The Toxicity and Polymorphism of β-Amyloid Oligomers


Abstract
1. Introduction
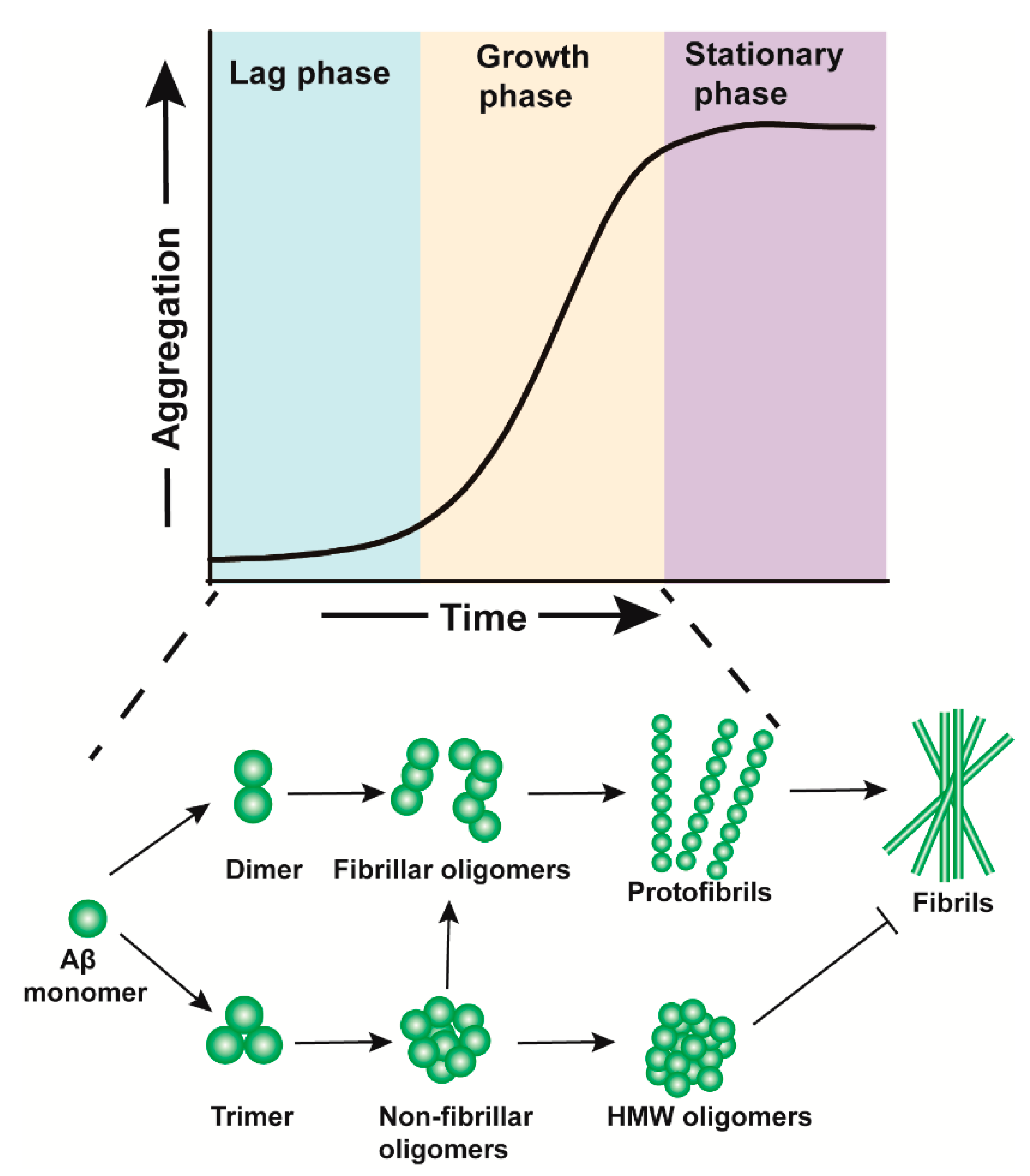
2. The Formation of Aβos
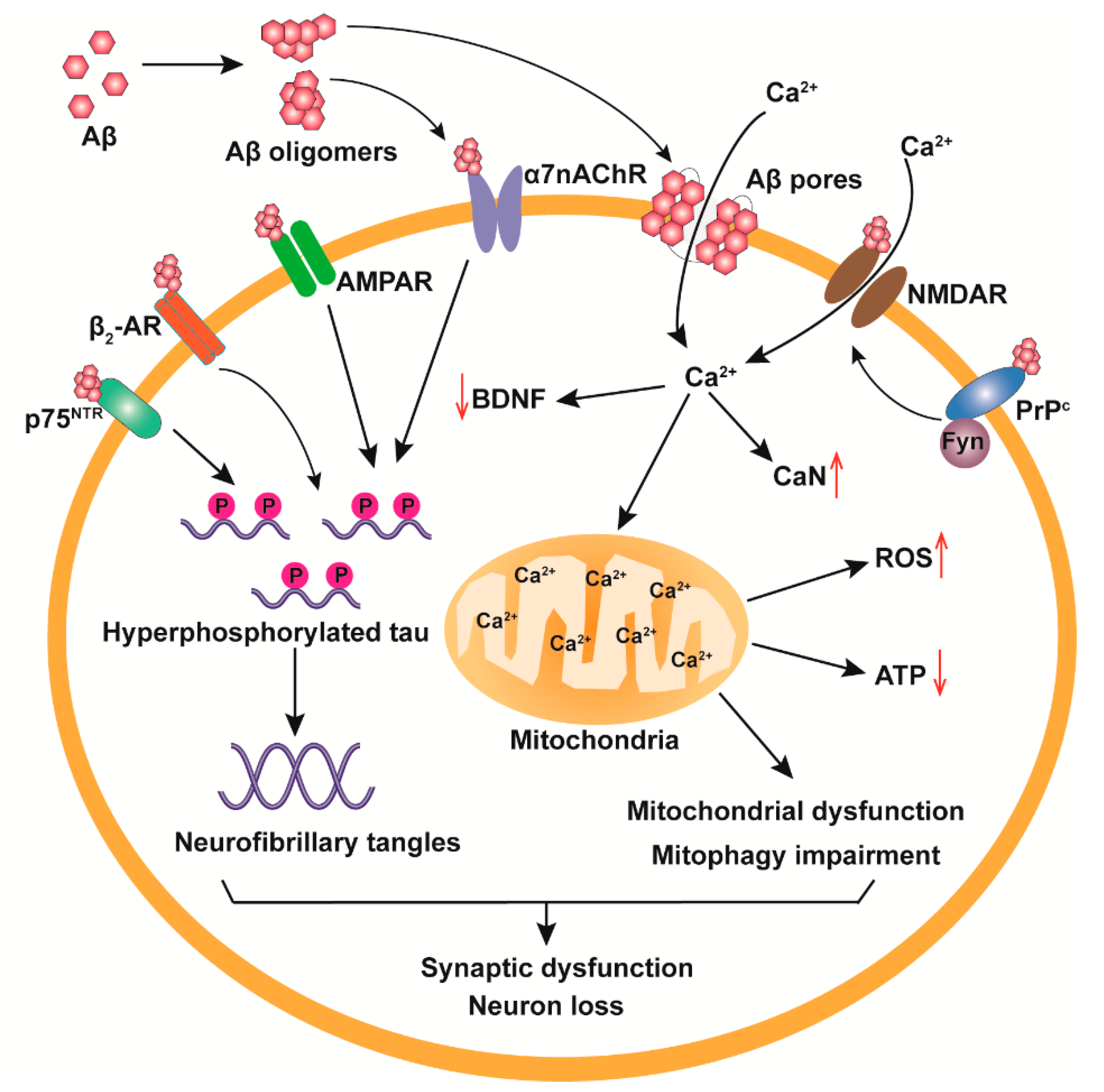
3. The Neurotoxicological Mechanisms of Aβos
3.1. Receptor-Mediated Neurotoxicity of Aβos
3.2. Cell Membrane Destruction
3.3. Mitochondrial Damage and Ca2+ Homeostasis Dysregulation
3.4. Tau Pathologies
4. The Polymorphism of Aβos
4.1. Aβ Dimers
4.2. Aβ Trimers
4.3. Aβ*56
4.4. Spherical Oligomers
4.4.1. ASPD
4.4.2. ADDLs
5. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβos | β-Amyloid oligomers |
| AD | Alzheimer’s Disease |
| Aβ | β-Amyloid |
| APP | Amyloid precursor protein |
| LTP | Long-term potentiation |
| ROS | Reactive oxygen species |
| PrPc | Prion protein |
| β2-AR | β2-Adrenergic receptor and |
| p75NTR | p75 Neurotrophin receptor |
| α7nAChR | α7-Nicotinic acetylcholine receptor |
| AICD | APP intracellular domain |
| NMDAR | N-methyl-d-aspartate receptor |
| AMPAR | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| PKA | Protein kinase A |
| MW | Molecular weight |
| cBF | Cholinergic basal forebrain |
| BACE1 | Beta-site amyloid precursor protein cleaving enzyme-1 |
| PirB | Paired immunoglobulin-like receptor B |
| mGluR5 | Metabotropic glutamate receptor 5 |
| GSK3β | Glycogen synthase kinase 3β |
| BDNF | Brain-derived neurotrophic factor |
| MPTP | Mitochondrial permeability transition pores |
| SEC | Size exclusion chromatography |
| SDS-PAGE | SDS gel electrophoresis |
| KLC1 | Kinesin-1 light chain |
| CaMKIIα | Ca2+-dependent calmodulin kinase IIα |
| ASPD | Amylospheroids |
| TEM | Transmission electron microscopic |
| NAKα3 | Na+/K+-ATPase α3 subunit |
| ADDLs | Aβ-derived diffusible aggregates |
References
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra372. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Koike, H.; Seki, H.; Kouchi, Z.; Ito, M.; Kinouchi, T.; Tomioka, S.; Sorimachi, H.; Saido, T.C.; Maruyama, K.; Suzuki, K.; et al. Thimet oligopeptidase cleaves the full-length Alzheimer amyloid precursor protein at a beta-secretase cleavage site in COS cells. J. Biochem. 1999, 126, 235–242. [Google Scholar] [CrossRef]
- Levitan, D.; Lee, J.; Song, L.; Manning, R.; Wong, G.; Parker, E.; Zhang, L. PS1 N- and C-terminal fragments form a complex that functions in APP processing and Notch signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 12186–12190. [Google Scholar] [CrossRef]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010, 13, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Whitson, J.S.; Selkoe, D.J.; Cotman, C.W. Amyloid beta protein enhances the survival of hippocampal neurons in vitro. Science 1989, 243, 1488–1490. [Google Scholar] [CrossRef]
- Koo, E.H.; Park, L.; Selkoe, D.J. Amyloid beta-protein as a substrate interacts with extracellular matrix to promote neurite outgrowth. Proc. Natl. Acad. Sci. USA 1993, 90, 4748–4752. [Google Scholar] [CrossRef]
- Eimer, W.A.; Kumar, D.K.V.; Shanmugam, N.K.N.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; Gyorgy, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated beta-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63.e3. [Google Scholar] [CrossRef] [PubMed]
- Bourgade, K.; Le Page, A.; Bocti, C.; Witkowski, J.M.; Dupuis, G.; Frost, E.H.; Fulop, T., Jr. Protective Effect of Amyloid-beta Peptides Against Herpes Simplex Virus-1 Infection in a Neuronal Cell Culture Model. J. Alzheimers Dis. 2016, 50, 1227–1241. [Google Scholar] [CrossRef]
- Hubin, E.; Deroo, S.; Schierle, G.K.; Kaminski, C.; Serpell, L.; Subramaniam, V.; van Nuland, N.; Broersen, K.; Raussens, V.; Sarroukh, R. Two distinct beta-sheet structures in Italian-mutant amyloid-beta fibrils: A potential link to different clinical phenotypes. Cell. Mol. Life Sci. 2015, 72, 4899–4913. [Google Scholar] [CrossRef]
- Luo, J.; Warmlander, S.K.; Graslund, A.; Abrahams, J.P. Reciprocal Molecular Interactions between the Abeta Peptide Linked to Alzheimer’s Disease and Insulin Linked to Diabetes Mellitus Type II. ACS Chem. Neurosci. 2016, 7, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Tornquist, M.; Michaels, T.C.T.; Sanagavarapu, K.; Yang, X.; Meisl, G.; Cohen, S.I.A.; Knowles, T.P.J.; Linse, S. Secondary nucleation in amyloid formation. Chem. Commun. 2018, 54, 8667–8684. [Google Scholar] [CrossRef]
