Generation of Hematopoietic-Like Stem Cells from Adult Human Peripheral Blood Following Treatment with Platelet-Derived Mitochondria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
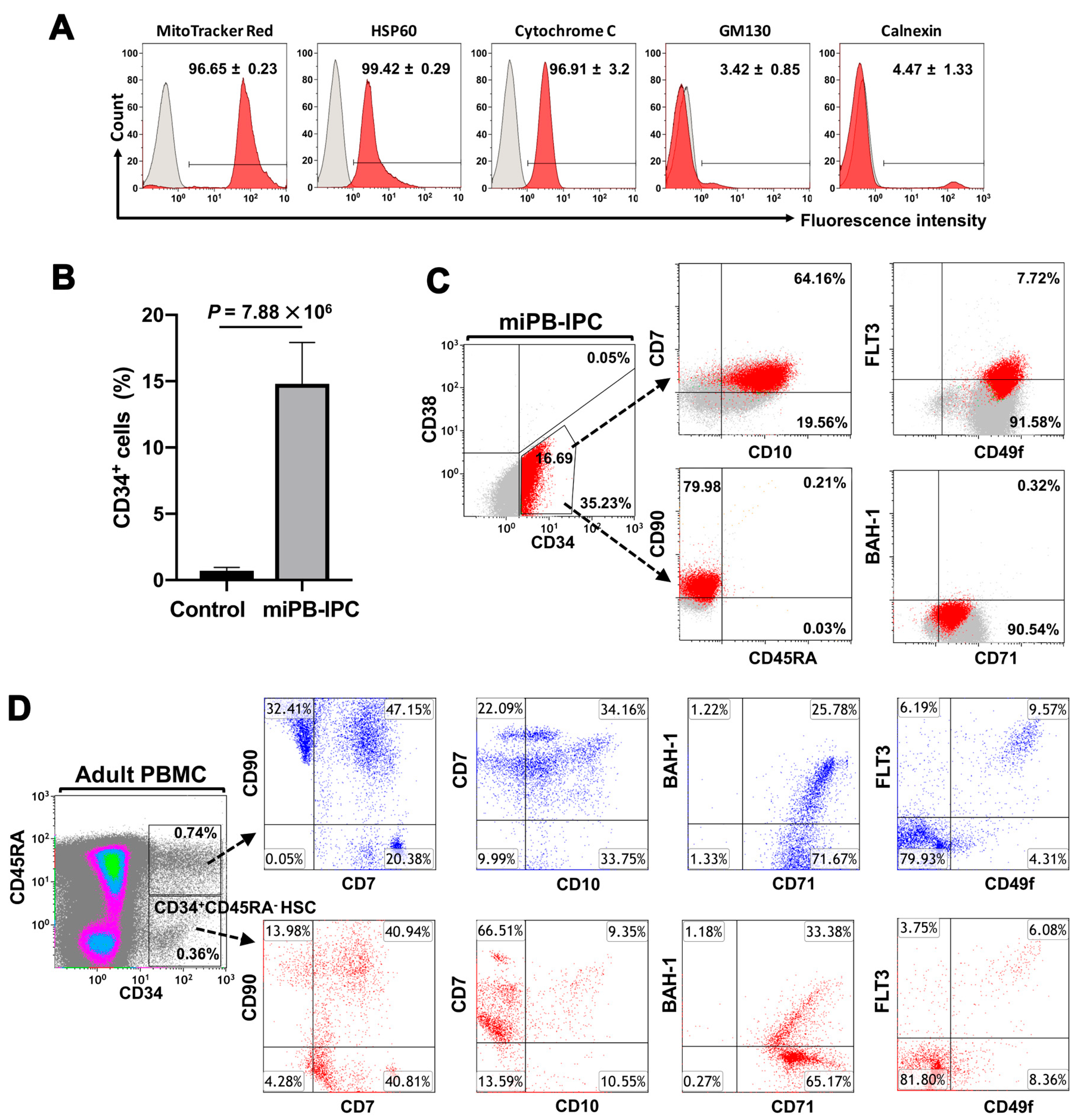
2.1. Ex Vivo Differentiation of Mitochondrion-Induced PB-IPCs (miPB-IPCs) into the Mitochondrion-Induced CD34+-HSC-Like Cells (miCD34+ HSCs) after Treatment with Platelet-Derived Mitochondria
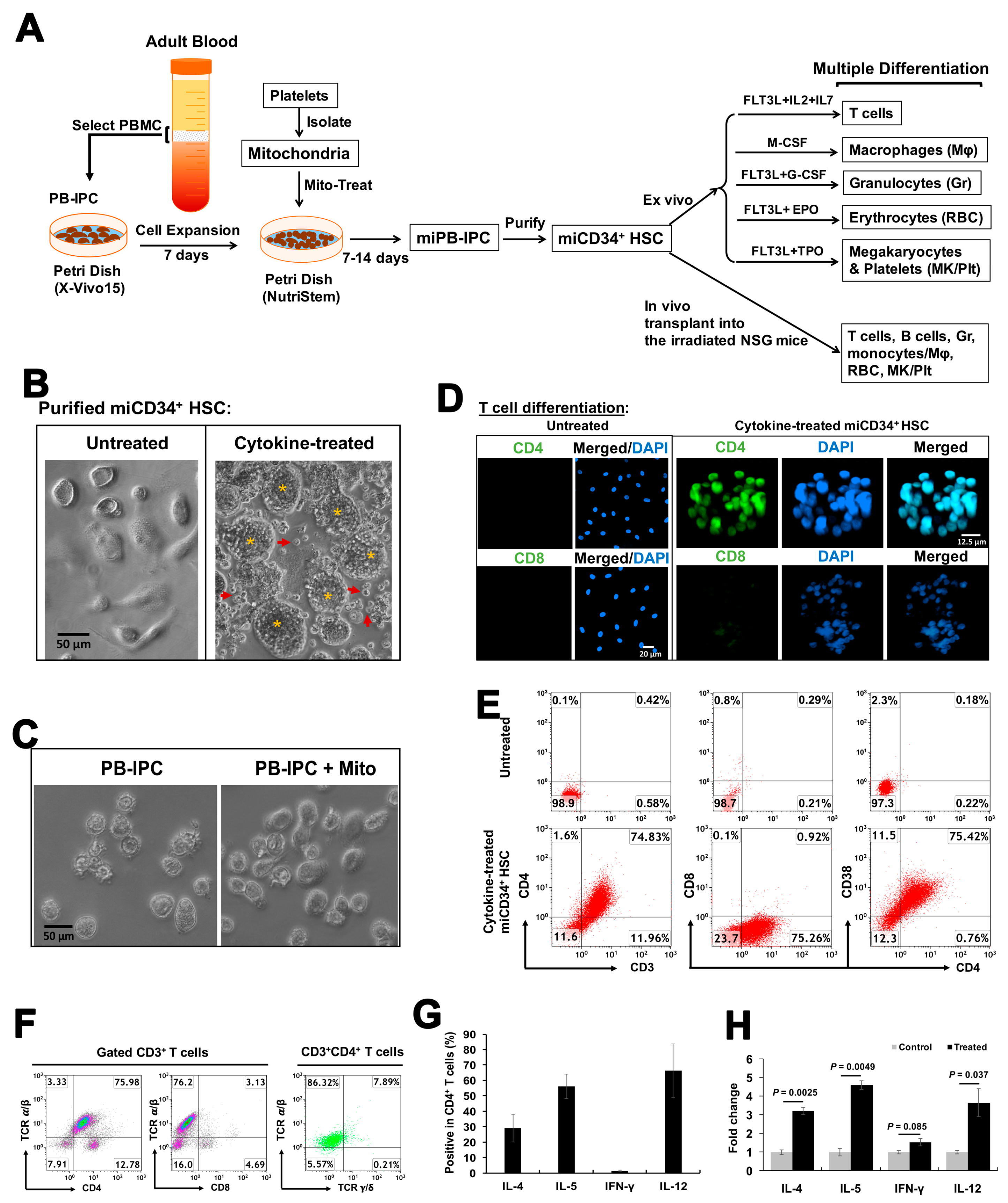
2.2. Differentiation of miCD34+ HSCs into T Cells
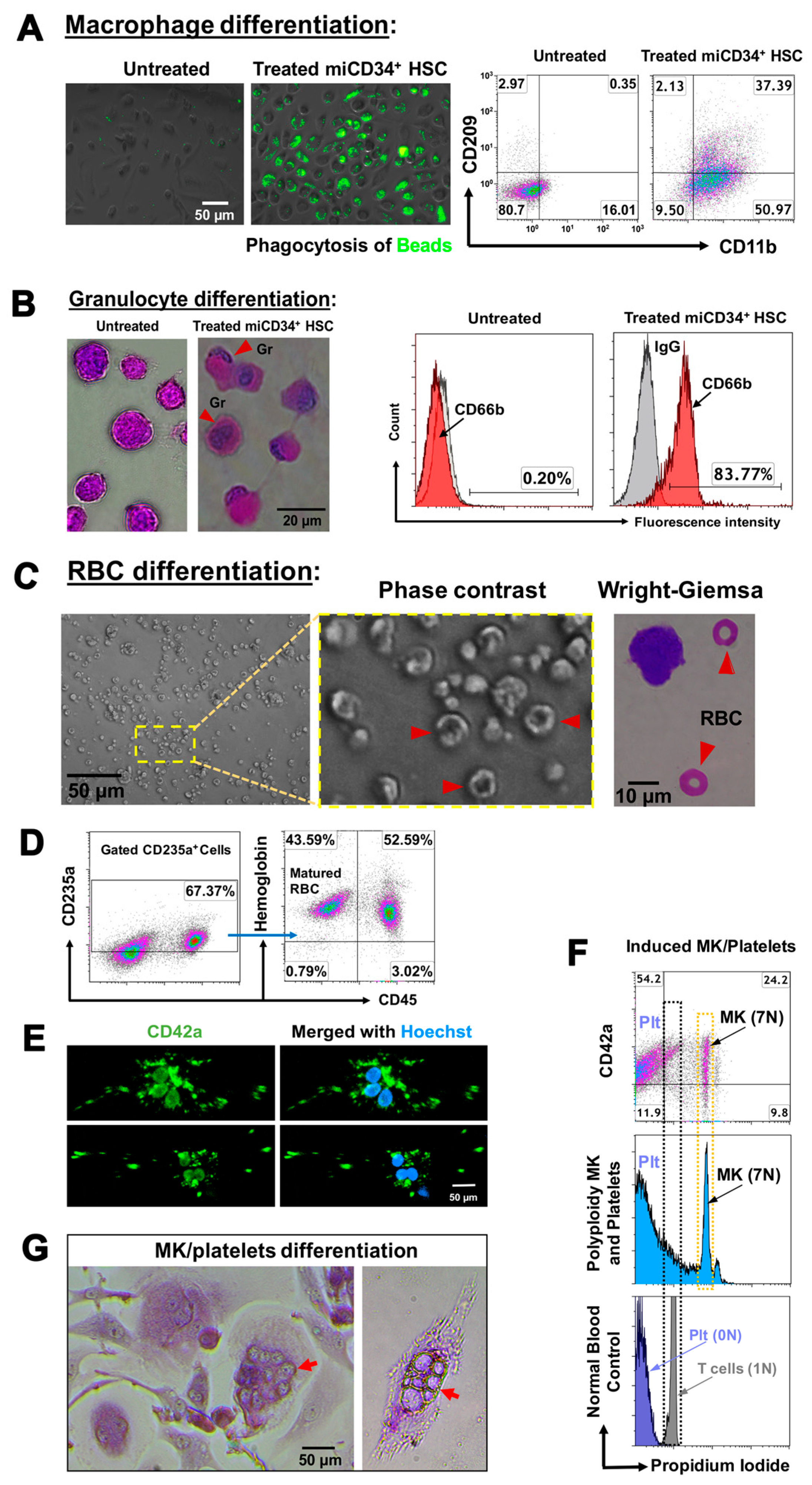
2.3. Ex Vivo Differentiation of miCD34+ HSCs into Other Hematopoietic Lineages
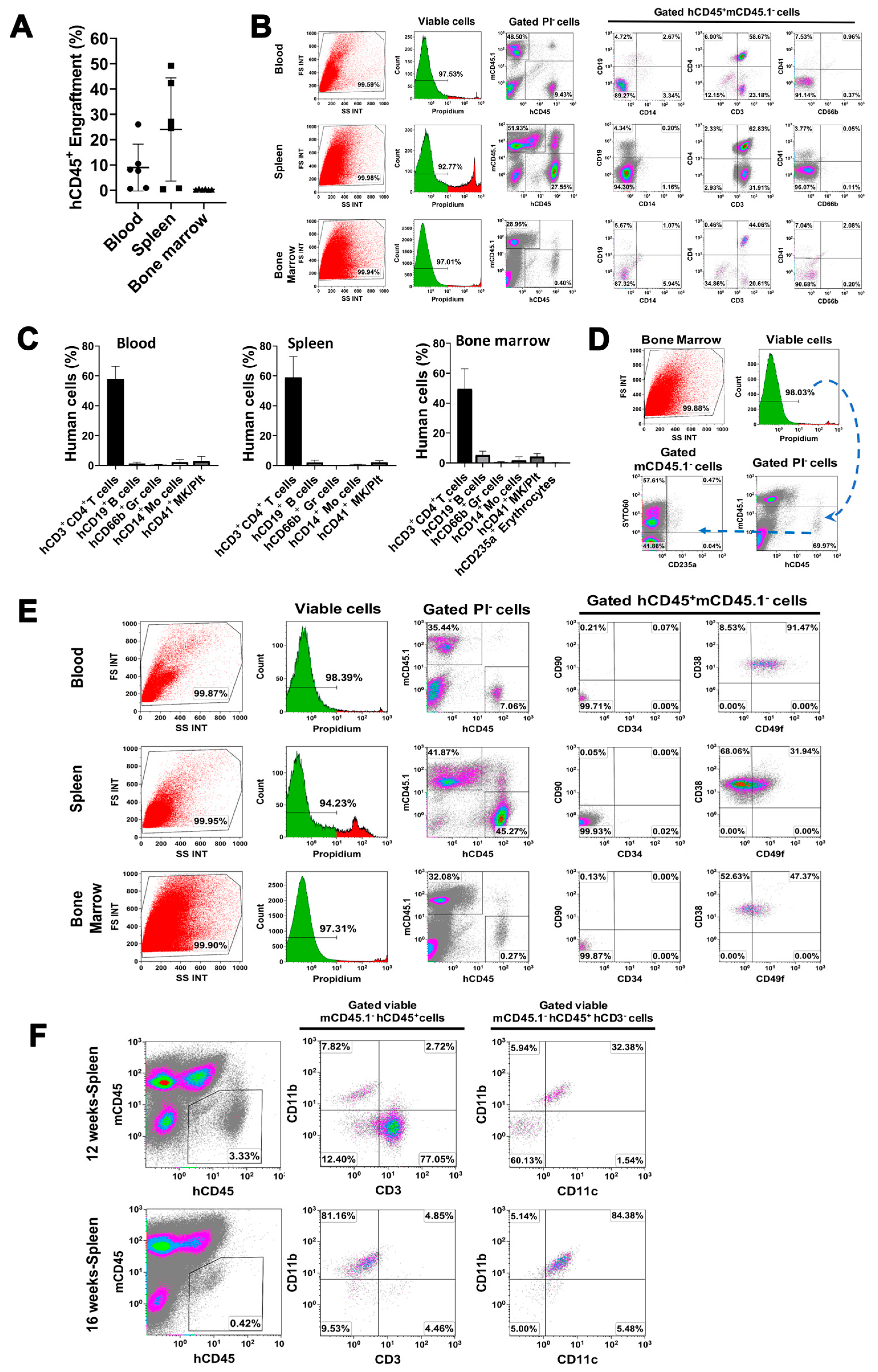
2.4. In Vivo Differentiation of miCD34+ HSCs into Other Hematopoietic Lineages after Transplant into NSG Mice
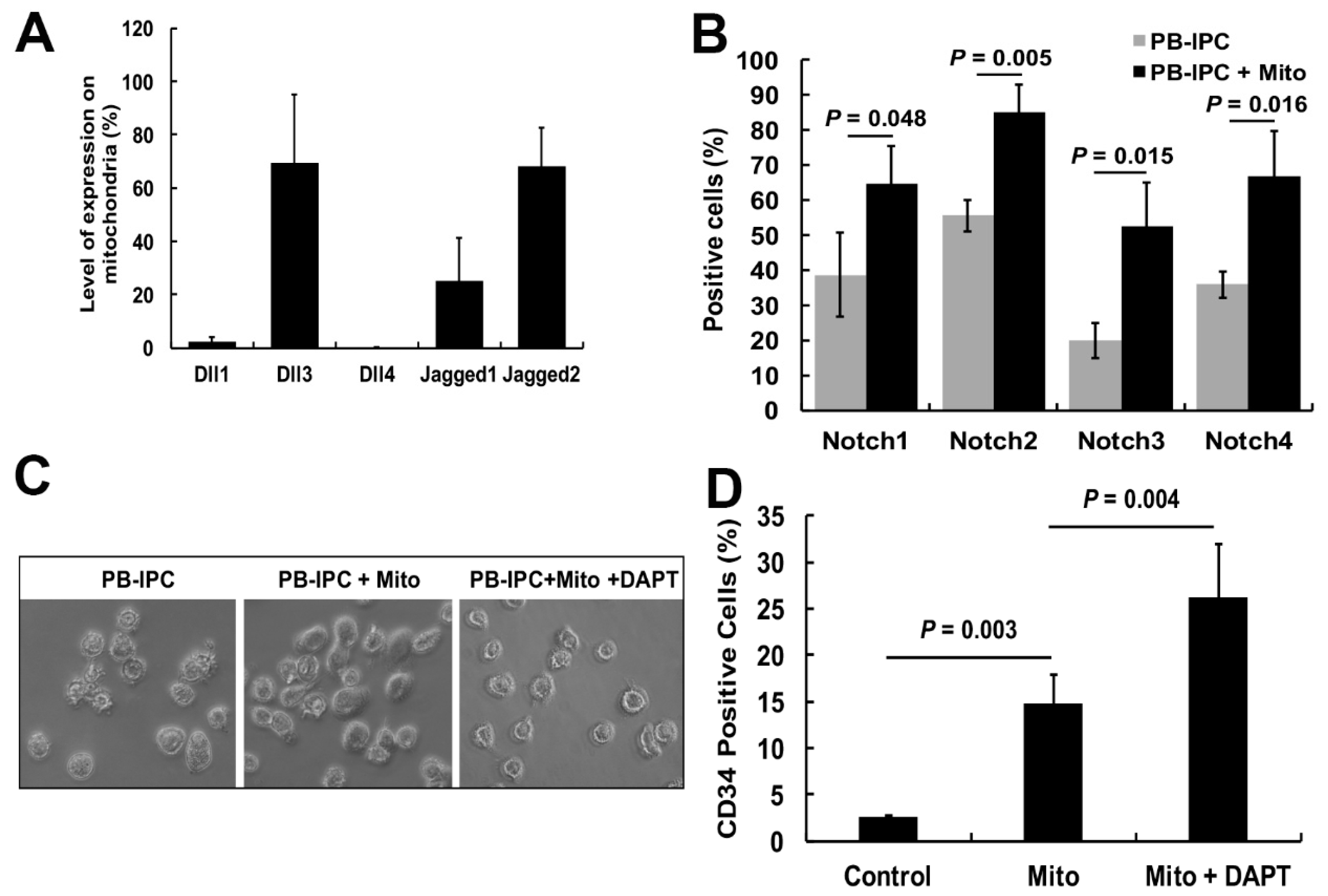
2.5. Notch Signaling Pathway Contributed to the miCD34+ HSC Differentiation after Treatment with Platelet-Derived Mitochondria
3. Discussion
4. Materials and Methods
4.1. PB-IPC Cell Culture
4.2. Isolation of Mitochondria from Platelets
4.3. In Vitro Differentiation of PB-IPCs into miCD34+ HSCs
4.4. Flow Cytometry
4.5. Multiple Differentiations of miCD34+ HSCs
4.6. Animal Study and Engraftment of miCD34+ HSCs into Irradiated NSG Mice
