Pleiotropic Role of Notch Signaling in Human Skin Diseases

 ,
,  , ,
, ,  , and
, and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Core Components of Notch Signaling
1.2. Notch Signaling
1.2.1. Canonical Notch Signaling
1.2.2. Non-Canonical Notch Signaling
2. Skin
2.1. Notch Signaling and Skin
2.2. Notch and Skin
2.3. The Main Proteins Related to Notch and Skin
3. Notch Signaling and Skin Diseases
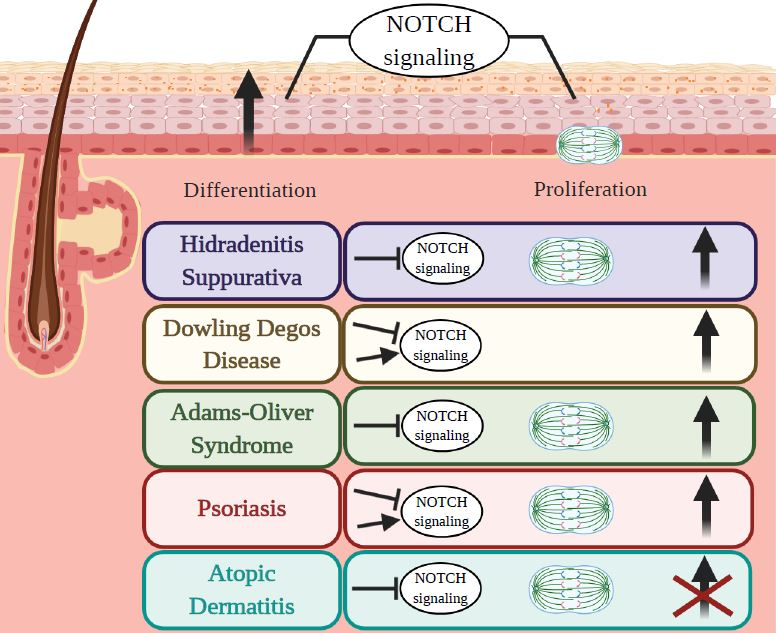
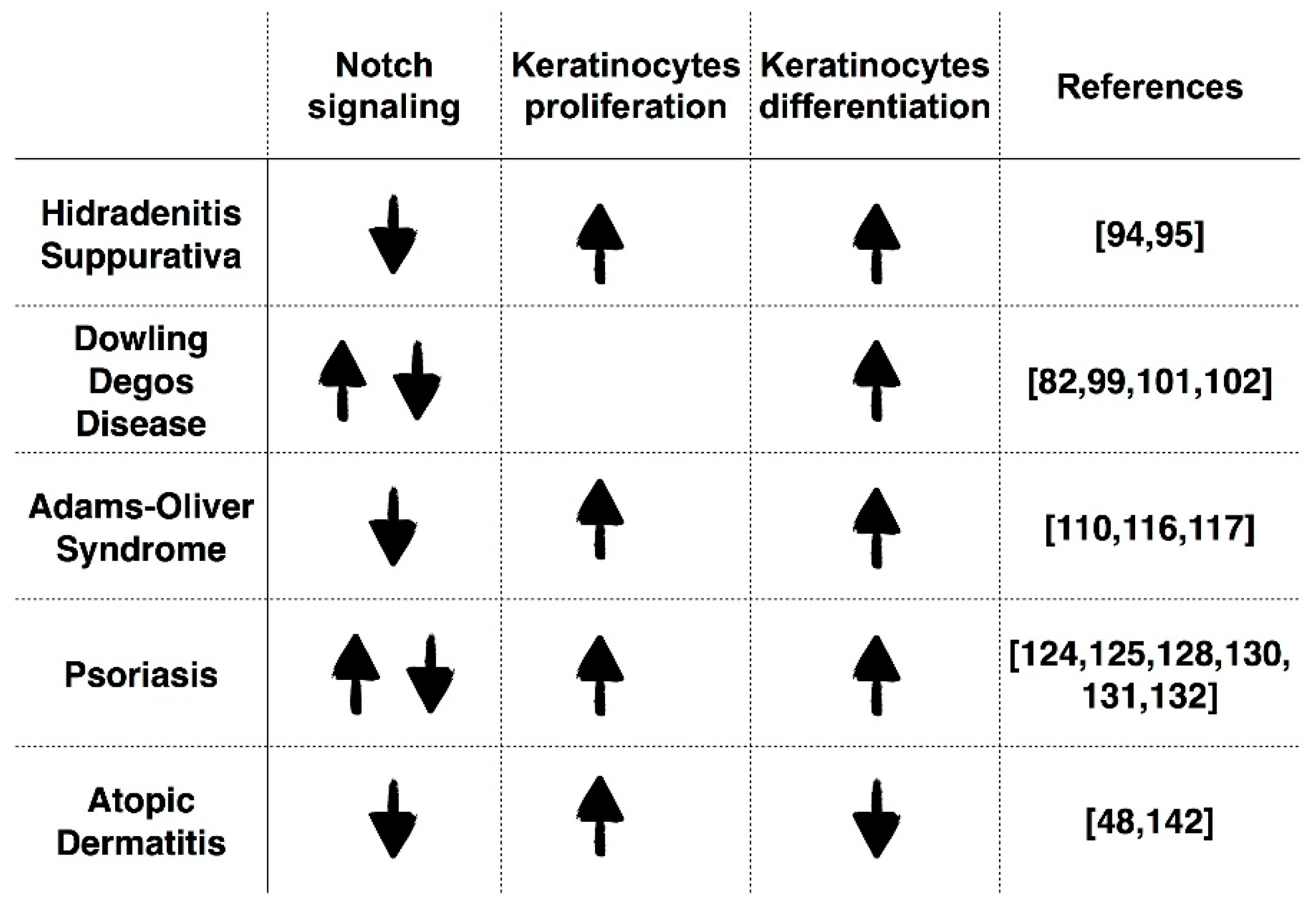
3.1. Hidradenitis Suppurativa (HS)
3.2. Dowling Degos Disease (DDD)
3.3. Adams–Oliver Syndrome (AOS)
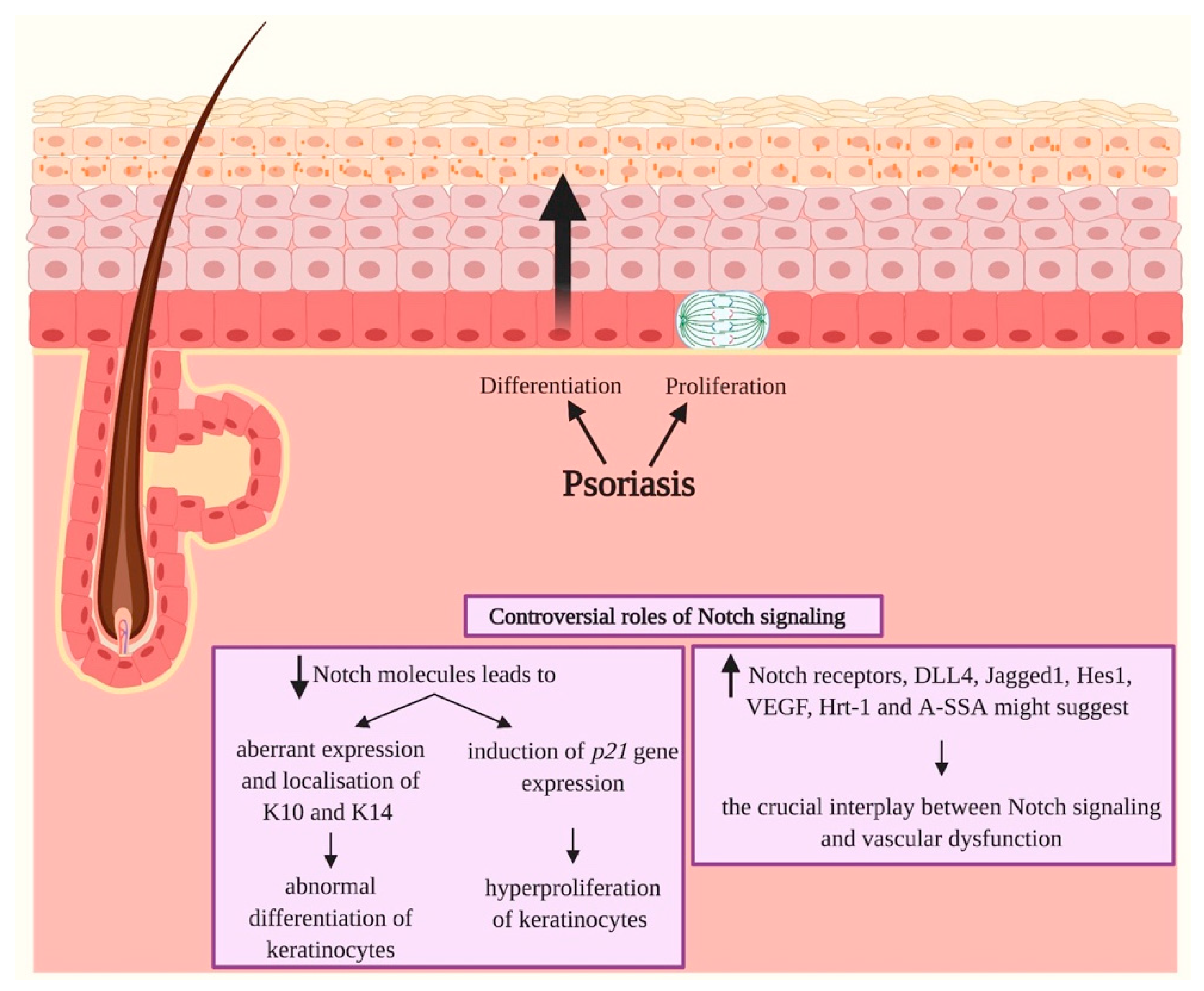
3.4. Psoriasis
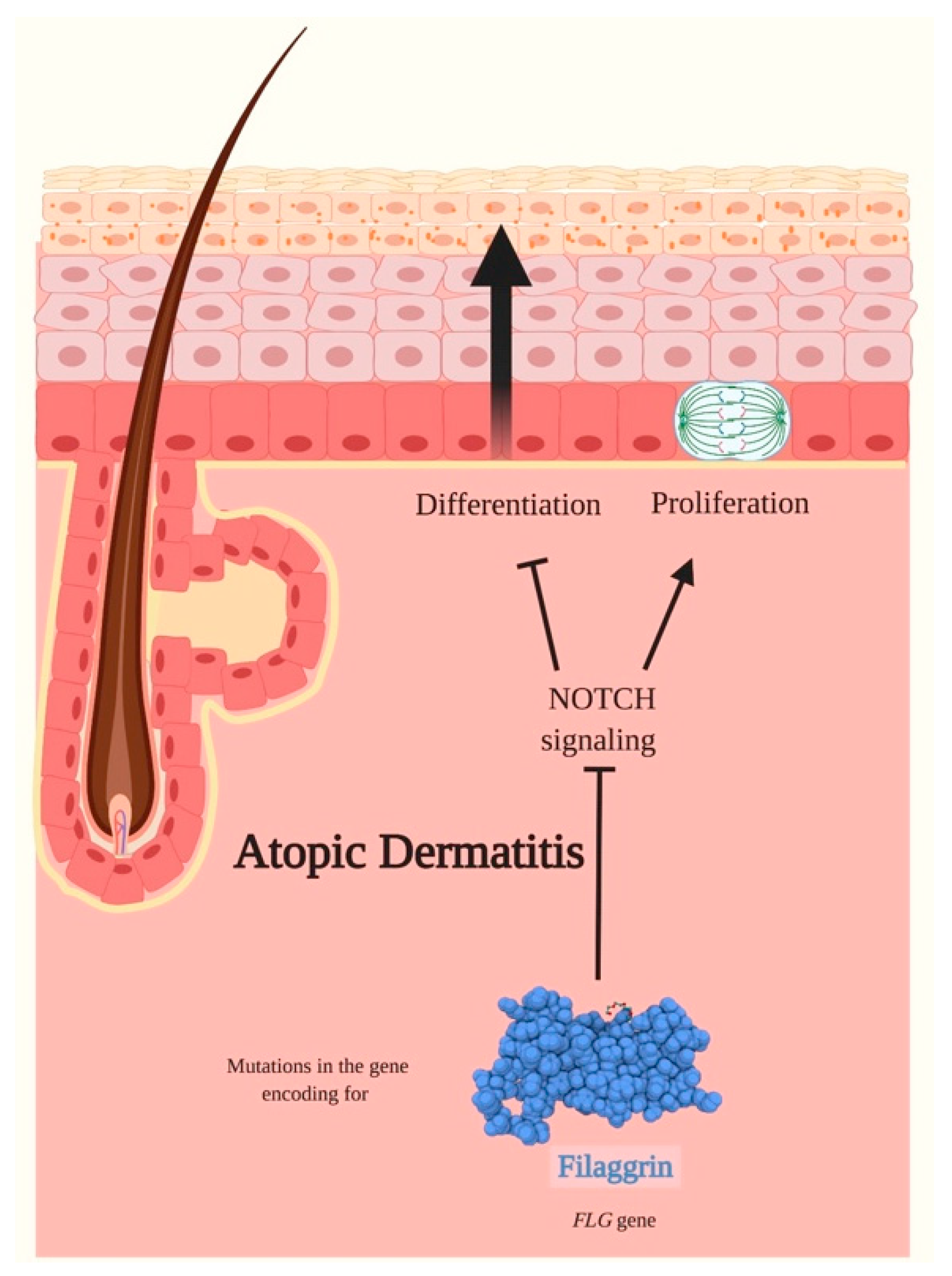
3.5. Atopic Dermatitis (AD)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NECD | Notch extracellular domain |
| EGF-like | Epidermal growth factor-like |
| NRR | Negative regulatory region |
| LNR | Cysteine-rich Lin12-Notch repeats |
| HD | Heterodimerization domain |
| TMD | Transmembrane domain |
| NICD | Intracellular domain |
| RBP-Jκ | Recombination Signal Binding Protein for Immunoglobulin Kappa J Region |
| RAM | Recombination Signal Binding Protein for Immunoglobulin Kappa J Region association module |
| ANK | Seven ankyrin repeats |
| NLS | Nuclear localization signals |
| TAD | Transactivation domain |
| PEST | Proline/glutamic acid/serine/threonine-rich motifs |
| DSL | Delta/Serrate/Lag-2 |
| DLL1 | Delta-like 1 ligand |
| DLL3 | Delta-like 3 ligand |
| DLL4 | Delta-like 4 ligand |
| NTD | N-terminal domain |
| DOS | Delta and OSM-11-like region |
| CRD | Cysteine-rich domain |
| PDZ | PSD-95/Dlg/ZO-1 |
| GPI | Glycosylphosphatidylinositol |
| NTMIC | Notch transmembrane and intracellular domain |
| NEXT | Notch extracellular truncation |
| Co-R | Co-repressor |
| MAML | Mastermind-like |
| NTC | Notch transcription complex |
| HES | Hairy/enhancer of split |
| HEY | Hairy/enhancer of split with YRPW motif |
| CDK-8 | Cyclin-dependent kinase 8 |
| MVBs | Multivesicular bodies |
| HIF | Hypoxia-inducible factor |
| Mef2 | Monocyte enhancer factor-2 |
| IFE | Interfollicular epidermis |
| ESC | Epidermal stem cells |
| K1 | Keratin 1 |
| K5 | Keratin 5 |
| K10 | Keratin 10 |
| PPARγ | Peroxisome-proliferator activated receptor |
| IVL | Involucrin |
| TGM | Transglutaminase |
| PPL | Periplakin |
| PSEN | Presenilin |
| PSENEN | Presenilin enhancer-2 |
| NCSTN | Nicastrin |
| APH1 | Anterior pharynx defective-1 |
| POFUT1 | GDP-fucose protein O-fucosyltransferase 1 |
| EOGT | Epidermal growth factor domain-specific O-linked N-acetylglucosamine transferase |
| POGLUT1 | Protein O-glucosyltransferase 1 |
| O-GlcNAc | O-linked N-acetylglucosamine |
| FILA | Filaggrin |
| FLG | Filaggrin gene |
| HS | Hidradenitis Suppurativa |
| PI3K | Phosphoinositide 3-kinase |
| AKT | Protein Kinase B |
| DDD | Dowling Degos Disease |
| KRT5 | Keratin 5 gene |
| K14 | Ketatin 14 |
| AOS | Adams–Oliver Syndrome |
| ACC | Aplasia cutis congenital |
| TTLD | Terminal transverse limb defects |
| CTMC | Cutis marmorata telangiectatica congenita |
| ARHGAP31 | Rho GTPase Activating Protein 31 |
| DOCK6 | Dedicator of Cytokinesis Protein 6 |
| GEF | Guanine nucleotide exchange factor |
| Hrt-1 | Ring-box protein HRT1 |
| A-SAA | Acute-phase Serum Amyloid A |
| AD | Atopic Dermatitis |
| DAPT | N-S-phenylglycine t-butylester |
References
- Axelrod, J.D.; Matsuno, K.; Artavanis-Tsakonas, S.; Perrimon, N. Interaction Between Wingless and Notch Signaling Pathways Mediated by Dishevelled. Science 1996, 271, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Talora, C.; Campese, A.F.; Bellavia, D.; Felli, M.P.; Vacca, A.; Gulino, A.; Screpanti, I. Notch signaling and diseases: An evolutionary journey from a simple beginning to complex outcomes. Biochimica et Biophysica Acta (BBA)-Mol. Basis Dis. 2008, 1782, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, F.; Osborne, B.A. Non-Canonical Notch Signaling in Cancer and Immunity. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Suresh, S.; Irvine, A.E. The NOTCH signaling pathway in normal and malignant blood cell production. J. Cell Commun. Signal. 2015, 9, 5–13. [Google Scholar] [CrossRef]