- Michaels, T.C.T.; Saric, A.; Curk, S.; Bernfur, K.; Arosio, P.; Meisl, G.; Dear, A.J.; Cohen, S.I.A.; Dobson, C.M.; Vendruscolo, M.; et al. Dynamics of oligomer populations formed during the aggregation of Alzheimer’s Abeta42 peptide. Nat. Chem. 2020, 12, 445–451. [Google Scholar] [CrossRef]
- Szczepankiewicz, O.; Linse, B.; Meisl, G.; Thulin, E.; Frohm, B.; Sala Frigerio, C.; Colvin, M.T.; Jacavone, A.C.; Griffin, R.G.; Knowles, T.; et al. N-Terminal Extensions Retard Abeta42 Fibril Formation but Allow Cross-Seeding and Coaggregation with Abeta42. J. Am. Chem. Soc. 2015, 137, 14673–14685. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef]
- Fandrich, M. Oligomeric intermediates in amyloid formation: Structure determination and mechanisms of toxicity. J. Mol. Biol. 2012, 421, 427–440. [Google Scholar] [CrossRef]
- Matsumura, S.; Shinoda, K.; Yamada, M.; Yokojima, S.; Inoue, M.; Ohnishi, T.; Shimada, T.; Kikuchi, K.; Masui, D.; Hashimoto, S.; et al. Two distinct amyloid beta-protein (Abeta) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology, and toxicity analyses. J. Biol. Chem. 2011, 286, 11555–11562. [Google Scholar] [CrossRef]
- Gellermann, G.P.; Byrnes, H.; Striebinger, A.; Ullrich, K.; Mueller, R.; Hillen, H.; Barghorn, S. Abeta-globulomers are formed independently of the fibril pathway. Neurobiol. Dis. 2008, 30, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Tabaton, M.; Nunzi, M.G.; Xue, R.; Usiak, M.; Autilio-Gambetti, L.; Gambetti, P. Soluble amyloid beta-protein is a marker of Alzheimer amyloid in brain but not in cerebrospinal fluid. Biochem. Biophys. Res. Commun. 1994, 200, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Frackowiak, J.; Zoltowska, A.; Wisniewski, H.M. Non-fibrillar beta-amyloid protein is associated with smooth muscle cells of vessel walls in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1994, 53, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid beta Oligomers (AbetaOs) in Alzheimer’s Disease. Int J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef]
- Vincent, B.; Sunyach, C.; Orzechowski, H.D.; St George-Hyslop, P.; Checler, F. p53-Dependent transcriptional control of cellular prion by presenilins. J. Neurosci. 2009, 29, 6752–6760. [Google Scholar] [CrossRef]
- Younan, N.D.; Chen, K.F.; Rose, R.S.; Crowther, D.C.; Viles, J.H. Prion protein stabilizes amyloid-beta (Abeta) oligomers and enhances Abeta neurotoxicity in a Drosophila model of Alzheimer’s disease. J. Biol. Chem. 2018, 293, 13090–13099. [Google Scholar] [CrossRef]
- Pagano, K.; Galante, D.; D’Arrigo, C.; Corsaro, A.; Nizzari, M.; Florio, T.; Molinari, H.; Tomaselli, S.; Ragona, L. Effects of Prion Protein on Abeta42 and Pyroglutamate-Modified AbetapEpsilon3-42 Oligomerization and Toxicity. Mol. Neurobiol. 2019, 56, 1957–1971. [Google Scholar] [CrossRef]
- Haas, L.T.; Salazar, S.V.; Kostylev, M.A.; Um, J.W.; Kaufman, A.C.; Strittmatter, S.M. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain 2016, 139, 526–546. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Corbett, G.T.; Wang, Z.; Hong, W.; Colom-Cadena, M.; Rose, J.; Liao, M.; Asfaw, A.; Hall, T.C.; Ding, L.; DeSousa, A.; et al. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Qi, Y.; Klyubin, I.; Ondrejcak, T.; Sarell, C.J.; Cuello, A.C.; Collinge, J.; Rowan, M.J. Targeting glutamatergic and cellular prion protein mechanisms of amyloid beta-mediated persistent synaptic plasticity disruption: Longitudinal studies. Neuropharmacology 2017, 121, 231–246. [Google Scholar] [CrossRef]
- Li, F.; Tsien, J.Z. Memory and the NMDA receptors. N. Engl. J. Med. 2009, 361, 302–303. [Google Scholar] [CrossRef]
- Muller, M.K.; Jacobi, E.; Sakimura, K.; Malinow, R.; von Engelhardt, J. NMDA receptors mediate synaptic depression, but not spine loss in the dentate gyrus of adult amyloid Beta (Abeta) overexpressing mice. Acta Neuropathol. Commun. 2018, 6, 110. [Google Scholar] [CrossRef]
- Liang, J.; Kulasiri, D.; Samarasinghe, S. Computational investigation of Amyloid-beta-induced location- and subunit-specific disturbances of NMDAR at hippocampal dendritic spine in Alzheimer’s disease. PLoS ONE 2017, 12, e0182743. [Google Scholar] [CrossRef]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, O.; Huo, Y.; Wang, G.; Man, H.Y. Amyloid-beta Induces AMPA Receptor Ubiquitination and Degradation in Primary Neurons and Human Brains of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1789–1801. [Google Scholar] [CrossRef]
