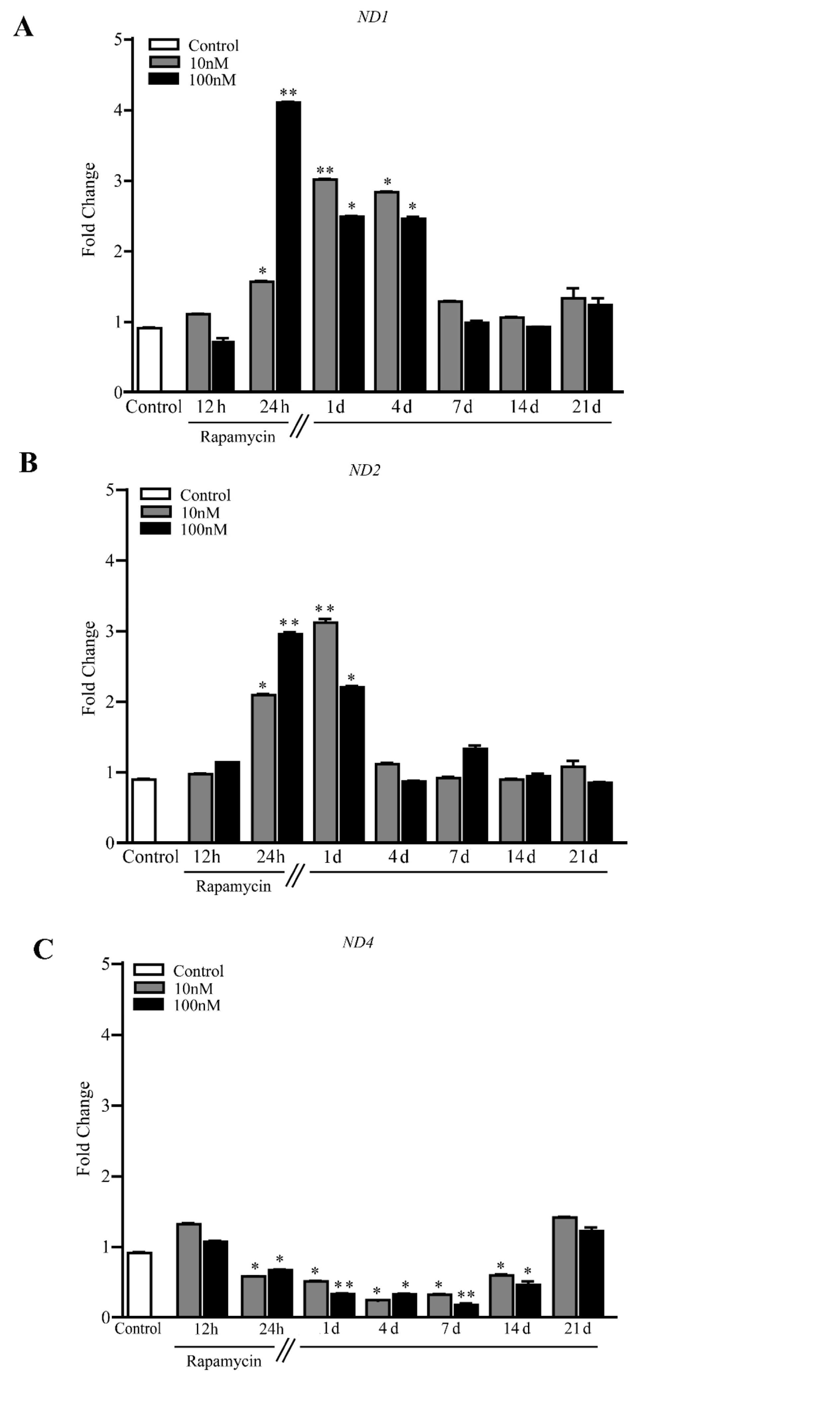
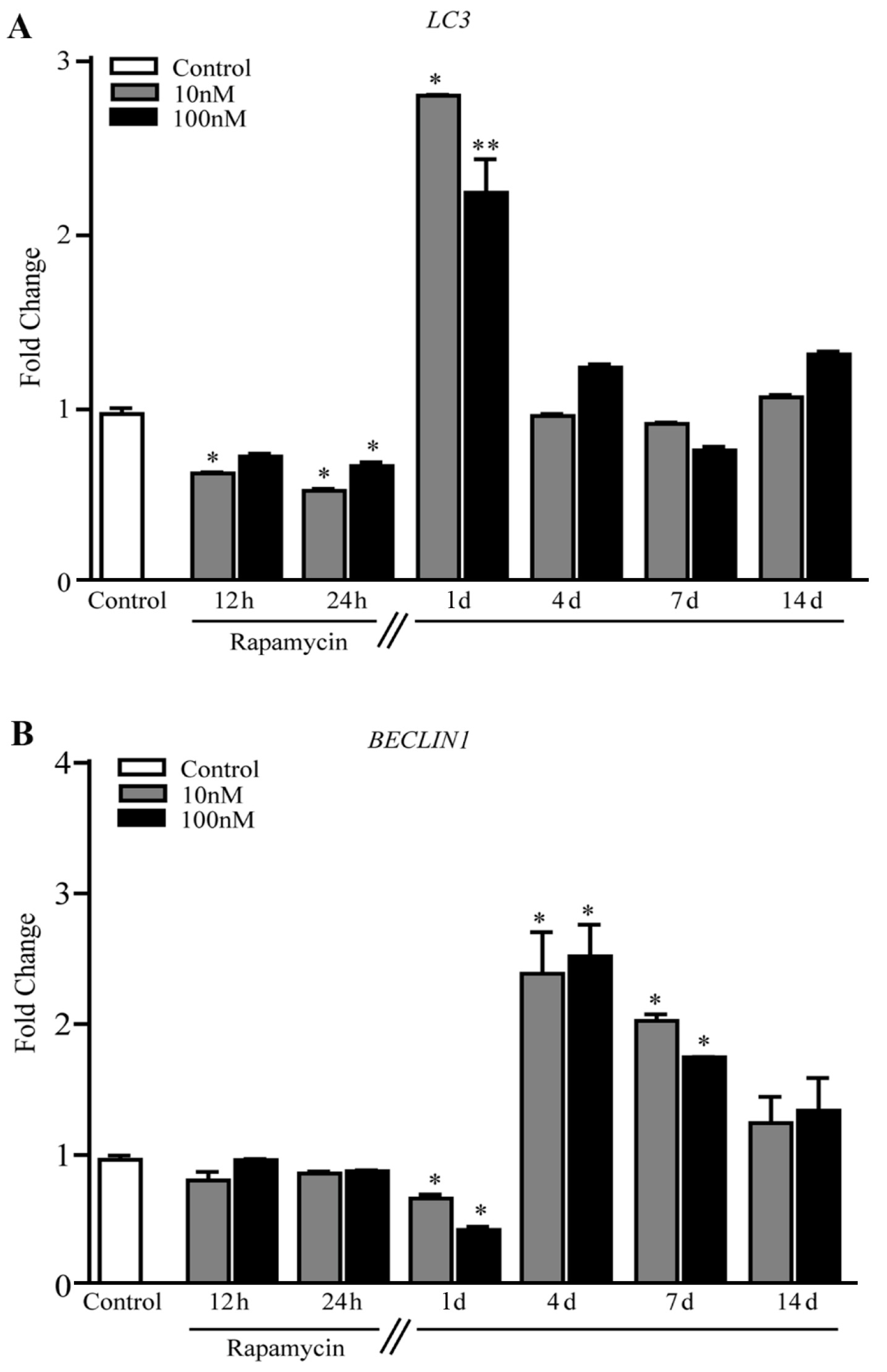
2.4. Rapamycin Increases the Expression of Mitochondriogenesis-Related Genes and Constitutive Mitochondrial Genes
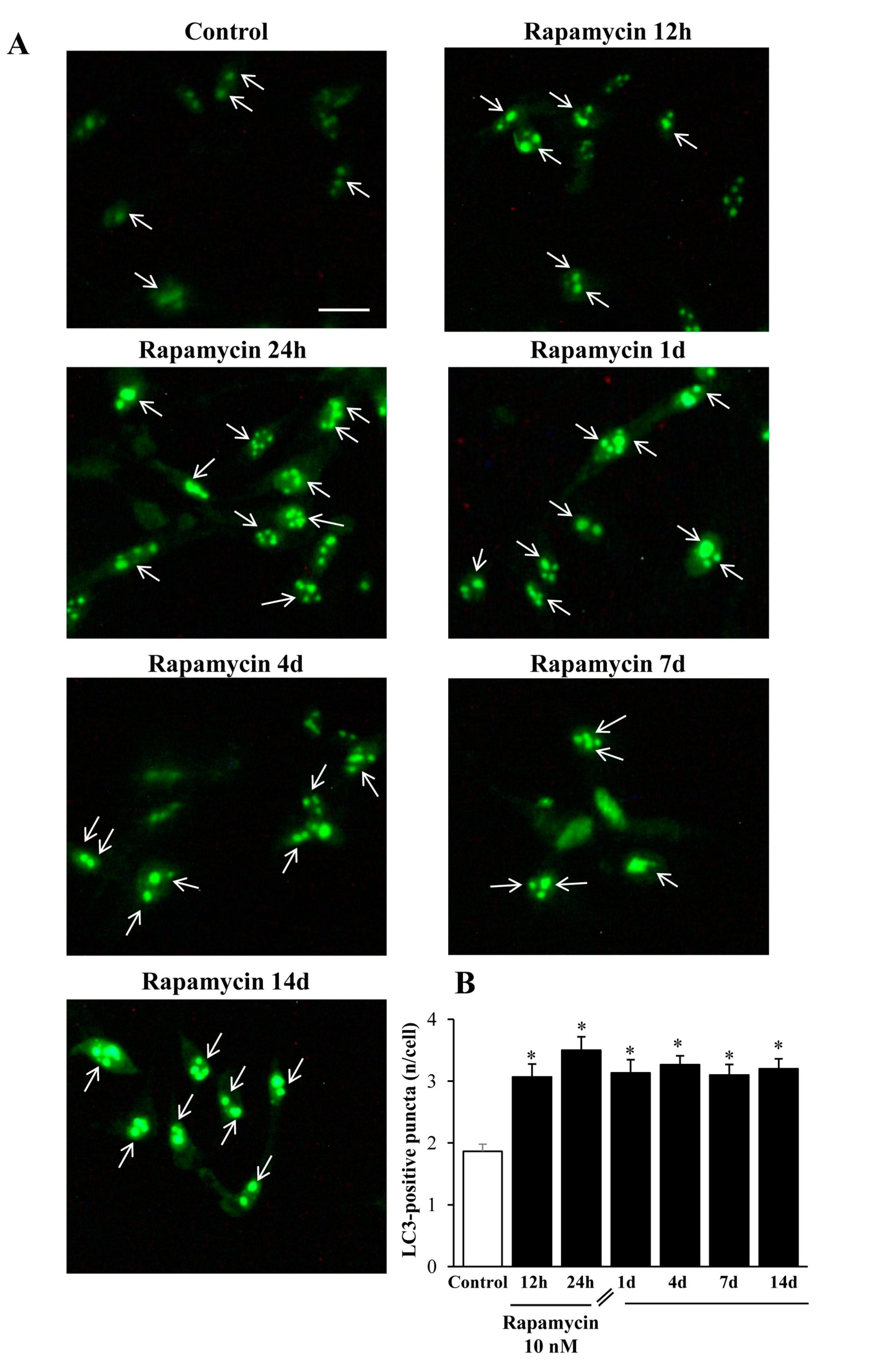
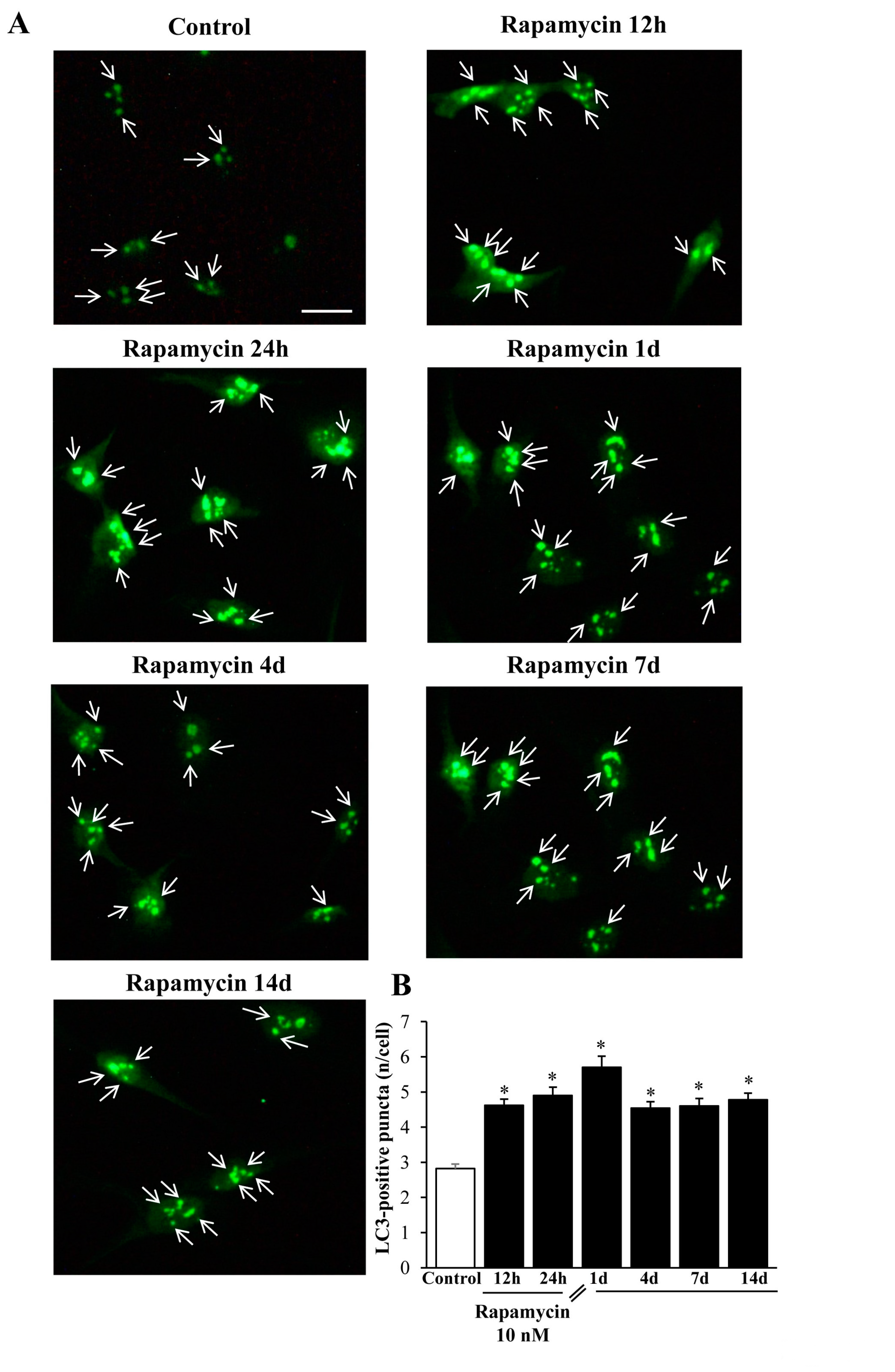
2.5. Immune-Fluorescence of LC3 within U87MG and A172 Cells
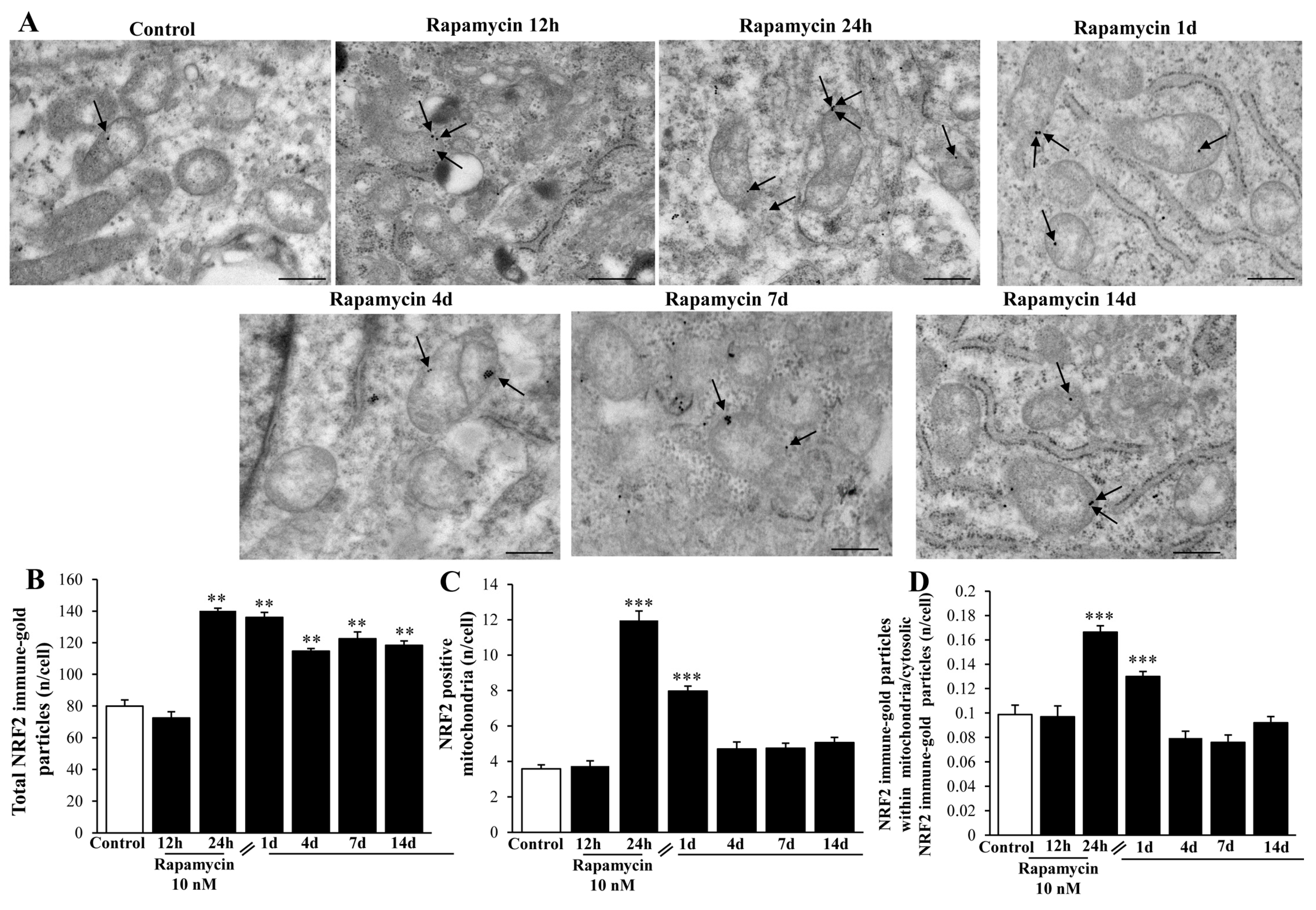
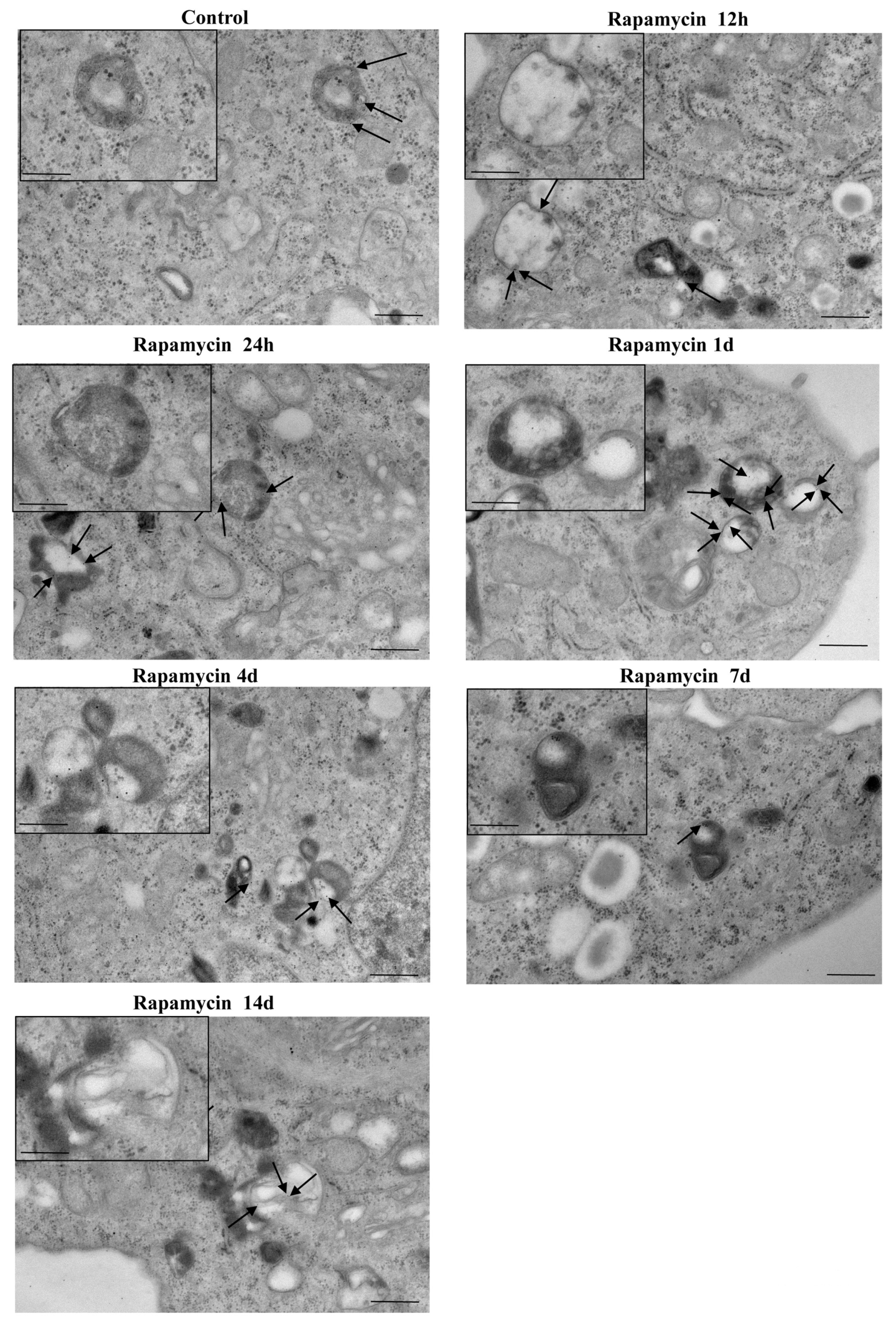
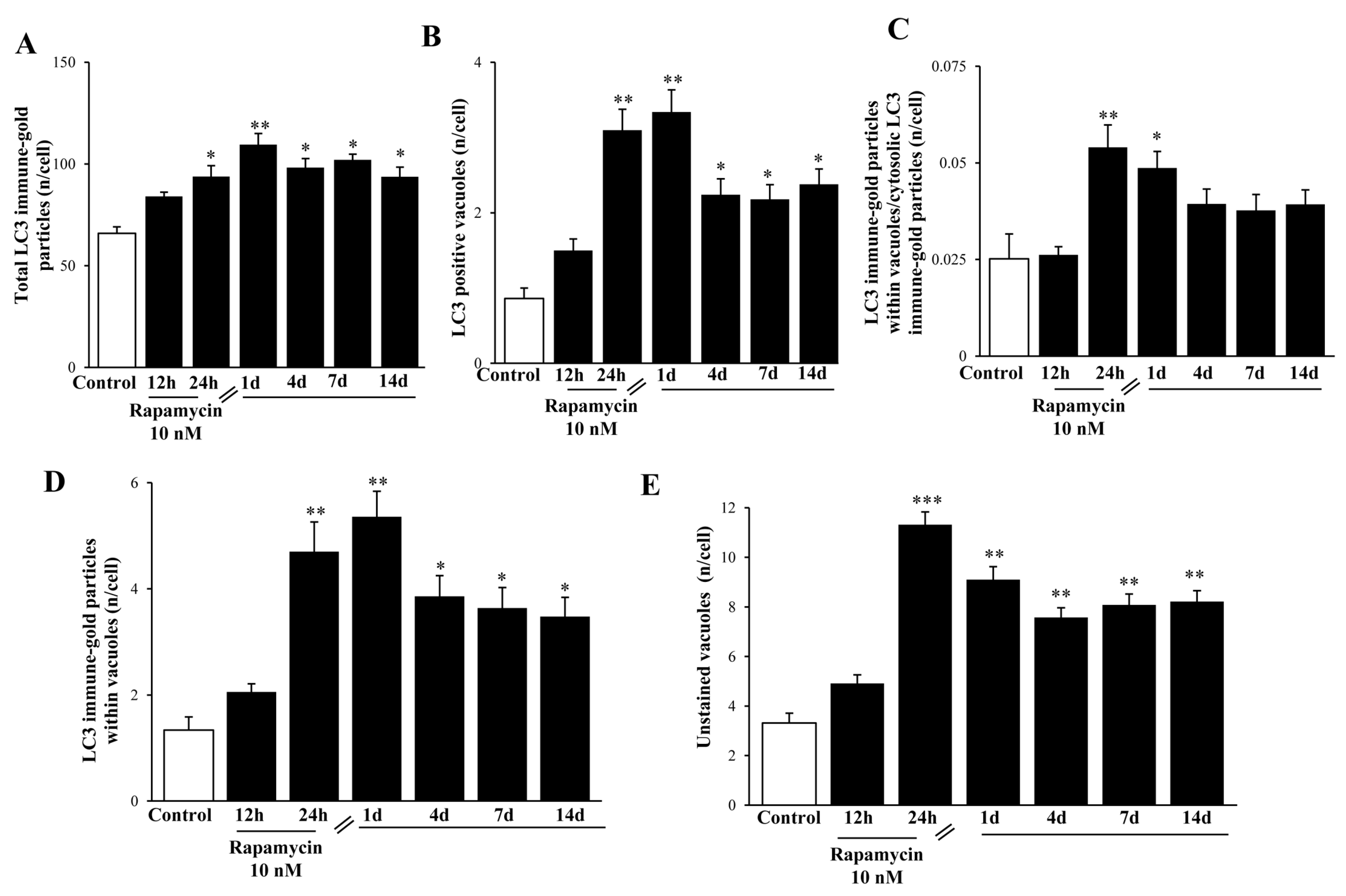
2.6. In Situ Immune-Gold-Based Assessment of Autophagy
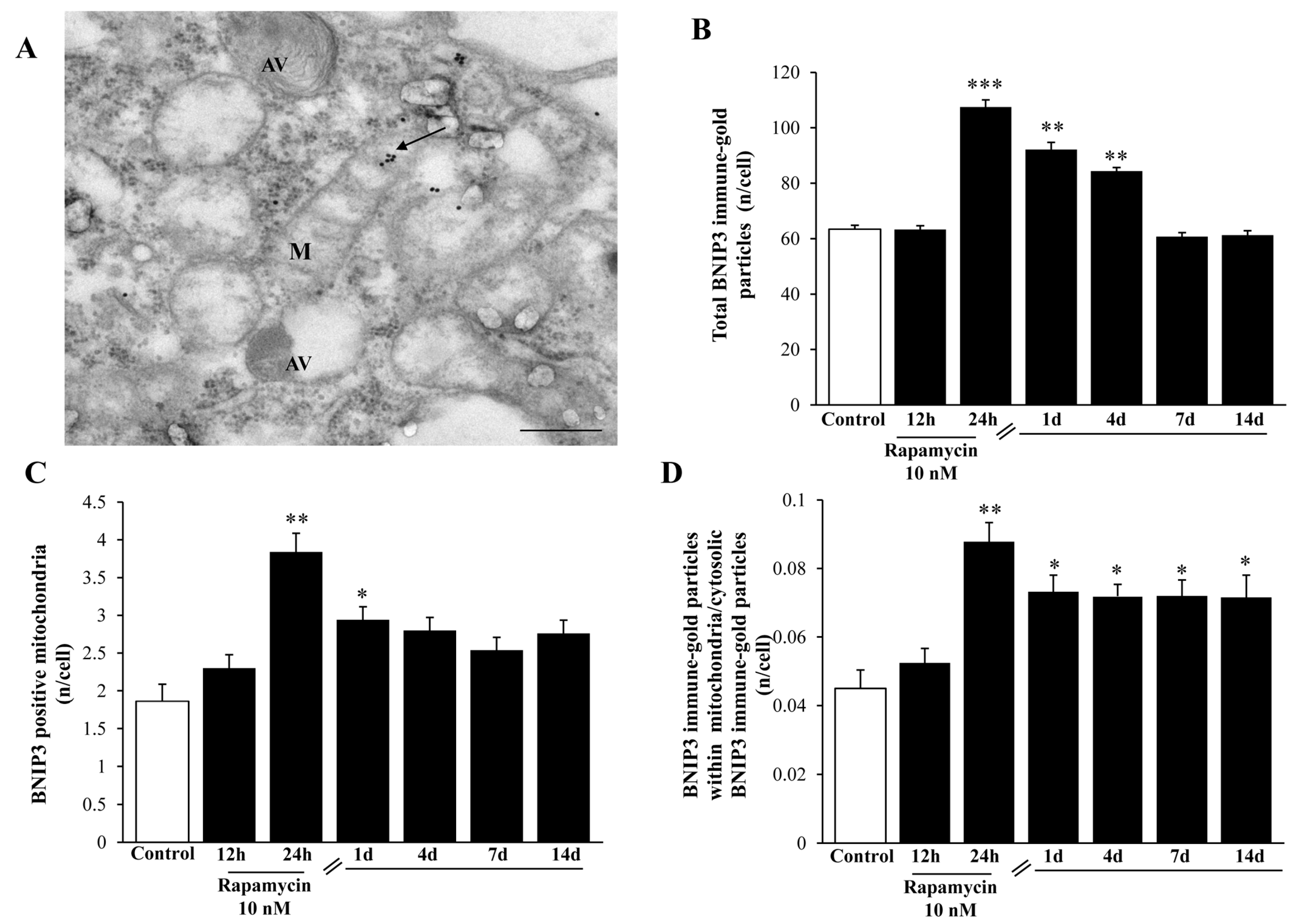
2.7. Mitophagy Copes with Autophagy Activation
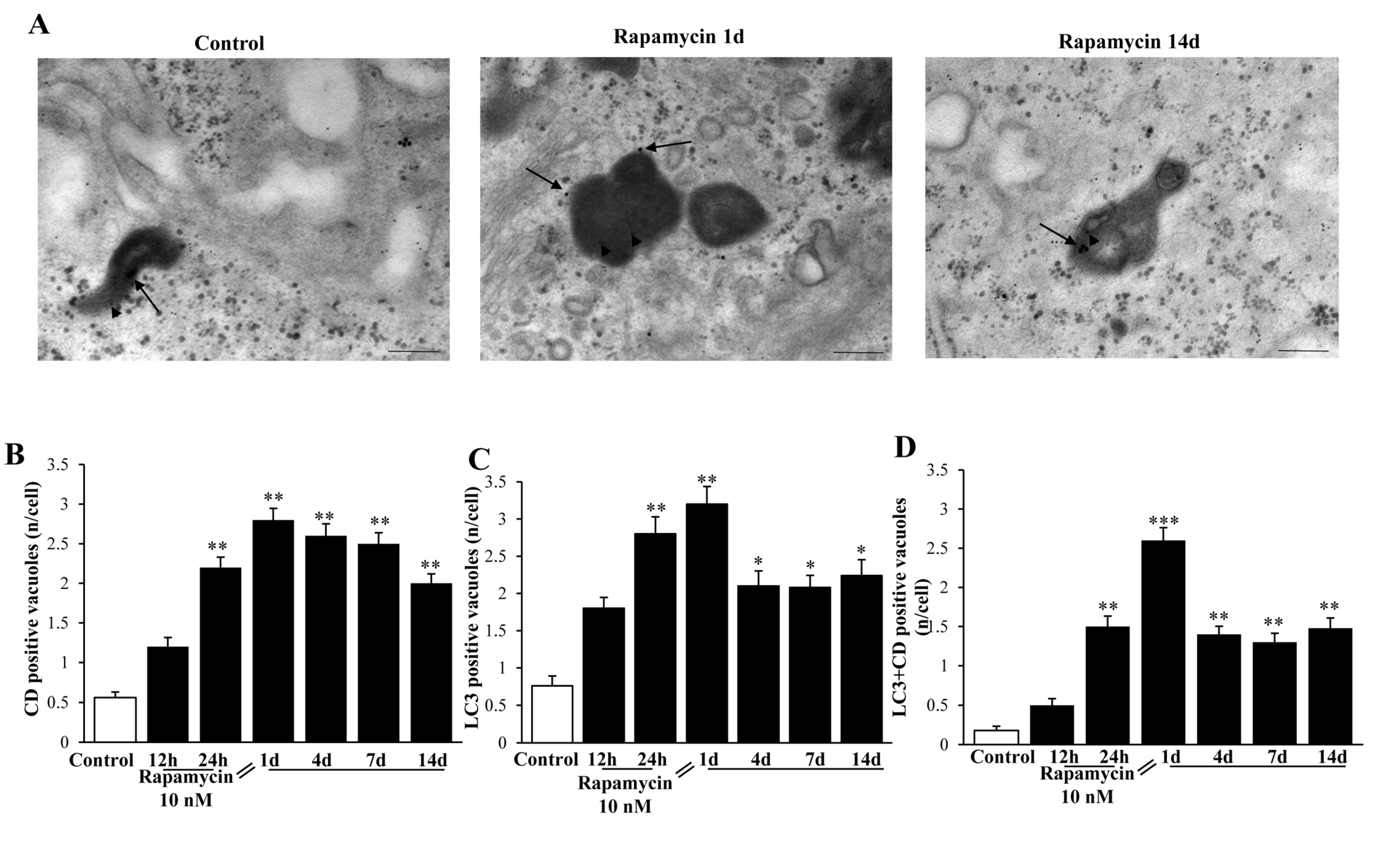
2.8. Autophagy Activation Includes Merging with Lysosomes
2.9. The Role of mTORC1 in the Effects Induced by Rapamycin in GBM Cell Lines
3. Discussion and Conclusions
4. Materials and Methods
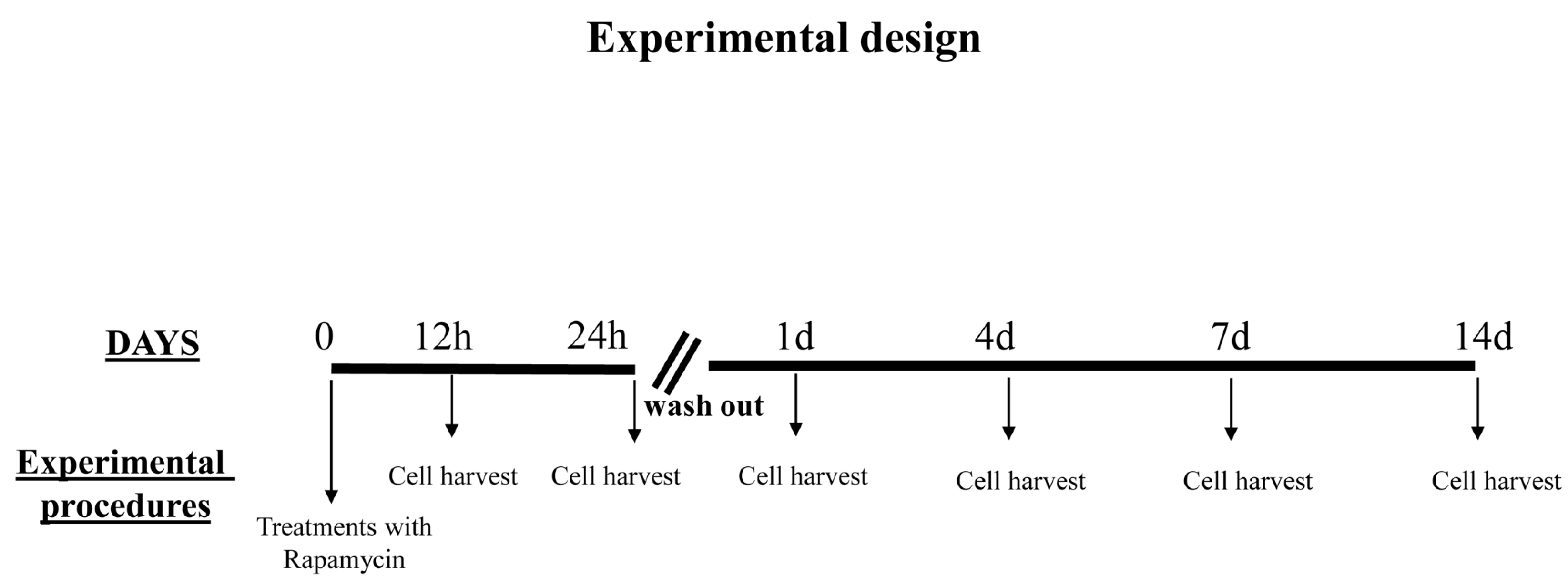
4.1. Experimental Design
Considerations on Pilot Experiments Assessing the Effects of Various Doses and Times of Rapamycin on the Mitochondrial Number
4.2. RNA Extraction
4.3. qReal-Time-PCR
4.4. Transmission Electron Microscopy (TEM)
4.4.1. Post-Embedding Immune-Electron Microscopy
4.4.2. Ultra-Structural Analysis of Mitochondria
4.5. Light Microscopy for MitoTracker Dye
4.6. Light Microscopy for LC3
4.7. SDS-PAGE Immune-Blotting
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jiang, H.; White, E.J.; Conrad, C.; Gomez-Manzano, C.; Fueyo, J. Autophagy pathways in glioblastoma. Methods Enzymol. 2009, 453, 273–286. [Google Scholar] [PubMed]
- Zhao, Y.; Huang, Q.; Yang, J.; Lou, M.; Wang, A.; Dong, J.; Qin, Z.; Zhang, T. Autophagy impairment inhibits differentiation of glioma stem/progenitor cells. Brain Res. 2010, 1313, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Bai, H.M.; Chen, L.; Li, B.; Lu, Y.C. Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors. J. Clin. Neurosci. 2010, 17, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer. 2011, 129, 2720–2731. [Google Scholar] [CrossRef]