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Doulatov, S.; Notta, F.; Laurenti, E.; Dick, J.E. Hematopoiesis: A human perspective. Cell Stem. Cell 2012, 10, 120–136. [Google Scholar] [CrossRef] [PubMed]
- Wahlster, L.; Daley, G.Q. Progress towards generation of human haematopoietic stem cells. Nat. Cell Biol. 2016, 18, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Swart, J.F.; Delemarre, E.M.; van Wijk, F.; Boelens, J.J.; Kuball, J.; van Laar, J.M.; Wulffraat, N.M. Haematopoietic stem cell transplantation for autoimmune diseases. Nat. Rev. Rheumatol. 2017, 13, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Gambell, P.; Herbert, K.; Dickinson, M.; Stokes, K.; Bressel, M.; Wall, D.; Harrison, S.; Prince, H.M. Peripheral blood CD34+ cell enumeration as a predictor of apheresis yield: An analysis of more than 1000 collections. Biol. Blood Marrow Transplant. 2012, 18, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Chabannon, C.; Kuball, J.; Bondanza, A.; Dazzi, F.; Pedrazzoli, P.; Toubert, A.; Ruggeri, A.; Fleischhauer, K.; Bonini, C. Hematopoietic stem cell transplantation in its 60s: A platform for cellular therapies. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Kanakry, C.G.; Fuchs, E.J.; Luznik, L. Modern approaches to HLA-haploidentical blood or marrow transplantation. Nat. Rev. Clin. Oncol. 2016, 13, 132. [Google Scholar] [CrossRef]