- Miller, E.C.; Teravskis, P.J.; Dummer, B.W.; Zhao, X.; Huganir, R.L.; Liao, D. Tau phosphorylation and tau mislocalization mediate soluble Abeta oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci. 2014, 39, 1214–1224. [Google Scholar] [CrossRef]
- Wang, D.; Fu, Q.; Zhou, Y.; Xu, B.; Shi, Q.; Igwe, B.; Matt, L.; Hell, J.W.; Wisely, E.V.; Oddo, S.; et al. Beta2 adrenergic receptor, protein kinase A (PKA) and c-Jun N-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J. Biol. Chem. 2013, 288, 10298–10307. [Google Scholar] [CrossRef]
- Wang, D.; Govindaiah, G.; Liu, R.; De Arcangelis, V.; Cox, C.L.; Xiang, Y.K. Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. FASEB J. 2010, 24, 3511–3521. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid beta-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2017, 37, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.R.; Wang, J.; Liu, Y.H.; Hu, G.L.; Gao, C.Y.; Wang, Y.J.; Zhou, X.F.; Zeng, F. Cysteine-Rich Repeat Domains 2 and 4 are Amyloid-beta Binding Domains of Neurotrophin Receptor p75NTR and Potential Targets to Block Amyloid-beta Neurotoxicity. J. Alzheimers Dis. 2018, 63, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L.; Li, W.W.; Xu, Y.L.; Gao, S.H.; Xu, M.Y.; Bu, X.L.; Liu, Y.H.; Wang, J.; Zhu, J.; Zeng, F.; et al. Neurotrophin receptor p75 mediates amyloid beta-induced tau pathology. Neurobiol. Dis. 2019, 132, 104567. [Google Scholar] [CrossRef]
- Manucat-Tan, N.B.; Shen, L.L.; Bobrovskaya, L.; Al-Hawwas, M.; Zhou, F.H.; Wang, Y.J.; Zhou, X.F. Knockout of p75 neurotrophin receptor attenuates the hyperphosphorylation of Tau in pR5 mouse model. Aging Albany Ny 2019, 11, 6762–6791. [Google Scholar]
- Qian, L.; Milne, M.R.; Shepheard, S.; Rogers, M.L.; Medeiros, R.; Coulson, E.J. Removal of p75 Neurotrophin Receptor Expression from Cholinergic Basal Forebrain Neurons Reduces Amyloid-beta Plaque Deposition and Cognitive Impairment in Aged APP/PS1 Mice. Mol. Neurobiol. 2019, 56, 4639–4652. [Google Scholar] [CrossRef] [PubMed]
- Saadipour, K.; Manucat-Tan, N.B.; Lim, Y.; Keating, D.J.; Smith, K.S.; Zhong, J.H.; Liao, H.; Bobrovskaya, L.; Wang, Y.J.; Chao, M.V.; et al. p75 neurotrophin receptor interacts with and promotes BACE1 localization in endosomes aggravating amyloidogenesis. J. Neurochem. 2018, 144, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.Q.; Jiao, S.S.; Saadipour, K.; Zeng, F.; Wang, Q.H.; Zhu, C.; Shen, L.L.; Zeng, G.H.; Liang, C.R.; Wang, J.; et al. p75NTR ectodomain is a physiological neuroprotective molecule against amyloid-beta toxicity in the brain of Alzheimer’s disease. Mol. Psychiatry 2015, 20, 1301–1310. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Yang, C.; Zhao, H.; Jian, C. Involvement of p75NTR in the effects of Abeta on L-type Ca(2+) channel in cultured neuronal networks. Life Sci. 2020, 243, 117293. [Google Scholar] [CrossRef]
- Cecon, E.; Dam, J.; Luka, M.; Gautier, C.; Chollet, A.M.; Delagrange, P.; Danober, L.; Jockers, R. Quantitative assessment of oligomeric amyloid beta peptide binding to alpha7 nicotinic receptor. Br. J. Pharm. 2019, 176, 3475–3488. [Google Scholar] [CrossRef]
- Bencherif, M.; Lippiello, P.M. Alpha7 neuronal nicotinic receptors: The missing link to understanding Alzheimer’s etiopathology? Med. Hypotheses 2010, 74, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Barykin, E.P.; Garifulina, A.I.; Kruykova, E.V.; Spirova, E.N.; Anashkina, A.A.; Adzhubei, A.A.; Shelukhina, I.V.; Kasheverov, I.E.; Mitkevich, V.A.; Kozin, S.A.; et al. Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the alpha7 Nicotinic Receptor and Promotes Neurotoxicity. Cells 2019, 8, 771. [Google Scholar] [CrossRef]
- Takata, K.; Amamiya, T.; Mizoguchi, H.; Kawanishi, S.; Kuroda, E.; Kitamura, R.; Ito, A.; Saito, Y.; Tawa, M.; Nagasawa, T.; et al. Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Abeta accumulation through suppression of neuronal gamma-secretase activity and promotion of microglial amyloid-beta phagocytosis and ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 62, 197–209. [Google Scholar] [PubMed]