- Liu, R.; Li, J.; Zhang, T.; Zou, L.; Chen, Y.; Wang, K.; Lei, Y.; Yuan, K.; Li, Y.; Lan, J.; et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy 2014, 10, 1241–1255. [Google Scholar] [CrossRef]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, piiE3824. [Google Scholar] [CrossRef]
- Eskelinen, E. Maturation of autophagic vacuoles in mammalian cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef]
- Lum, J.J.; DeBerardinis, R.J.; Thompson, C.B. Autophagy in metazoans: Cell survival in the land of plenty. Nat. Rev. Mol. Cell. Biol. 2005, 6, 439–448. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell. Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam, D.H.; Wang, J. ULK1⋅ATG13⋅FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef]
- Neufeld, T.P. TOR-dependent control of autophagy: Biting the hand that feeds. Curr. Opin. Cell. Biol. 2010, 22, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Zhai, G.; Suzuki, Y.; Sarkesh, S.; Black, P.M.; Muzikansky, A.; Loeffler, J.S. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J. Clin. Oncol. 2004, 22, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Sunayama, J.; Sato, A.; Matsuda, K.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro. Oncol. 2010, 12, 1205–1219. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhang, L.Q.; Zhang, X.G.; Li, X.; Ren, Y.B.; Ma, X.Y.; Li, X.G.; Wang, L.X. Association between AKT/mTOR signalling pathway and malignancy grade of human gliomas. J. Neuroncol. 2011, 103, 453–458. [Google Scholar] [CrossRef]
- Fan, Q.W.; Weiss, W.A. Autophagy and Akt promote survival in glioma. Autophagy 2011, 7, 536–538. [Google Scholar] [CrossRef]
- Galan-Moya, E.M.; Le Guelte, A.; Lima Fernandes, E.; Thirant, C.; Dwyer, J.; Bidere, N.; Couraud, P.O.; Scott, M.G.; Junier, M.P.; Chneiweiss, H.; et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. 2011, 12, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing mTOR complexes in regulation of glioblastoma multiforme and its stem cells. Adv. Biol. Regul. 2013, 53, 202–210. [Google Scholar] [CrossRef]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Suppression of the invasive potential of glioblastoma cells by mTOR inhibitors involves modulation of NFκB and PKC-α signaling. Sci. Rep. 2016, 6, 22455. [Google Scholar] [CrossRef]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef]