- Chillakuri, C.R.; Sheppard, D.; Lea, S.M.; Handford, P.A. Notch receptor–ligand binding and activation: Insights from molecular studies. Semin. Cell Dev. Biol. 2012, 23, 421–428. [Google Scholar] [CrossRef]
- Fiuza, U.-M.; Arias, A.M. Cell and molecular biology of Notch. J. Endocrinol. 2007, 194, 459–474. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Xu, X.; Choi, S.H.; Hu, T.; Tiyanont, K.; Habets, R.; Groot, A.J.; Vooijs, M.; Aster, J.C.; Chopra, R.; Fryer, C.; et al. Insights into Autoregulation of Notch3 from Structural and Functional Studies of Its Negative Regulatory Region. Structure 2015, 23, 1227–1235. [Google Scholar] [CrossRef]
- Canalis, E. Notch in skeletal physiology and disease. Osteoporos. Int. 2018, 29, 2611–2621. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Taniguchi, Y.; Minoguchi, S.; Sakai, T.; Tun, T.; Furukawa, T.; Honjo, T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-Jκ/Su(H). Curr. Biol. 1995, 5, 1416–1423. [Google Scholar] [CrossRef]
- Huenniger, K.; Krämer, A.; Soom, M.; Chang, I.; Köhler, M.; Depping, R.; Kehlenbach, R.H.; Kaether, C. Notch1 signaling is mediated by importins alpha 3, 4, and 7. Cell. Mol. Life Sci. 2010, 67, 3187–3196. [Google Scholar] [CrossRef]
- Cave, J.W. Selective repression of Notch pathway target gene transcription. Dev. Biol. 2011, 360, 123–131. [Google Scholar] [CrossRef][Green Version]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev. Cell 2017, 41, 228–241. [Google Scholar] [CrossRef]
- Barolo, S. Three habits of highly effective signaling pathways: Principles of transcriptional control by developmental cell signaling. Genes Dev. 2002, 16, 1167–1181. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Nandagopal, N.; Santat, L.A.; LeBon, L.; Sprinzak, D.; Bronner, M.E.; Elowitz, M.B. Dynamic Ligand Discrimination in the Notch Signaling Pathway. Cell 2018, 172, 869–880.e19. [Google Scholar] [CrossRef]
- Handford, P.A.; Korona, B.; Suckling, R.; Redfield, C.; Lea, S.M. Structural Insights into Notch Receptor-Ligand Interactions. In Molecular Mechanisms of Notch Signaling; Borggrefe, T., Giaimo, B.D., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; pp. 33–46. ISBN 978-3-319-89511-6. [Google Scholar]
- Komatsu, H.; Chao, M.Y.; Larkins-Ford, J.; Corkins, M.E.; Somers, G.A.; Tucey, T.; Dionne, H.M.; White, J.Q.; Wani, K.; Boxem, M.; et al. OSM-11 Facilitates LIN-12 Notch Signaling during Caenorhabditis elegans Vulval Development. PLoS Biol. 2008, 6, e196. [Google Scholar] [CrossRef]
- D’Souza, B.; Meloty-Kapella, L.; Weinmaster, G. Canonical and Non-Canonical Notch Ligands. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 73–129. ISBN 978-0-12-380914-8. [Google Scholar]
- Morrissette, J.J.D. Defective intracellular transport and processing of JAG1 missense mutations in Alagille syndrome. Hum. Mol. Genet. 2001, 10, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Pintar, A.; De Biasio, A.; Popovic, M.; Ivanova, N.; Pongor, S. The intracellular region of Notch ligands: Does the tail make the difference? Biol. Direct 2007, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Hu, S.; Wang, J.; He, S.; Zhang, Y. DLL4+ dendritic cells: Key regulators of Notch Signaling in effector T cell responses. Pharmacol. Res. 2016, 113, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Andrawes, M.B.; Xu, X.; Liu, H.; Ficarro, S.B.; Marto, J.A.; Aster, J.C.; Blacklow, S.C. Intrinsic Selectivity of Notch 1 for Delta-like 4 Over Delta-like 1. J. Biol. Chem. 2013, 288, 25477–25489. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Taniguchi, K.; Hamamoto, H.; Ito, Y.; Futaki, S.; Inomata, Y.; Shima, T.; Asakuma, M.; Lee, S.; Tanaka, K.; et al. Delta-like 3 localizes to neuroendocrine cells and plays a pivotal role in gastrointestinal neuroendocrine malignancy. Cancer Sci. 2019, 110, 3122–3131. [Google Scholar] [CrossRef]
- Spino, M.; Kurz, S.C.; Chiriboga, L.; Serrano, J.; Zeck, B.; Sen, N.; Patel, S.; Shen, G.; Vasudevaraja, V.; Tsirigos, A.; et al. Cell Surface Notch Ligand DLL3 is a Therapeutic Target in Isocitrate Dehydrogenase–mutant Glioma. Clin. Cancer Res. 2019, 25, 1261–1271. [Google Scholar] [CrossRef]
- Andersen, P.; Uosaki, H.; Shenje, L.T.; Kwon, C. Non-canonical Notch signaling: Emerging role and mechanism. Trends Cell Biol. 2012, 22, 257–265. [Google Scholar] [CrossRef]