- Chang, K.W.; Zong, H.F.; Rizvi, M.Y.; Ma, K.G.; Zhai, W.; Wang, M.; Yang, W.N.; Ji, S.F.; Qian, Y.H. Modulation of the MAPKs pathways affects Abeta-induced cognitive deficits in Alzheimer’s disease via activation of alpha7nAChR. Neurobiol. Learn. Mem 2020, 168, 107154. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.W.; Zong, H.F.; Ma, K.G.; Zhai, W.Y.; Yang, W.N.; Hu, X.D.; Xu, J.H.; Chen, X.L.; Ji, S.F.; Qian, Y.H. Activation of alpha7 nicotinic acetylcholine receptor alleviates Abeta1-42-induced neurotoxicity via downregulation of p38 and JNK MAPK signaling pathways. Neurochem. Int. 2018, 120, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Vidal, G.S.; Djurisic, M.; William, C.M.; Birnbaum, M.E.; Garcia, K.C.; Hyman, B.T.; Shatz, C.J. Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 2013, 341, 1399–1404. [Google Scholar] [CrossRef]
- Cao, Q.; Shin, W.S.; Chan, H.; Vuong, C.K.; Dubois, B.; Li, B.; Murray, K.A.; Sawaya, M.R.; Feigon, J.; Black, D.L.; et al. Inhibiting amyloid-beta cytotoxicity through its interaction with the cell surface receptor LilrB2 by structure-based design. Nat. Chem. 2018, 10, 1213–1221. [Google Scholar] [CrossRef]
- Miyamoto, T.; Kim, D.; Knox, J.A.; Johnson, E.; Mucke, L. Increasing the Receptor Tyrosine Kinase EphB2 Prevents Amyloid-beta-induced Depletion of Cell Surface Glutamate Receptors by a Mechanism That Requires the PDZ-binding Motif of EphB2 and Neuronal Activity. J. Biol. Chem. 2016, 291, 1719–1734. [Google Scholar] [CrossRef]
- Hu, R.; Wei, P.; Jin, L.; Zheng, T.; Chen, W.Y.; Liu, X.Y.; Shi, X.D.; Hao, J.R.; Sun, N.; Gao, C. Overexpression of EphB2 in hippocampus rescues impaired NMDA receptors trafficking and cognitive dysfunction in Alzheimer model. Cell Death Dis. 2017, 8, e2717. [Google Scholar] [CrossRef]
- Lee, M.; Lee, H.J.; Park, I.S.; Park, J.A.; Kwon, Y.J.; Ryu, Y.H.; Kim, C.H.; Kang, J.H.; Hyun, I.Y.; Lee, K.C.; et al. Abeta pathology downregulates brain mGluR5 density in a mouse model of Alzheimer. Neuropharmacology 2018, 133, 512–517. [Google Scholar] [CrossRef]
- Brody, A.H.; Strittmatter, S.M. Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer’s Disease Through Prion Protein and mGluR5. Adv. Pharm. 2018, 82, 293–323. [Google Scholar]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis. 2013, 33, S67–S78. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion Channel Formation by Amyloid-beta42 Oligomers but Not Amyloid-beta40 in Cellular Membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.C.; Freeley, M.; Nield, J.; Palma, M.; Viles, J.H. Amyloid-β oligomers have a profound detergent-like effect on lipid membrane bilayers, imaged by atomic force and electron microscopy. J. Biol. Chem. 2019, 294, 7566–7572. [Google Scholar] [CrossRef]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe-Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High molecular weight amyloid beta1-42 oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef]
- Dong, X.; Sun, Y.; Wei, G.; Nussinov, R.; Ma, B. Binding of protofibrillar Abeta trimers to lipid bilayer surface enhances Abeta structural stability and causes membrane thinning. Phys. Chem. Chem. Phys. 2017, 19, 27556–27569. [Google Scholar] [CrossRef]
- Jang, H.; Connelly, L.; Arce, F.T.; Ramachandran, S.; Kagan, B.L.; Lal, R.; Nussinov, R. Mechanisms for the Insertion of Toxic, Fibril-like beta-Amyloid Oligomers into the Membrane. J. Chem. Theory Comput. 2013, 9, 822–833. [Google Scholar] [CrossRef]
- Serra-Batiste, M.; Ninot-Pedrosa, M.; Bayoumi, M.; Gairi, M.; Maglia, G.; Carulla, N. A beta 42 assembles into specific beta-barrel pore-forming oligomers in membrane-mimicking environments. Proc. Natl. Acad. Sci. USA 2016, 113, 10866–10871. [Google Scholar] [CrossRef]
- Fernandez-Perez, E.J.; Sepulveda, F.J.; Peters, C.; Bascunan, D.; Riffo-Lepe, N.O.; Gonzalez-Sanmiguel, J.; Sanchez, S.A.; Peoples, R.W.; Vicente, B.; Aguayo, L.G. Effect of Cholesterol on Membrane Fluidity and Association of Abeta Oligomers and Subsequent Neuronal Damage: A Double-Edged Sword. Front. Aging Neurosci. 2018, 10, 226. [Google Scholar] [CrossRef]