- Ferrucci, M.; Biagioni, F.; Lenzi, P.; Gambardella, S.; Ferese, R.; Calierno, M.T.; Falleni, A.; Grimaldi, A.; Frati, A.; Esposito, V.; et al. Rapamycin promotes differentiation increasing βIII-tubulin, NeuN, and NeuroD while suppressing nestin expression in glioblastoma cells. Oncotarget 2017, 8, 29574–29599. [Google Scholar] [CrossRef]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. Biomed. Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Biagioni, F.; Frati, A.; Esposito, V.; Calierno, M.T.; Lenzi, P.; Fornai, F. The emerging role of m-TOR up-regulation in brain Astrocytoma. Histol. Histopathol. 2017, 32, 413–431. [Google Scholar] [PubMed]
- Arcella, A.; Biagioni, F.; Antonietta Oliva, M.; Bucci, D.; Frati, A.; Esposito, V.; Cantore, G.; Giangaspero, F.; Fornai, F. Rapamycin inhibits the growth of glioblastoma. Brain Res. 2013, 1495, 37–51. [Google Scholar] [CrossRef]
- Kim, E.H.; Jo, Y.; Sai, S.; Park, M.J.; Kim, J.Y.; Kim, J.S.; Lee, Y.J.; Cho, J.M.; Kwak, S.Y.; Baek, J.H.; et al. Tumor-treating fields induce autophagy by blocking the Akt2/miR29b axis in glioblastoma cells. Oncogene 2019, 38, 6630–6646. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Facchini, V.; Kiss, R. Proautophagic drugs: A novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist 2007, 12, 1395–1403. [Google Scholar] [CrossRef]
- Friedman, M.D.; Jeevan, D.S.; Tobias, M.; Murali, R.; Jhanwar-Uniyal, M. Targeting cancer stem cells in glioblastoma multiforme using mTOR inhibitors and the differentiating agent all-trans retinoic acid. Oncol. Rep. 2013, 30, 1645–1650. [Google Scholar] [CrossRef]
- Palumbo, S.; Tini, P.; Toscano, M.; Allavena, G.; Angeletti, F.; Manai, F.; Miracco, C.; Comincini, S.; Pirtoli, L. Combined EGFR and autophagy modulation impairs cell migration and enhances radiosensitivity in human glioblastoma cells. J. Cell. Physiol. 2014, 229, 1863–1873. [Google Scholar] [CrossRef]