- Fortini, M.E. Notch Signaling: The Core Pathway and Its Posttranslational Regulation. Dev. Cell 2009, 16, 633–647. [Google Scholar] [CrossRef]
- Vodovar, N.; Schweisguth, F. Functions of O-fucosyltransferase in Notch trafficking and signaling: Towards the end of a controversy? J. Biol. 2008, 7, 7. [Google Scholar] [CrossRef]
- Gordon, W.R.; Vardar-Ulu, D.; L’Heureux, S.; Ashworth, T.; Malecki, M.J.; Sanchez-Irizarry, C.; McArthur, D.G.; Histen, G.; Mitchell, J.L.; Aster, J.C.; et al. Effects of S1 Cleavage on the Structure, Surface Export, and Signaling Activity of Human Notch1 and Notch2. PLoS ONE 2009, 4, e6613. [Google Scholar] [CrossRef]
- Schweisguth, F. Regulation of Notch Signaling Activity. Curr. Biol. 2004, 14, R129–R138. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Canalis, E. Notch Signaling and the Skeleton. Endocr. Rev. 2016, 37, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Kovall, R.A. More complicated than it looks: Assembly of Notch pathway transcription complexes. Oncogene 2008, 27, 5099–5109. [Google Scholar] [CrossRef]
- Wang, H.; Zang, C.; Liu, X.S.; Aster, J.C. The Role of Notch Receptors in Transcriptional Regulation: Notch and transcription. J. Cell. Physiol. 2015, 230, 982–988. [Google Scholar] [CrossRef]
- Iso, T.; Kedes, L.; Hamamori, Y. HES and HERP families: Multiple effectors of the notch signaling pathway. J. Cell. Physiol. 2003, 194, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R. Notch Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011213. [Google Scholar] [CrossRef] [PubMed]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017, 552, 258–262. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, L.; Zhao, S.; Zhao, X.-Y.; Min, P.-X.; Ma, Y.-D.; Wang, Y.-Y.; Chen, Y.; Tang, S.-J.; Zhang, Y.-J.; et al. Non-canonical Notch Signaling Regulates Actin Remodeling in Cell Migration by Activating PI3K/AKT/Cdc42 Pathway. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Alfred, V.; Vaccari, T. Mechanisms of Non-canonical Signaling in Health and Disease: Diversity to Take Therapy up a Notch? In Molecular Mechanisms of Notch Signaling; Borggrefe, T., Giaimo, B.D., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; pp. 187–204. ISBN 978-3-319-89511-6. [Google Scholar]
- Baron, M. Endocytic routes to Notch activation. Semin. Cell Dev. Biol. 2012, 23, 437–442. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, Q. Plasma membrane-derived extracellular microvesicles mediate non-canonical intercellular NOTCH signaling. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Masiero, M.; Banham, A.H. Notch signaling: Its roles and therapeutic potential in hematological malignancies. Oncotarget 2016, 7, 29804–29823. [Google Scholar] [CrossRef] [PubMed]
- Gilaberte, Y.; Prieto-Torres, L.; Pastushenko, I.; Juarranz, Á. Anatomy and Function of the Skin. In Nanoscience in Dermatology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1–14. ISBN 978-0-12-802926-8. [Google Scholar]
- Watt, F.M. Mammalian skin cell biology: At the interface between laboratory and clinic. Science 2014, 346, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Skin stem cells: Rising to the surface. J. Cell Biol. 2008, 180, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Kao, C.-H.; Lin, K.M.-C.; Kaartinen, V.; Yang, L.-T. Notch Signaling Regulates Late-Stage Epidermal Differentiation and Maintains Postnatal Hair Cycle Homeostasis. PLoS ONE 2011, 6, e15842. [Google Scholar] [CrossRef]
- Mehrel, T.; Hohl, D.; Rothnagel, J.A.; Longley, M.A.; Bundman, D.; Cheng, C.; Lichti, U.; Bisher, M.E.; Steven, A.C.; Steinert, P.M.; et al. Identification of a major keratinocyte cell envelope protein, loricrin. Cell 1990, 61, 1103–1112. [Google Scholar] [CrossRef]
- Nickoloff, B.J.; Qin, J.-Z.; Chaturvedi, V.; Denning, M.F.; Bonish, B.; Miele, L. Jagged-1 mediated activation of notch signaling induces complete maturation of human keratinocytes through NF-κB and PPARγ. Cell Death Differ. 2002, 9, 842–855. [Google Scholar] [CrossRef]
- Fuchs, E.; Horsley, V. More than one way to skin. Genes Dev. 2008, 22, 976–985. [Google Scholar] [CrossRef]