- Woods, N.B.; Parker, A.S.; Moraghebi, R.; Lutz, M.K.; Firth, A.L.; Brennand, K.J.; Berggren, W.T.; Raya, A.; Izpisua Belmonte, J.C.; Gage, F.H.; et al. Brief report: Efficient generation of hematopoietic precursors and progenitors from human pluripotent stem cell lines. Stem. Cells 2011, 29, 1158–1164. [Google Scholar] [CrossRef]
- Gori, J.L.; Butler, J.M.; Chan, Y.Y.; Chandrasekaran, D.; Poulos, M.G.; Ginsberg, M.; Nolan, D.J.; Elemento, O.; Wood, B.L.; Adair, J.E.; et al. Vascular niche promotes hematopoietic multipotent progenitor formation from pluripotent stem cells. J. Clin. Investig. 2015, 125, 1243–1254. [Google Scholar] [CrossRef]
- Blaser, B.W.; Zon, L.I. Making HSCs in vitro: don’t forget the hemogenic endothelium. Blood 2018. [Google Scholar] [CrossRef]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem. Cells 2013, 31, 458–466. [Google Scholar] [CrossRef]
- Sugimura, R.; Jha, D.K.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Doulatov, S.; Vo, L.T.; Chou, S.S.; Kim, P.G.; Arora, N.; Li, H.; Hadland, B.K.; Bernstein, I.D.; Collins, J.J.; Zon, L.I.; et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem. Cell 2013, 13, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Baum, C.; Cowan, C.; Dzierzak, E.; Hacein-Bey-Abina, S.; Karlsson, S.; Lapidot, T.; Lemischka, I.; Mendez-Ferrer, S.; Mikkers, H.; et al. Stem cell self-renewal: Lessons from bone marrow, gut and iPS toward clinical applications. Leukemia 2011, 25, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mazzone, T. Human cord blood stem cells and the journey to a cure for type 1 diabetes. Autoimmun. Rev. 2010, 10, 103–107. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, Z.; Zhao, T.; Ye, M.; Hu, C.; Yin, Z.; Li, H.; Zhang, Y.; Diao, Y.; Li, Y.; et al. Reversal of type 1 diabetes via islet beta cell regeneration following immune modulation by cord blood-derived multipotent stem cells. BMC Med. 2012, 10, 3. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, Z.; Zhao, T.; Ye, M.; Hu, C.; Zhou, H.; Yin, Z.; Chen, Y.; Zhang, Y.; Wang, S.; et al. Targeting insulin resistance in type 2 diabetes via immune modulation of cord blood-derived multipotent stem cells (CB-SCs) in stem cell educator therapy: Phase I/II clinical trial. BMC Med. 2013, 11, 160. [Google Scholar] [CrossRef]