- Vahed, M.; Neya, S.; Matsuzaki, K.; Hoshino, T. Analysis of Physicochemical Interaction of Abeta40 with a GM1 Ganglioside-Containing Lipid Membrane. J. Phys. Chem. B 2018, 122, 3771–3781. [Google Scholar] [CrossRef]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Abeta oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, C.; Klimov, D.K. Cholesterol Changes the Mechanisms of Abeta Peptide Binding to the DMPC Bilayer. J. Chem. Inf. Model. 2017, 57, 2554–2565. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Perez, E.J.; Sepulveda, F.J.; Peoples, R.; Aguayo, L.G. Role of membrane GM1 on early neuronal membrane actions of Abeta during onset of Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3105–3116. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, M.; Shi, X.; Wang, K.; Gao, G.; Shen, L.; Sun, T. Kinetic study of Abeta(1-42) amyloidosis in the presence of ganglioside-containing vesicles. Colloids Surf. B Biointerfaces 2020, 185, 110615. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Klein, W.L. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef]
- Sun, J.L.; Stokoe, S.A.; Roberts, J.P.; Sathler, M.F.; Nip, K.A.; Shou, J.; Ko, K.; Tsunoda, S.; Kim, S. Co-activation of selective nicotinic acetylcholine receptors is required to reverse beta amyloid-induced Ca(2+) hyperexcitation. Neurobiol. Aging 2019, 84, 166–177. [Google Scholar] [CrossRef]
- Gan, K.J.; Silverman, M.A. Dendritic and axonal mechanisms of Ca2+ elevation impair BDNF transport in Abeta oligomer-treated hippocampal neurons. Mol. Biol. Cell 2015, 26, 1058–1071. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De, S.; Meir, A. The Mitochondrial Voltage-Dependent Anion Channel 1, Ca(2+) Transport, Apoptosis, and Their Regulation. Front. Oncol 2017, 7, 60. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, T.; Ge, X.; Chen, J.; Zhao, Y.; Fu, J. Parkin overexpression attenuates Abeta-induced mitochondrial dysfunction in HEK293 cells by restoring impaired mitophagy. Life Sci. 2020, 244, 117322. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Yan, Q.W.; Xia, J.; Zhang, X.L.; Li, B.X.; Yin, L.Y.; Xu, B. Treadmill Exercise Attenuates Abeta-Induced Mitochondrial Dysfunction and Enhances Mitophagy Activity in APP/PS1 Transgenic Mice. Neurochem. Res. 2020, 45, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Pratico, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol 2019, 76, 915–924. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Smith, R.; Ohlsson, T.; Strandberg, O.; Mattsson, N.; Insel, P.S.; Palmqvist, S.; Hansson, O. Associations between tau, A beta, and cortical thickness with cognition in Alzheimer disease. Neurology 2019, 92, E601–E612. [Google Scholar] [CrossRef]
- Hurtado, D.E.; Molina-Porcel, L.; Iba, M.; Aboagye, A.K.; Paul, S.M.; Trojanowski, J.Q.; Lee, V.M. A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 2010, 177, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.; et al. Alzheimer’s disease-like pathology induced by amyloid-beta oligomers in nonhuman primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-beta plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Pontecorvo, M.J.; Devous, M.D.; Kennedy, I.; Navitsky, M.; Lu, M.; Galante, N.; Salloway, S.; Doraiswamy, P.M.; Southekal, S.; Arora, A.K.; et al. A multicentre longitudinal study of flortaucipir (18F) in normal ageing, mild cognitive impairment and Alzheimer’s disease dementia. Brain 2019, 142, 1723–1735. [Google Scholar] [CrossRef]
- Zhang, F.; Gannon, M.; Chen, Y.; Yan, S.; Zhang, S.; Feng, W.; Tao, J.; Sha, B.; Liu, Z.; Saito, T.; et al. beta-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3beta/tau cascade. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Baek, M.S.; Cho, H.; Lee, H.S.; Choi, J.Y.; Lee, J.H.; Ryu, Y.H.; Lee, M.S.; Lyoo, C.H. Temporal trajectories of in vivo tau and amyloid-beta accumulation in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020. [Google Scholar] [CrossRef]
- Liu, P.; Reed, M.N.; Kotilinek, L.A.; Grant, M.K.O.; Forster, C.L.; Qiang, W.; Shapiro, S.L.; Reichl, J.H.; Chiang, A.C.A.; Jankowsky, J.L.; et al. Quaternary Structure Defines a Large Class of Amyloid-beta Oligomers Neutralized by Sequestration. Cell Rep. 2015, 11, 1760–1771. [Google Scholar] [CrossRef]