- Fuchs, E. Epidermal differentiation: The bare essentials. J. Cell Biol. 1990, 111, 2807–2814. [Google Scholar] [CrossRef]
- Massi, D.; Panelos, J. Notch Signaling and the Developing Skin Epidermis. In Notch Signaling in Embryology and Cancer; Reichrath, J., Reichrath, S., Eds.; Advances in Experimental Medicine and Biology; Springer US: New York, NY, USA, 2012; pp. 131–141. ISBN 978-1-4614-0898-7. [Google Scholar]
- Roberson, E.D.O.; Bowcock, A.M. Psoriasis genetics: Breaking the barrier. Trends Genet. 2010, 26, 415–423. [Google Scholar] [CrossRef]
- Okuyama, R.; Tagami, H.; Aiba, S. Notch signaling: Its role in epidermal homeostasis and in the pathogenesis of skin diseases. J. Dermatol. Sci. 2008, 49, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, A.; Talora, C.; Okuyama, R.; Nicolas, M.; Mammucari, C.; Oh, H.; Aster, J.C.; Krishna, S.; Metzger, D.; Chambon, P.; et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001, 20, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Negri, V.A.; Logtenberg, M.E.W.; Renz, L.M.; Oules, B.; Walko, G.; Watt, F.M. Delta-like 1-mediated cis-inhibition of Jagged1/2 signalling inhibits differentiation of human epidermal cells in culture. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Lowry, W.E.; Pasolli, H.A.; Fuchs, E. Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 2006, 20, 3022–3035. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M.; Estrach, S.; Ambler, C.A. Epidermal Notch signalling: Differentiation, cancer and adhesion. Curr. Opin. Cell Biol. 2008, 20, 171–179. [Google Scholar] [CrossRef]
- Yeo, S.-Y.; Chitnis, A.B. Jagged-mediated Notch signaling maintains proliferating neural progenitors and regulates cell diversity in the ventral spinal cord. Proc. Natl. Acad. Sci. USA 2007, 104, 5913–5918. [Google Scholar] [CrossRef]
- Yang, R.-H.; Qi, S.-H.; Shu, B.; Ruan, S.-B.; Lin, Z.-P.; Lin, Y.; Shen, R.; Zhang, F.-G.; Chen, X.-D.; Xie, J.-L. Epidermal stem cells (ESCs) accelerate diabetic wound healing via the Notch signalling pathway. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef]
- Lowell, S.; Jones, P.; Le Roux, I.; Dunne, J.; Watt, F.M. Stimulation of human epidermal differentiation by Delta–Notch signalling at the boundaries of stem-cell clusters. Curr. Biol. 2000, 10, 491–500. [Google Scholar] [CrossRef]
- Palazzo, E.; Morandi, P.; Lotti, R.; Saltari, A.; Truzzi, F.; Schnebert, S.; Dumas, M.; Marconi, A.; Pincelli, C. Notch Cooperates with Survivin to Maintain Stemness and to Stimulate Proliferation in Human Keratinocytes during Ageing. Int. J. Mol. Sci. 2015, 16, 26291–26302. [Google Scholar] [CrossRef]
- Okuyama, R.; Nguyen, B.-C.; Talora, C.; Ogawa, E.; di Vignano, A.T.; Lioumi, M.; Chiorino, G.; Tagami, H.; Woo, M.; Dotto, G.P. High Commitment of Embryonic Keratinocytes to Terminal Differentiation through a Notch1-caspase 3 Regulatory Mechanism. Dev. Cell 2004, 6, 551–562. [Google Scholar] [CrossRef]
- Mazur, P.K.; Grüner, B.M.; Nakhai, H.; Sipos, B.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Schmid, R.M.; Siveke, J.T. Identification of Epidermal Pdx1 Expression Discloses Different Roles of Notch1 and Notch2 in Murine KrasG12D-Induced Skin Carcinogenesis In Vivo. PLoS ONE 2010, 5, e13578. [Google Scholar] [CrossRef]
- Nguyen, B.-C. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef] [PubMed]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 Loss Promotes Skin Tumorigenesis by Impacting the Stromal Microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar] [PubMed]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Sundberg, J.P.; Beatus, P.; Lendahl, U.; Joutel, A.; Gridley, T. Characterization ofNotch3-deficient mice: Normal embryonic development and absence of genetic interactions with aNotch1 mutation. Genesis 2003, 37, 139–143. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, M.-H.; Tian, X.; Cheng, H.-T.; Gridley, T.; Shen, J.; Kopan, R. gamma-secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Dev. Cell 2004, 7, 731–743. [Google Scholar] [CrossRef]
- Conlon, R.A.; Reaume, A.G.; Rossant, J. Notch1 is required for the coordinate segmentation of somites. Development 1995, 121, 1533–1545. [Google Scholar]