- Ma, J.; Meng, F.; Li, S.; Liu, L.; Zhao, L.; Liu, Y.; Hu, Y.; Li, Z.; Yao, Y.; Xi, Z.; et al. Autophagy induction by endothelial-monocyte activating polypeptide II contributes to the inhibition of malignant biological behaviors by the combination of EMAP II with rapamycin in human glioblastoma. Front. Mol. Neurosci. 2015, 8, 74. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Andreieva, S.V.; Korets, K.V.; Mykytenko, D.O.; Baklaushev, V.P.; Chekhonin, V.P.; Dmitrenko, V.V. mTOR inhibitor temsirolimus and MEK1/2 inhibitor U0126 promote chromosomal instability and cell type-dependent phenotype changes of glioblastoma cells. Gene 2016, 579, 58–68. [Google Scholar] [CrossRef]
- Ren, Z.; Liang, J.; Zhang, P.; Chen, J.; Wen, J. Inhibition of human glioblastoma cell invasion involves PION@E6 mediated autophagy process. Cancer Manag. Res. 2019, 11, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, N. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35, 7S–14S. [Google Scholar] [CrossRef]
- Franz, D.N.; Leonard, J.; Tudor, C.; Chuck, G.; Care, M.; Sethuraman, G.; Dinopoulos, A.; Thomas, G.; Crone, K.R. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann. Neurol. 2006, 59, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef]
- Stenton, S.B.; Partovi, N.; Ensom, M.H. Sirolimus: The evidence for clinical pharmacokinetic monitoring. Clin. Pharmacokinet. 2005, 44, 769–786. [Google Scholar] [CrossRef]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell. Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coupling mitogenesis and mitophagy for longevity. Autophagy 2015, 11, 1428–1430. [Google Scholar] [CrossRef]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in gliomas and their vascular microenvironment. Biochim. Biophys. Acta. 2011, 1807, 602–608. [Google Scholar] [CrossRef]