- Wang, T.; Xie, X.X.; Ji, M.; Wang, S.W.; Zha, J.; Zhou, W.W.; Yu, X.L.; Wei, C.; Ma, S.; Xi, Z.Y.; et al. Naturally occurring autoantibodies against A beta oligomers exhibited more beneficial effects in the treatment of mouse model of Alzheimer’s disease than intravenous immunoglobulin. Neuropharmacology 2016, 105, 561–576. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, X.-X.; Xue, D.; Liu, D.-G.; Hu, X.-Y.; Zhao, M.; Yang, S.-G.; Yang, Y.; Xia, Y.-J.; Wang, Y.; et al. Conformation-dependent scFv antibodies specifically recognize the oligomers assembled from various amyloids and show colocalization of amyloid fibrils with oligomers in patients with amyloidoses. Biochim. Et Biophys. Acta BBA Proteins Proteom. 2011, 1814, 1703–1712. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, S.W.; Wang, Y.J.; Zhang, R.; Li, Y.N.; Su, Y.J.; Zhou, W.W.; Yu, X.L.; Liu, R.T. Pan-amyloid oligomer specific scFv antibody attenuates memory deficits and brain amyloid burden in mice with Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 69–78. [Google Scholar] [CrossRef]
- Brinkmalm, G.; Hong, W.; Wang, Z.; Liu, W.; O’Malley, T.T.; Sun, X.; Frosch, M.P.; Selkoe, D.J.; Portelius, E.; Zetterberg, H.; et al. Identification of neurotoxic cross-linked amyloid-beta dimers in the Alzheimer’s brain. Brain 2019, 142, 1441–1457. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hafiz, L.; Muller-Schiffmann, A.; Korth, C.; Fazari, B.; Chao, O.Y.; Nikolaus, S.; Schable, S.; Herring, A.; Keyvani, K.; Lamounier-Zepter, V.; et al. Abeta dimers induce behavioral and neurochemical deficits of relevance to early Alzheimer’s disease. Neurobiol. Aging 2018, 69, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zott, B.; Simon, M.M.; Hong, W.; Unger, F.; Chen-Engerer, H.J.; Frosch, M.P.; Sakmann, B.; Walsh, D.M.; Konnerth, A. A vicious cycle of beta amyloid-dependent neuronal hyperactivation. Science 2019, 365, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.K.; Cappai, R.; Pham, C.L.; Ciccotosto, G.D. Membrane-bound tetramer and trimer Abeta oligomeric species correlate with toxicity towards cultured neurons. J. Neurochem. 2016, 136, 594–608. [Google Scholar] [CrossRef]
- Crouch, P.J.; Hung, L.W.; Adlard, P.A.; Cortes, M.; Lal, V.; Filiz, G.; Perez, K.A.; Nurjono, M.; Caragounis, A.; Du, T.; et al. Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 381–386. [Google Scholar] [CrossRef]
- Sherman, M.A.; LaCroix, M.; Amar, F.; Larson, M.E.; Forster, C.; Aguzzi, A.; Bennett, D.A.; Ramsden, M.; Lesne, S.E. Soluble Conformers of Abeta and Tau Alter Selective Proteins Governing Axonal Transport. J. Neurosci. 2016, 36, 9647–9658. [Google Scholar] [CrossRef]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol 2006, 572, 477–492. [Google Scholar] [CrossRef]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Lesne, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain 2013, 136, 1383–1398. [Google Scholar] [CrossRef]
- Amar, F.; Sherman, M.A.; Rush, T.; Larson, M.; Boyle, G.; Chang, L.; Gotz, J.; Buisson, A.; Lesne, S.E. The amyloid-beta oligomer Abeta*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Nimmrich, V.; Striebinger, A.; Krantz, C.; Keller, P.; Janson, B.; Bahr, M.; Schmidt, M.; Bitner, R.S.; Harlan, J.; et al. Globular amyloid beta-peptide oligomer—A homogenous and stable neuropathological protein in Alzheimer’s disease. J. Neurochem. 2005, 95, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Yanazawa, M.; Sasahara, T.; Kitamura, Y.; Hiroaki, H.; Fukazawa, Y.; Kii, I.; Nishiyama, T.; Kakita, A.; Takeda, H.; et al. Na, K-ATPase alpha3 is a death target of Alzheimer patient amyloid-beta assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E4465–E4474. [Google Scholar] [CrossRef]
- Wen, J.; Fang, F.; Guo, S.H.; Zhang, Y.; Peng, X.L.; Sun, W.M.; Wei, X.R.; He, J.S.; Hung, T. Amyloid beta-Derived Diffusible Ligands (ADDLs) Induce Abnormal Autophagy Associated with Abeta Aggregation Degree. J. Mol. Neurosci. 2018, 64, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Teng, Z.; Cui, C.; Wang, R.; Liu, M.; Zhang, Y. Amyloid beta-derived diffusible ligands (ADDLs) induce abnormal expression of insulin receptors in rat hippocampal neurons. J. Mol. Neurosci. 2014, 52, 124–130. [Google Scholar] [CrossRef]
- Herzer, S.; Meldner, S.; Rehder, K.; Grone, H.J.; Nordstrom, V. Lipid microdomain modification sustains neuronal viability in models of Alzheimer’s disease. Acta Neuropathol. Commun. 2016, 4, 103. [Google Scholar] [CrossRef]