- Hamada, Y.; Kadokawa, Y.; Okabe, M.; Ikawa, M.; Coleman, J.R.; Tsujimoto, Y. Mutation in ankyrin repeats of the mouse Notch2 gene induces early embryonic lethality. Development 1999, 126, 3415–3424. [Google Scholar]
- Chigurupati, S.; Arumugam, T.V.; Son, T.G.; Lathia, J.D.; Jameel, S.; Mughal, M.R.; Tang, S.-C.; Jo, D.-G.; Camandola, S.; Giunta, M.; et al. Involvement of Notch Signaling in Wound Healing. PLoS ONE 2007, 2, e1167. [Google Scholar] [CrossRef]
- Na, J.; Shin, J.Y.; Jeong, H.; Lee, J.Y.; Kim, B.J.; Kim, W.S.; Yune, T.Y.; Ju, B.-G. JMJD3 and NF-κB-dependent activation of Notch1 gene is required for keratinocyte migration during skin wound healing. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Olsauskas-Kuprys, R.; Zlobin, A.; Osipo, C. Gamma secretase inhibitors of Notch signaling. OncoTargets Ther. 2013, 943. [Google Scholar] [CrossRef]
- Carroll, C.M.; Li, Y.-M. Physiological and pathological roles of the γ-secretase complex. Brain Res. Bull. 2016, 126, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y. The gamma-secretase complex: From structure to function. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Haltiwanger, R.S. Significance of glycosylation in Notch signaling. Biochem. Biophys. Res. Commun. 2014, 453, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Uemura, K.; Ge, C.; Shi, S.; Tashima, Y.; Stanley, P. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J. Biol. Chem. 2008, 283, 13638–13651. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Valdivia, R.; Takeuchi, H.; Samarghandi, A.; Lopez, M.; Leonardi, J.; Haltiwanger, R.S.; Jafar-Nejad, H. Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development 2011, 138, 1925–1934. [Google Scholar] [CrossRef]
- Li, M.; Cheng, R.; Liang, J.; Yan, H.; Zhang, H.; Yang, L.; Li, C.; Jiao, Q.; Lu, Z.; He, J.; et al. Mutations in POFUT1, Encoding Protein O-fucosyltransferase 1, Cause Generalized Dowling-Degos Disease. Am. J. Hum. Genet. 2013, 92, 895–903. [Google Scholar] [CrossRef]
- Basmanav, F.B.; Oprisoreanu, A.-M.; Pasternack, S.M.; Thiele, H.; Fritz, G.; Wenzel, J.; Größer, L.; Wehner, M.; Wolf, S.; Fagerberg, C.; et al. Mutations in POGLUT1, Encoding Protein O-Glucosyltransferase 1, Cause Autosomal-Dominant Dowling-Degos Disease. Am. J. Hum. Genet. 2014, 94, 135–143. [Google Scholar] [CrossRef]
- Ogawa, M.; Sawaguchi, S.; Kawai, T.; Nadano, D.; Matsuda, T.; Yagi, H.; Kato, K.; Furukawa, K.; Okajima, T. Impaired O-linked N-acetylglucosaminylation in the endoplasmic reticulum by mutated EGF domain-specific O-linked N-acetylglucosamine transferase found in Adams-Oliver syndrome. J. Biol. Chem. 2015, 290, 2137–2149. [Google Scholar] [CrossRef]
- Sawaguchi, S.; Varshney, S.; Ogawa, M.; Sakaidani, Y.; Yagi, H.; Takeshita, K.; Murohara, T.; Kato, K.; Sundaram, S.; Stanley, P.; et al. O-GlcNAc on NOTCH1 EGF repeats regulates ligand-induced Notch signaling and vascular development in mammals. eLife 2017, 6. [Google Scholar] [CrossRef]
- Sandilands, A.; Sutherland, C.; Irvine, A.D.; McLean, W.H.I. Filaggrin in the frontline: Role in skin barrier function and disease. J. Cell Sci. 2009, 122, 1285–1294. [Google Scholar] [CrossRef]
- Irvine, A.D.; McLean, W.H.I.; Leung, D.Y.M. Filaggrin Mutations Associated with Skin and Allergic Diseases. N. Engl. J. Med. 2011, 365, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Elkin, K.; Daveluy, S.; Avanaki, K. (Mohammad) Hidradenitis suppurativa: Current understanding, diagnostic and surgical challenges, and developments in ultrasound application. Skin Res. Technol. 2020, 26, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Liy-Wong, C.; Pope, E.; Lara-Corrales, I. Hidradenitis suppurativa in the pediatric population. J. Am. Acad. Dermatol. 2015, 73, S36–S41. [Google Scholar] [CrossRef] [PubMed]
- Von Laffert, M.; Helmbold, P.; Wohlrab, J.; Fiedler, E.; Stadie, V.; Marsch, W.C. Hidradenitis suppurativa (acne inversa): Early inflammatory events at terminal follicles and at interfollicular epidermis. Exp. Dermatol. 2010, 19, 533–537. [Google Scholar] [CrossRef]
- Von Laffert, M.; Stadie, V.; Wohlrab, J.; Marsch, W.C. Hidradenitis suppurativa/acne inversa: Bilocated epithelial hyperplasia with very different sequelae: HS with bilocated epithelial hyperplasia. Br. Dermatol. 2011, 164, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.C.; Desai, N.; Emtestam, L.; Hunger, R.E.; Ioannides, D.; Juhász, I.; Lapins, J.; Matusiak, L.; Prens, E.P.; Revuz, J.; et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 619–644. [Google Scholar] [CrossRef]