- Arismendi-Morillo, G.; Castellano-Ramírez, A.; Seyfried, T.N. Ultrastructural characterization of the Mitochondria-associated membranes abnormalities in human astrocytomas: Functional and therapeutics implications. Ultrastruct. Pathol. 2017, 41, 234–244. [Google Scholar] [CrossRef]
- Deighton, R.F.; Le Bihan, T.; Martin, S.F.; Gerth, A.M.J.; McCulloch, M.; Edgar, J.M.; Kerr, L.E.; Whittle, I.R.; McCulloch, J. Interactions among mitochondrial proteins altered in glioblastoma. J. Neurooncol. 2014, 118, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.C.; Grootveld, M. The importance of mitochondria in the tumourigenic phenotype: Gliomasas the paradigm. Int. J. Mol. Med. 2011, 27, 159–171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ruffoli, R.; Bartalucci, A.; Frati, A.; Fornai, F. Ultrastructural studies of ALS mitochondria connect altered function and permeability with defects of mitophagy and mitochondriogenesis. Front. Cell Neurosci. 2015, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, M.; Kaidery, N.A.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian., D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef]
- Huang, D.D.; Fan, S.D.; Chen, X.Y.; Yan, X.L.; Zhang, X.Z.; Ma, B.W.; Yu, D.Y.; Xiao, W.Y.; Zhuang, C.L.; Yu, Z. Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner. Exp. Gerontol. 2019, 119, 61–73. [Google Scholar] [CrossRef]
- Kitaoka, Y.; Tamura, Y.; Takahashi, K.; Takeda, K.; Takemasa, T.; Hatta, H. Effects of Nrf2 deficiency on mitochondrial oxidative stress in aged skeletal muscle. Physiol. Rep. 2019, 7, e13998. [Google Scholar] [CrossRef]
- Ghadially, F.N. Ultrastructural Pathology of the Cell and Matrix, 3rd ed.; Wellington Butterworths: London, UK, 1988; pp. 191–328. [Google Scholar]
- Fornai, F.; Longone, P.; Ferrucci, M.; Lenzi, P.; Isidoro, C.; Ruggieri, S.; Paparelli, A. Autophagy and amyotrophic lateral sclerosis: The multiple roles of lithium. Autophagy 2008, 4, 527–530. [Google Scholar] [CrossRef]