- Shi, X.D.; Sun, K.; Hu, R.; Liu, X.Y.; Hu, Q.M.; Sun, X.Y.; Yao, B.; Sun, N.; Hao, J.R.; Wei, P.; et al. Blocking the Interaction between EphB2 and ADDLs by a Small Peptide Rescues Impaired Synaptic Plasticity and Memory Deficits in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2016, 36, 11959–11973. [Google Scholar] [CrossRef]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef]
- Mc Donald, J.M.; Savva, G.M.; Brayne, C.; Welzel, A.T.; Forster, G.; Shankar, G.M.; Selkoe, D.J.; Ince, P.G.; Walsh, D.M.; on behalf of the Medical Research Council Cognitive Function and Ageing Study. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain 2010, 133, 1328–1341. [Google Scholar] [CrossRef] [PubMed]
- Mc Donald, J.M.; O’Malley, T.T.; Liu, W.; Mably, A.J.; Brinkmalm, G.; Portelius, E.; Wittbold, W.M., 3rd; Frosch, M.P.; Walsh, D.M. The aqueous phase of Alzheimer’s disease brain contains assemblies built from approximately 4 and approximately 7 kDa Abeta species. Alzheimers Dement. 2015, 11, 1286–1305. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Leissring, M.A.; Adame, A.; Sun, X.; Spooner, E.; Masliah, E.; Selkoe, D.J.; Lemere, C.A.; Walsh, D.M. Biochemical and immunohistochemical analysis of an Alzheimer’s disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol. Dis. 2009, 36, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Vazquez de la Torre, A.; Gay, M.; Vilaprinyo-Pascual, S.; Mazzucato, R.; Serra-Batiste, M.; Vilaseca, M.; Carulla, N. Direct Evidence of the Presence of Cross-Linked Abeta Dimers in the Brains of Alzheimer’s Disease Patients. Anal. Chem. 2018, 90, 4552–4560. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, T.T.; Witbold, W.M., 3rd; Linse, S.; Walsh, D.M. The Aggregation Paths and Products of Abeta42 Dimers Are Distinct from Those of the Abeta42 Monomer. Biochemistry 2016, 55, 6150–6161. [Google Scholar] [CrossRef]
- Meilandt, W.J.; Cisse, M.; Ho, K.; Wu, T.; Esposito, L.A.; Scearce-Levie, K.; Cheng, I.H.; Yu, G.Q.; Mucke, L. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Abeta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J. Neurosci. 2009, 29, 1977–1986. [Google Scholar] [CrossRef]
- Kreutzer, A.G.; Yoo, S.; Spencer, R.K.; Nowick, J.S. Stabilization, Assembly, and Toxicity of Trimers Derived from Abeta. J. Am. Chem. Soc. 2017, 139, 966–975. [Google Scholar] [CrossRef]
- Reed, M.N.; Hofmeister, J.J.; Jungbauer, L.; Welzel, A.T.; Yu, C.; Sherman, M.A.; Lesne, S.; LaDu, M.J.; Walsh, D.M.; Ashe, K.H.; et al. Cognitive effects of cell-derived and synthetically derived Abeta oligomers. Neurobiol. Aging 2011, 32, 1784–1794. [Google Scholar] [CrossRef]
- Cheng, I.H.; Scearce-Levie, K.; Legleiter, J.; Palop, J.J.; Gerstein, H.; Bien-Ly, N.; Puolivali, J.; Lesne, S.; Ashe, K.H.; Muchowski, P.J.; et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 2007, 282, 23818–23828. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Tran, L.; Lambert, M.P.; Glabe, C.G.; Klein, W.L.; LaFerla, F.M. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J. Biol. Chem. 2006, 281, 1599–1604. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.J.; Lee, J.H.; Kim, S.Y.; Son, G.; Kim, J.Y.; Cho, B.; Yu, S.W.; Chang, K.A.; Suh, Y.H.; Moon, C. Differential spatial expression of peripheral olfactory neuron-derived BACE1 induces olfactory impairment by region-specific accumulation of beta-amyloid oligomer. Cell Death Dis. 2017, 8, e2977. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Sato, M.; Matsumoto, S.; Noguchi, A.; Yasutake, K.; Yoshida, N.; Sato, K. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc. Natl. Acad. Sci. USA 2003, 100, 6370–6375. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Inoue, M.; Xiao, Y.; Matsumura, Y.; Nabeshima, Y.; Hoshi, M.; Ishii, Y. Structural Insight into an Alzheimer’s Brain-Derived Spherical Assembly of Amyloid beta by Solid-State NMR. J. Am. Chem. Soc. 2015, 137, 6480–6483. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, A.; Matsumura, S.; Dezawa, M.; Tada, M.; Yanazawa, M.; Ito, A.; Akioka, M.; Kikuchi, S.; Sato, M.; Ideno, S.; et al. Isolation and characterization of patient-derived, toxic, high mass amyloid beta-protein (Abeta) assembly from Alzheimer disease brains. J. Biol. Chem. 2009, 284, 32895–32905. [Google Scholar] [CrossRef]