- Delgado, E.; Perez-Basterrechea, M.; Suarez-Alvarez, B.; Zhou, H.; Revuelta, E.M.; Garcia-Gala, J.M.; Perez, S.; Alvarez-Viejo, M.; Menendez, E.; Lopez-Larrea, C.; et al. Modulation of Autoimmune T-Cell Memory by Stem Cell Educator Therapy: Phase 1/2 Clinical Trial. EBioMedicine 2015, 2, 2024–2036. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, Z.; Delgado, E.; Li, H.; Zhou, H.; Hu, W.; Perez-Basterrechea, M.; Janostakova, A.; Tan, Q.; Wang, J.; et al. Platelet-Derived Mitochondria Display Embryonic Stem Cell Markers and Improve Pancreatic Islet beta-cell Function in Humans. Stem. Cells Transl. Med. 2017, 6, 1684–1697. [Google Scholar] [CrossRef]
- Li, Y.; Yan, B.; Wang, H.; Li, H.; Li, Q.; Zhao, D.; Chen, Y.; Zhang, Y.; Li, W.; Zhang, J.; et al. Hair regrowth in alopecia areata patients following Stem Cell Educator therapy. BMC Med. 2015, 13, 87. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, Z.; Lazzarini, P.; Wang, Y.; Di, A.; Chen, M. A unique human blood-derived cell population displays high potential for producing insulin. Biochem. Biophys. Res. Commun. 2007, 360, 205–211. [Google Scholar] [CrossRef]
- Yu, H.; Hu, W.; Song, X.; Zhao, Y. Generation of Multipotent Stem Cells from Adult Human Peripheral Blood Following the Treatment with Platelet-Derived Mitochondria. Cells 2020, 9, 1350. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116. [Google Scholar] [CrossRef] [PubMed]