- Struewing, I.T.; Barnett, C.D.; Tang, T.; Mao, C.D. Lithium Increases PGC-1alpha Expression and Mitochondrial Biogenesis in Primary Bovine Aortic Endothelial Cells. FEBS J. 2007, 274, 2749–2765. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Chan, J.Y.; Cheung, M.C.; Moi, P.; Chan, K.; Kan, Y.W. Chromosomal localization of the human NF-E2 family of bZIP transcription factors by fluorescence in situ hybridization. Hum. Genet. 1995, 95, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell. Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell. Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and Neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef]
- Dupuis, A.; Chevallet, M.; Darrouzet, E.; Duborjal, H.; Lunardi, J.; Issartel, J.P. The complex I from Rhodobacter capsulatus. Biochem. Biophys. Acta. 1998, 1364, 147–165. [Google Scholar] [CrossRef][Green Version]
- Chen, T.; He, J.; Shen, L.; Fang, H.; Nie, H.; Jin, T.; Wei, X.; Xin, Y.; Jiang, Y.; Li, H.; et al. The mitochondrial DNA 4977-bp deletion and its implication in copy number alteration in colorectal cancer. BMC Med. Genet. 2011, 12, 8. [Google Scholar] [CrossRef]
- Pasquali, L.; Longone, P.; Isidoro, C.; Ruggieri, S.; Paparelli, A.; Fornai, F. Autophagy, lithium, and amyotrophic lateral sclerosis. Muscle Nerve. 2009, 40, 173–194. [Google Scholar] [CrossRef]
- Schefe, J.H.; Lehmann, K.E.; Buschmann, I.R.; Unger, T.; Funke-Kaiser, H. Quantitative real-time RT-PCR data analysis: Current concepts and the novel “gene expression’s CT difference” formula. J. Mol. Med. (Berl). 2006, 84, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.S.; Reed, A.; Chen, F.; Stewart, C.N., Jr. Statistical analysis of real-time PCR data. BMC Bioinform. 2006, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Marongiu, R.; Falleni, A.; Gelmetti, V.; Busceti, C.L.; Michiorri, S.; Valente, E.M.; Fornai, F. A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 2012, 150, 194–217. [Google Scholar] [PubMed]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome a Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell. Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef]