- Komura, H.; Kakio, S.; Sasahara, T.; Arai, Y.; Takino, N.; Sato, M.; Satomura, K.; Ohnishi, T.; Nabeshima, Y.I.; Muramatsu, S.I.; et al. Alzheimer Abeta Assemblies Accumulate in Excitatory Neurons upon Proteasome Inhibition and Kill Nearby NAKalpha3 Neurons by Secretion. iScience 2019, 13, 452–477. [Google Scholar] [CrossRef]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef]
- Chang, L.; Bakhos, L.; Wang, Z.; Venton, D.L.; Klein, W.L. Femtomole immunodetection of synthetic and endogenous amyloid-beta oligomers and its application to Alzheimer’s disease drug candidate screening. J. Mol. Neurosci. 2003, 20, 305–313. [Google Scholar] [CrossRef]
- Lambert, M.P.; Velasco, P.T.; Chang, L.; Viola, K.L.; Fernandez, S.; Lacor, P.N.; Khuon, D.; Gong, Y.S.; Bigio, E.H.; Shaw, P.; et al. Monoclonal antibodies that target pathological assemblies of A beta. J. Neurochem. 2007, 100, 23–35. [Google Scholar] [CrossRef]
- Xie, Z.; Shapiro, L.P.; Cahill, M.E.; Russell, T.A.; Lacor, P.N.; Klein, W.L.; Penzes, P. Kalirin-7 prevents dendritic spine dysgenesis induced by amyloid beta-derived oligomers. Eur. J. Neurosci. 2019, 49, 1091–1101. [Google Scholar] [CrossRef]
- Henley, D.; Raghavan, N.; Sperling, R.; Aisen, P.; Raman, R.; Romano, G. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1483–1485. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.Y.; Thomas, R.G.; et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. New Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.R.; Buckland, G.R.; Harrison, C.H.; Page, A.; Harris, S.; Love, S.; Neal, J.W.; Holmes, C.; Boche, D. Persistent neuropathological effects 14 years following amyloid-beta immunization in Alzheimer’s disease. Brain 2019, 142, 2113–2126. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Aβos | Structure | Toxicity | The Type of Research | References |
|---|---|---|---|---|
| Dimers | Covalently coupled by monomers | Reducing neuron length, inhibiting LTP response | Ex vivo | [101] |
| Synaptic damage and tau hyperphosphorylation | Ex vivo | [102] | ||
| Reducing hippocampal acetylcholine and serotonin turnover rates, causing neurotransmitter dysfunction and behavioral impairments | In vivo | [103] | ||
| Neuronal overactivation by inhibiting the glutamate reuptake process | In vivo and ex vivo | [104] | ||
| Trimers | __ | Nerve cell death | In vitro | [105] |
| Cognitive impairment | In vivo | [106] | ||
| Reducing KLC1, changing tau conformation and disrupting axonal transport | Ex vivo and in vivo | [107] | ||
| Inhibiting LTP response | Ex vivo | [108] | ||
| Aβ*56 | __ | Memory impairment | In vivo | [109] |
| Elevating NMDAR-dependent intracellular Ca2+ concentration, activating CaMKIIα, tau hyperphosphorylation | Ex vivo | [110,111] | ||
| Inactivating ubiquitin-dependent and ubiquitin-independent proteasome | In vitro | [112] | ||
| ASPD | Spherical oligomers with diameter being ∼11.9 ± 1.7 nm | Synaptic damage and inhibition of LTP | In vivo and ex vivo | [113] |
| Impairing NAKα3-specific activity, activating N-type voltage-gated calcium channels, increasing the levels of cytoplasmic Ca2+, mitochondrial calcium dyshomeostasis, mature neuron death and cognitive impairment | Ex vivo | [20,114] | ||
| ADDLs | Small sphere with a diameter of 5–6 nm | Abnormal autophagy of neurons | Ex vivo | [115] |
| Reducing the level of insulin receptor and disrupting normal insulin signaling | In vitro and in vivo | [116,117] | ||
| Inducing the degradation of EPhB2 receptor and the phosphorylation of NMDAR, impairing synaptic plasticity | Ex vivo and in vivo | [118] | ||
| Abnormal localization of tau to dendrites by activating tyrosine kinase Fyn | Ex vivo and in vivo | [119,120] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-r.; Liu, R.-t. The Toxicity and Polymorphism of β-Amyloid Oligomers. Int. J. Mol. Sci. 2020, 21, 4477. https://doi.org/10.3390/ijms21124477
Huang Y-r, Liu R-t. The Toxicity and Polymorphism of β-Amyloid Oligomers. International Journal of Molecular Sciences. 2020; 21(12):4477. https://doi.org/10.3390/ijms21124477
Chicago/Turabian StyleHuang, Ya-ru, and Rui-tian Liu. 2020. "The Toxicity and Polymorphism of β-Amyloid Oligomers" International Journal of Molecular Sciences 21, no. 12: 4477. https://doi.org/10.3390/ijms21124477
APA StyleHuang, Y.-r., & Liu, R.-t. (2020). The Toxicity and Polymorphism of β-Amyloid Oligomers. International Journal of Molecular Sciences, 21(12), 4477. https://doi.org/10.3390/ijms21124477