- Wortsman, X.; Jemec, G. A 3D ultrasound study of sinus tract formation in hidradenitis suppurativa. Dermatol. Online J. 2013, 19, 18564. [Google Scholar]
- Tricarico, P.M.; Boniotto, M.; Genovese, G.; Zouboulis, C.C.; Marzano, A.V.; Crovella, S. An Integrated Approach to Unravel Hidradenitis Suppurativa Etiopathogenesis. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. γ-Secretase Gene Mutations in Familial Acne Inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef]
- Xiao, X.; He, Y.; Li, C.; Zhang, X.; Xu, H.; Wang, B. Nicastrin mutations in familial acne inversa impact keratinocyte proliferation and differentiation through the Notch and phosphoinositide 3-kinase/AKT signalling pathways. Br. J. Dermatol. 2016, 174, 522–532. [Google Scholar] [CrossRef]
- Demehri, S.; Kopan, R. Notch signaling in bulge stem cells is not required for selection of hair follicle fate. Development 2009, 136, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.H.; Mahajan, S.A.; Khopkar, U.S.; Kharkar, V.D. Follicular Dowling-Degos Disease: A Rare Pigmentary Dermatosis. Indian Dermatol. Online J. 2017, 8, 487–489. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.S.; Cook, C. Dowling Degos Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Betz, R.C.; Planko, L.; Eigelshoven, S.; Hanneken, S.; Pasternack, S.M.; Bussow, H.; Van Den Bogaert, K.; Wenzel, J.; Braun-Falco, M.; Rutten, A.; et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am. J. Hum. Genet. 2006, 78, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Davis, M.D.; Schanbacher, C.F.; Su, W.P. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): A clinical and histopathologic study of 6 cases. J. Am. Acad. Dermatol. 1999, 40, 462–467. [Google Scholar] [CrossRef]
- Herrmann, H.; Aebi, U. Intermediate filaments: Molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu. Rev. Biochem. 2004, 73, 749–789. [Google Scholar] [CrossRef]
- Alam, H.; Sehgal, L.; Kundu, S.T.; Dalal, S.N.; Vaidya, M.M. Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. Mol. Biol. Cell 2011, 22, 4068–4078. [Google Scholar] [CrossRef]
- Ralser, D.J.; Basmanav, F.B.Ü.; Tafazzoli, A.; Wititsuwannakul, J.; Delker, S.; Danda, S.; Thiele, H.; Wolf, S.; Busch, M.; Pulimood, S.A.; et al. Mutations in γ-secretase subunit–encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J. Clin. Investig. 2017, 127, 1485–1490. [Google Scholar] [CrossRef]
- Yao, D.; Huang, Y.; Huang, X.; Wang, W.; Yan, Q.; Wei, L.; Xin, W.; Gerson, S.; Stanley, P.; Lowe, J.B.; et al. Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood 2011, 117, 5652–5662. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; He, Y.; Lin, L.; Li, C. Comorbidities or different entities? Phenotype variability associated with PSENEN mutations. Br. J. Dermatol. 2019, 180, 221–222. [Google Scholar] [CrossRef] [PubMed]
- Garcovich, S.; Tricarico, P.M.; Meddour, C.N.; Giovanardi, G.; Peris, K.; Crovella, S.; Boniotto, M. Novel nicastrin mutation in hidradenitis suppurativa-Dowling Degos disease clinical phenotype: More than just clinical overlap? Br. J. Dermatol. 2020. [Google Scholar] [CrossRef]
- Ilagan, M.X.G.; Kopan, R. SnapShot: Notch signaling pathway. Cell 2007, 128, 1246. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.N.; Southgate, L.; Stittrich, A.-B.; Venselaar, H.; Beekmans, S.J.A.; den Hollander, N.; Bijlsma, E.K.; Helderman-van den Enden, A.; Verheij, J.B.G.M.; Glusman, G.; et al. Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome. Am. J. Hum. Genet. 2015, 97, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Hassed, S.; Li, S.; Mulvihill, J.; Aston, C.; Palmer, S. Adams-Oliver syndrome review of the literature: Refining the diagnostic phenotype: Adams-Oliver Syndrome: Refining the Phenotype. Am. J. Med Genet. Part A 2017, 173, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.N.; Sukalo, M.; Schröder, K.C.; Schanze, D.; Baynam, G.; Borck, G.; Bramswig, N.C.; Duman, D.; Gilbert-Dussardier, B.; Holder-Espinasse, M.; et al. Elucidating the genetic architecture of Adams-Oliver syndrome in a large European cohort. Hum. Mutat. 2018, 39, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Bartels, S.J.J.; Spruijt, C.G.; Brinkman, A.B.; Jansen, P.W.T.C.; Vermeulen, M.; Stunnenberg, H.G. A SILAC-Based Screen for Methyl-CpG Binding Proteins Identifies RBP-J as a DNA Methylation and Sequence-Specific Binding Protein. PLoS ONE 2011, 6, e25884. [Google Scholar] [CrossRef]