- Stirling, J.W.; Graff, P.S. Antigen unmasking for immunoelectron microscopy: Labeling is improved by treating with sodium ethoxide or sodium metaperiodate, then heating on retrieval medium. J. Histochem. Cytochem. 1995, 43, 115–123. [Google Scholar] [CrossRef]
- Pendergrass, W.; Wolf, N.; Poot, M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry A 2004, 61, 162–169. [Google Scholar] [CrossRef]
- Gautam, N.; Sankaran, S.; Yason, J.A.; Tan, K.S.W.; Gascoigne, N.R.J. A high content imaging flow cytometry approach to study mitochondria in T cells: MitoTracker Green FM dye concentration optimization. Methods 2018, 134–135, 11–19. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Target Sequence Number | Primer Sequences |
|---|---|---|
| PGC1α | NM_001330752 | FW 5′- GAGCTTCTCAAATATCTGAC -3′ RW 5′- GGTCAGAGGAAGAGATAAA -3′ |
| TFAM | NM_003201.3 | FW 5′- AAGTCAGATTATGTCTTTGG -3′ RW 5′-TTCCAAGCAGTGTTAACTC-3′ |
| NRF2 | NM_006164.5 | FW 5′- ACCATTTCAGATGAAACTTCAG -3′ RW 5′- AAACCACCCAATGCAGGAC -3′ |
| ND1 | LC178901.1 | FW 5′- ATACCCATGGCCAACCTCCTACTCCTC -3′ RW 5′- GGTATGGGGAGGGGGGTTCATAG -3′, |
| ND2 | LC178901.1 | FW 5′- CCCTTACCACGCTACTCCTA -3′ RW 5′- GGCGGGAGAAGTAGATTGAA -3′ |
| ND4 | LC178901.1 | FW 5′-AAAACTAATCGTCCCAACAATT -3′ RW 5′- ATAAGTGGCGTTGGCTTGCCATGATTG -3′ |
| ATP6 | LC178901.1 | FW 5′ - CCTTCCACAAGGAACTCCAA -3′ RW 5′- GGTAGCTGTTGGTGGGCTAA -3′ |
| Cyt b | LC178901.1 | FW 5′- ATTCCTTCATGTCGGGACGAG -3′ RW 5′- GGGATGGCTGATAGGAGGTT -3′ |
| LC3 | NM_032514.4 | FW 5′ -CATGAGCGAGTTGGTCAAGA-3′ RW 5′ -CCATGCTGTGCTGGTTCA- 3′ |
| Beclin1 | NM_001313998.2 | FW 5′ -GGATGGTGTCTCTCGCAGAT- 3′ RW 5′ -TTGGCACTTTCTGTGGACAT- 3′ |
| β–Actin | NM_001101.3 | FW 5′-GTGCGTGACATTAAGGAG-3′ RW 5′-GCCAGACAGCACTGTGT-3′ |
| β-Globin | NM_000518.4 | FW 5′-CTAAGGTGAAGGCTCATG-3′ RW 5′- GATAGGCAGCCTGCACT-3 |
| Antibody | Purchaser | Cat# | Research Resource Identifier (RRID) |
|---|---|---|---|
| Rabbit anti-NRF2 | Abcam | ab137550 | AB_2687540 |
| Rabbit anti-LC3 | Abcam | ab128025 | AB_11143008 |
| Rabbit anti-BNIP3 | Thermo Fisher Scientific | 701696 | AB_2607981 |