- Calloni, R.; Cordero, E.A.; Henriques, J.A.; Bonatto, D. Reviewing and updating the major molecular markers for stem cells. Stem. Cells Dev. 2013, 22, 1455–1476. [Google Scholar] [CrossRef] [PubMed]
- Krebsbach, P.H.; Villa-Diaz, L.G. The Role of Integrin alpha6 (CD49f) in Stem Cells: More than a Conserved Biomarker. Stem. Cells Dev. 2017, 26, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.L.; Zhu, J.; Price, M.A.; Payne, K.J.; Barsky, L.W.; Crooks, G.M. Identification of a novel, human multilymphoid progenitor in cord blood. Blood 2001, 97, 3683–3690. [Google Scholar] [CrossRef]
- Yamamoto, R.; Morita, Y.; Ooehara, J.; Hamanaka, S.; Onodera, M.; Rudolph, K.L.; Ema, H.; Nakauchi, H. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 2013, 154, 1112–1126. [Google Scholar] [CrossRef]
- Butko, E.; Pouget, C.; Traver, D. Complex regulation of HSC emergence by the Notch signaling pathway. Dev. Biol. 2016, 409, 129–138. [Google Scholar] [CrossRef]
- De Obaldia, M.E.; Bhandoola, A. Transcriptional regulation of innate and adaptive lymphocyte lineages. Annual. Rev. Immunol. 2015, 33, 607–642. [Google Scholar] [CrossRef]