- Shaheen, R.; Aglan, M.; Keppler-Noreuil, K.; Faqeih, E.; Ansari, S.; Horton, K.; Ashour, A.; Zaki, M.S.; Al-Zahrani, F.; Cueto-González, A.M.; et al. Mutations in EOGT Confirm the Genetic Heterogeneity of Autosomal-Recessive Adams-Oliver Syndrome. Am. J. Hum. Genet. 2013, 92, 598–604. [Google Scholar] [CrossRef]
- Sakaidani, Y.; Nomura, T.; Matsuura, A.; Ito, M.; Suzuki, E.; Murakami, K.; Nadano, D.; Matsuda, T.; Furukawa, K.; Okajima, T. O-Linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell–matrix interactions. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef]
- Stittrich, A.-B.; Lehman, A.; Bodian, D.L.; Ashworth, J.; Zong, Z.; Li, H.; Lam, P.; Khromykh, A.; Iyer, R.K.; Vockley, J.G.; et al. Mutations in NOTCH1 Cause Adams-Oliver Syndrome. Am. J. Hum. Genet. 2014, 95, 275–284. [Google Scholar] [CrossRef]
- Meester, J.A.N.; Verstraeten, A.; Alaerts, M.; Schepers, D.; Van Laer, L.; Loeys, B.L. Overlapping but distinct roles for NOTCH receptors in human cardiovascular disease. Clin. Genet. 2019, 95, 85–94. [Google Scholar] [CrossRef]
- Brzozowa, M.; Wojnicz, R.; Kowalczyk-Ziomek, G.; Helewski, K. Reviews The Notch ligand Delta-like 4 (DLL4) as a target in angiogenesis-based cancer therapy? Współczesna Onkologia 2013, 3, 234–237. [Google Scholar] [CrossRef]
- Southgate, L.; Sukalo, M.; Karountzos, A.S.V.; Taylor, E.J.; Collinson, C.S.; Ruddy, D.; Snape, K.M.; Dallapiccola, B.; Tolmie, J.L.; Joss, S.; et al. Haploinsufficiency of the NOTCH1 Receptor as a Cause of Adams–Oliver Syndrome With Variable Cardiac Anomalies. Circ. Cardiovasc. Genet. 2015, 8, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Yamauchi, J.; Sanbe, A.; Tanoue, A. Dock6, a Dock-C subfamily guanine nucleotide exchanger, has the dual specificity for Rac1 and Cdc42 and regulates neurite outgrowth. Exp. Cell Res. 2007, 313, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.-H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Chang, H.-W.; Yan, D.; Singh, R.; Liu, J.; Lu, X.; Ucmak, D.; Lee, K.; Afifi, L.; Fadrosh, D.; Leech, J.; et al. Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef]
- Benhadou, F.; Mintoff, D.; del Marmol, V. Psoriasis: Keratinocytes or Immune Cells–Which Is the Trigger? Dermatology 2019, 235, 91–100. [Google Scholar] [CrossRef]
- Calonje, J.E.; Brenn, T.; Lazar, A.J.; McKee, P.H. Pathology of the Skin; Elsevier Health Sciences UK: London, UK, 2011; ISBN 978-0-7234-3718-5. [Google Scholar]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay Between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Arul, S.; Dayalan, H.; Jegadeesan, M.; Damodharan, P. Induction of differentiation in psoriatic keratinocytes by propylthiouracil and fructose. BBA Clin. 2016, 6, 82–86. [Google Scholar] [CrossRef]
- Albanesi, C.; De Pità, O.; Girolomoni, G. Resident skin cells in psoriasis: A special look at the pathogenetic functions of keratinocytes. Clin. Dermatol. 2007, 25, 581–588. [Google Scholar] [CrossRef]
- Kim, J.; Krueger, J.G. The Immunopathogenesis of Psoriasis. Dermatol. Clin. 2015, 33, 13–23. [Google Scholar] [CrossRef]
- Thélu, J.; Rossio, P.; Favier, B. Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma, psoriasis and wound healing. BMC Dermatol. 2002, 2. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Takekoshi, S.; Takagi, T.; Kitatani, K.; Toriumi, K.; Kojima, T.; Kato, M.; Ikoma, N.; Mabuchi, T.; Ozawa, A. Notch Signaling May Be Involved in the Abnormal Differentiation of Epidermal Keratinocytes in Psoriasis. Acta Histochemica et Cytochemica 2014, 47, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Bowcock, A.M.; Krueger, J.G. Pathogenesis and therapy of psoriasis. Nature 2007, 445, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Skarmoutsou, E.; Trovato, C.; Granata, M.; Rossi, G.A.; Mosca, A.; Longo, V.; Gangemi, P.; Pettinato, M.; D’Amico, F.; Mazzarino, M.C. Biological therapy induces expression changes in Notch pathway in psoriasis. Arch. Dermatol. Res. 2015, 307, 863–873. [Google Scholar] [CrossRef]