| Mouse anti-Cathepsin-D | Sigma-Aldrich | C0715 | AB_258707 |
| EM Goat anti-Rabbit IgG, gold conjugate antibody | BBInternational | EM.GAR 10 | AB_1769128 |
| EM Goat anti-Rabbit IgG, gold conjugate antibody | BBInternational | EM.GAR 20 | AB_1769136 |
| Goat Anti-Mouse IgG (H+L) | BBInternational | EM.GMHL20/0.25 | AB_1769164 |
| Mouse anti-Phospho-p70 S6 Kinase | Cell Signaling | 9025 | AB_2734746 |
| Alexa Fluor 488 | Thermo Fisher Scientific | A-11008 | AB_143165 |
| Goat anti-Mouse IgG, H+L chain specific peroxidase conjugate antibody | Millipore | AP308P | AB_11215796 |
| Goat anti-Rabbit IgG, H+L chain specific peroxidase conjugate antibody | Millipore | AP307P | AB_92641 |
| Rabbit anti-beta Actin antibody | Abcam | ab34731 | AB_722539 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferese, R.; Lenzi, P.; Fulceri, F.; Biagioni, F.; Fabrizi, C.; Gambardella, S.; Familiari, P.; Frati, A.; Limanaqi, F.; Fornai, F. Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells. Int. J. Mol. Sci. 2020, 21, 4570. https://doi.org/10.3390/ijms21134570
Ferese R, Lenzi P, Fulceri F, Biagioni F, Fabrizi C, Gambardella S, Familiari P, Frati A, Limanaqi F, Fornai F. Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells. International Journal of Molecular Sciences. 2020; 21(13):4570. https://doi.org/10.3390/ijms21134570
Chicago/Turabian StyleFerese, Rosangela, Paola Lenzi, Federica Fulceri, Francesca Biagioni, Cinzia Fabrizi, Stefano Gambardella, Pietro Familiari, Alessandro Frati, Fiona Limanaqi, and Francesco Fornai. 2020. "Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells" International Journal of Molecular Sciences 21, no. 13: 4570. https://doi.org/10.3390/ijms21134570
APA StyleFerese, R., Lenzi, P., Fulceri, F., Biagioni, F., Fabrizi, C., Gambardella, S., Familiari, P., Frati, A., Limanaqi, F., & Fornai, F. (2020). Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells. International Journal of Molecular Sciences, 21(13), 4570. https://doi.org/10.3390/ijms21134570