- Weber, D.; Wiese, C.; Gessler, M. Hey bHLH transcription factors. Curr. Top. Dev. Biol. 2014, 110, 285–315. [Google Scholar] [CrossRef]
- Anjos-Afonso, F.; Currie, E.; Palmer, H.G.; Foster, K.E.; Taussig, D.C.; Bonnet, D. CD34(-) cells at the apex of the human hematopoietic stem cell hierarchy have distinctive cellular and molecular signatures. Cell Stem. Cell 2013, 13, 161–174. [Google Scholar] [CrossRef]
- Akashi, K.; Traver, D.; Miyamoto, T.; Weissman, I.L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 2000, 404, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Fukuda, A.; Hisatake, K. Mechanisms of the Metabolic Shift during Somatic Cell Reprogramming. Int. J. Mol. Sci. 2019, 20, 2254. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Glesne, D.; Huberman, E. A human peripheral blood monocyte-derived subset acts as pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2003, 100, 2426–2431. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, H.; Mazzone, T. Identification of stem cells from human umbilical cord blood with embryonic and hematopoietic characteristics. Exp. Cell Res. 2006, 312, 2454–2464. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Hu, W.; Song, X.; Descalzi-Montoya, D.; Yang, Z.; Korngold, R.; Zhao, Y. Generation of Hematopoietic-Like Stem Cells from Adult Human Peripheral Blood Following Treatment with Platelet-Derived Mitochondria. Int. J. Mol. Sci. 2020, 21, 4249. https://doi.org/10.3390/ijms21124249
Yu H, Hu W, Song X, Descalzi-Montoya D, Yang Z, Korngold R, Zhao Y. Generation of Hematopoietic-Like Stem Cells from Adult Human Peripheral Blood Following Treatment with Platelet-Derived Mitochondria. International Journal of Molecular Sciences. 2020; 21(12):4249. https://doi.org/10.3390/ijms21124249
Chicago/Turabian StyleYu, Haibo, Wei Hu, Xiang Song, Dante Descalzi-Montoya, Zheng Yang, Robert Korngold, and Yong Zhao. 2020. "Generation of Hematopoietic-Like Stem Cells from Adult Human Peripheral Blood Following Treatment with Platelet-Derived Mitochondria" International Journal of Molecular Sciences 21, no. 12: 4249. https://doi.org/10.3390/ijms21124249
APA StyleYu, H., Hu, W., Song, X., Descalzi-Montoya, D., Yang, Z., Korngold, R., & Zhao, Y. (2020). Generation of Hematopoietic-Like Stem Cells from Adult Human Peripheral Blood Following Treatment with Platelet-Derived Mitochondria. International Journal of Molecular Sciences, 21(12), 4249. https://doi.org/10.3390/ijms21124249