- Abdou, A.G.; Maraee, A.H.; Sharaf, A.; Elnaidany, N.F. Up-regulation of Notch-1 in psoriasis: An immunohistochemical study. Ann. Diagn. Pathol. 2012, 16, 177–184. [Google Scholar] [CrossRef]
- Jiao, Z.; Wang, W.; Guo, M.; Zhang, T.; Chen, L.; Wang, Y.; You, H.; Li, J. Expression analysis of Notch-related molecules in peripheral blood T helper cells of patients with rheumatoid arthritis. Scand. J. Rheumatol. 2010, 39, 26–32. [Google Scholar] [CrossRef]
- Rooney, P.; Connolly, M.; Gao, W.; McCormick, J.; Biniecka, M.; Sullivan, O.; Kirby, B.; Sweeney, C.; Molloy, E.; Markham, T.; et al. Notch-1 mediates endothelial cell activation and invasion in psoriasis. Exp. Dermatol. 2014, 23, 113–118. [Google Scholar] [CrossRef]
- Kowalska-Olędzka, E.; Czarnecka, M.; Baran, A. Epidemiology of atopic dermatitis in Europe. J. Drug Assess. 2019, 8, 126–128. [Google Scholar] [CrossRef]
- Fishbein, A.B.; Silverberg, J.I.; Wilson, E.J.; Ong, P.Y. Update on Atopic Dermatitis: Diagnosis, Severity Assessment, and Treatment Selection. J. Allergy Clin. Immunol. Pract. 2020, 8, 91–101. [Google Scholar] [CrossRef]
- Dalgard, F.J.; Gieler, U.; Tomas-Aragones, L.; Lien, L.; Poot, F.; Jemec, G.B.E.; Misery, L.; Szabo, C.; Linder, D.; Sampogna, F.; et al. The Psychological Burden of Skin Diseases: A Cross-Sectional Multicenter Study among Dermatological Out-Patients in 13 European Countries. J. Investig. Dermatol. 2015, 135, 984–991. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Nograles, K.E.; Krueger, J.G. Contrasting pathogenesis of atopic dermatitis and psoriasis—Part II: Immune cell subsets and therapeutic concepts. J. Allergy Clin. Immunol. 2011, 127, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Proksch, E.; Brasch, J. Abnormal epidermal barrier in the pathogenesis of contact dermatitis. Clin. Dermatol. 2012, 30, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Apfelbacher, C.J.; Diepgen, T.L.; Schmitt, J. Determinants of eczema: Population-based cross-sectional study in Germany: Determinants of eczema. Allergy 2011, 66, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.M. New Insights into Atopic Dermatitis: Role of Skin Barrier and Immune Dysregulation. Allergol. Int. 2013, 62, 151–161. [Google Scholar] [CrossRef]
- Dumortier, A.; Durham, A.-D.; Di Piazza, M.; Vauclair, S.; Koch, U.; Ferrand, G.; Ferrero, I.; Demehri, S.; Song, L.L.; Farr, A.G.; et al. Atopic Dermatitis-Like Disease and Associated Lethal Myeloproliferative Disorder Arise from Loss of Notch Signaling in the Murine Skin. PLoS ONE 2010, 5, e9258. [Google Scholar] [CrossRef]
- Ma, L.; Xue, H.; Qi, R.; Wang, Y.; Yuan, L. Effect of γ-secretase inhibitor on Th17 cell differentiation and function of mouse psoriasis-like skin inflammation. J. Transl. Med. 2018, 16. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gratton, R.; Tricarico, P.M.; Moltrasio, C.; Lima Estevão de Oliveira, A.S.; Brandão, L.; Marzano, A.V.; Zupin, L.; Crovella, S. Pleiotropic Role of Notch Signaling in Human Skin Diseases. Int. J. Mol. Sci. 2020, 21, 4214. https://doi.org/10.3390/ijms21124214
Gratton R, Tricarico PM, Moltrasio C, Lima Estevão de Oliveira AS, Brandão L, Marzano AV, Zupin L, Crovella S. Pleiotropic Role of Notch Signaling in Human Skin Diseases. International Journal of Molecular Sciences. 2020; 21(12):4214. https://doi.org/10.3390/ijms21124214
Chicago/Turabian StyleGratton, Rossella, Paola Maura Tricarico, Chiara Moltrasio, Ana Sofia Lima Estevão de Oliveira, Lucas Brandão, Angelo Valerio Marzano, Luisa Zupin, and Sergio Crovella. 2020. "Pleiotropic Role of Notch Signaling in Human Skin Diseases" International Journal of Molecular Sciences 21, no. 12: 4214. https://doi.org/10.3390/ijms21124214
APA StyleGratton, R., Tricarico, P. M., Moltrasio, C., Lima Estevão de Oliveira, A. S., Brandão, L., Marzano, A. V., Zupin, L., & Crovella, S. (2020). Pleiotropic Role of Notch Signaling in Human Skin Diseases. International Journal of Molecular Sciences, 21(12), 4214. https://doi.org/10.3390/ijms21124214