CCR5-Mediated Signaling is Involved in Invasion of Glioblastoma Cells in Its Microenvironment


 , ,
, ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
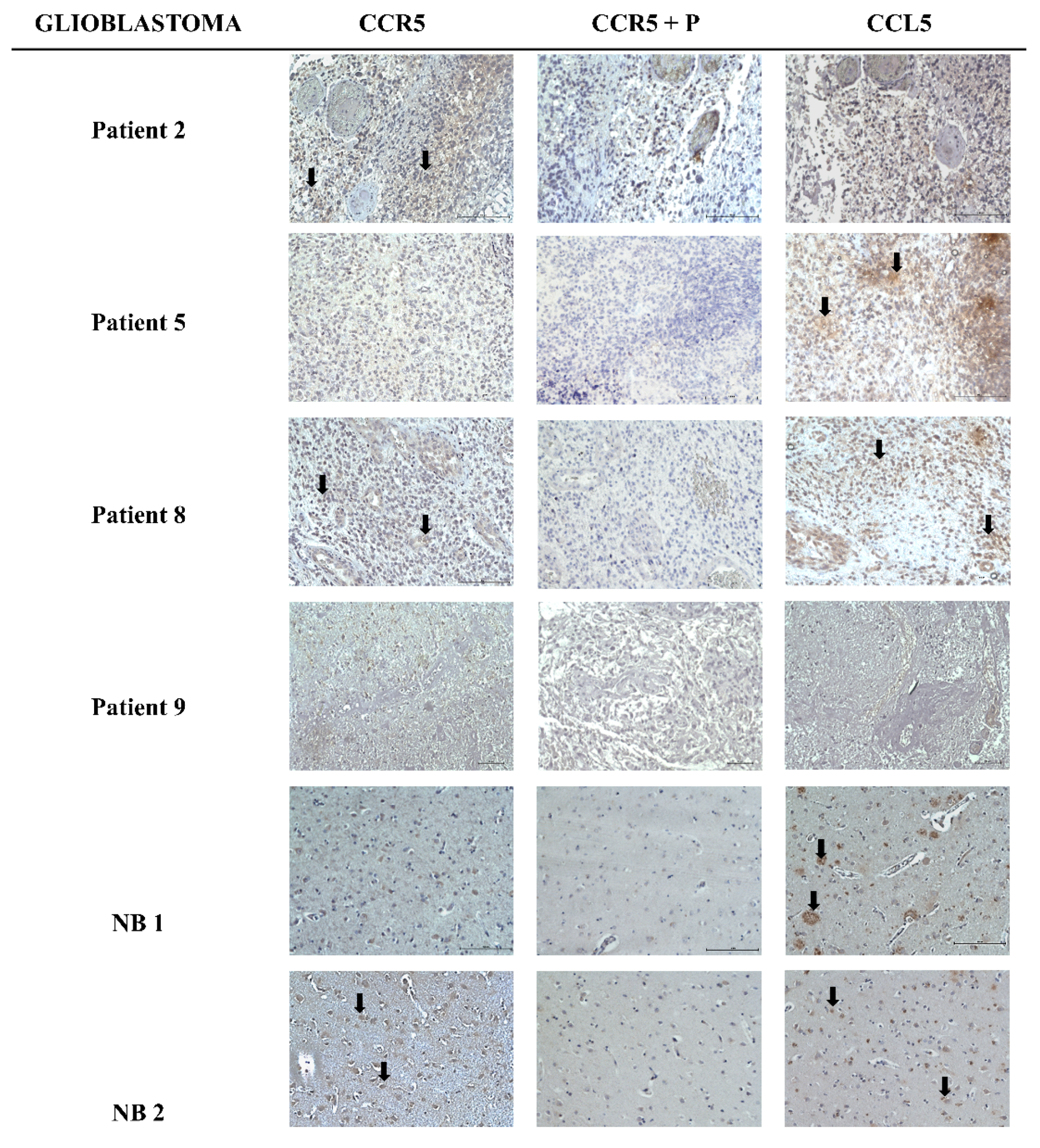
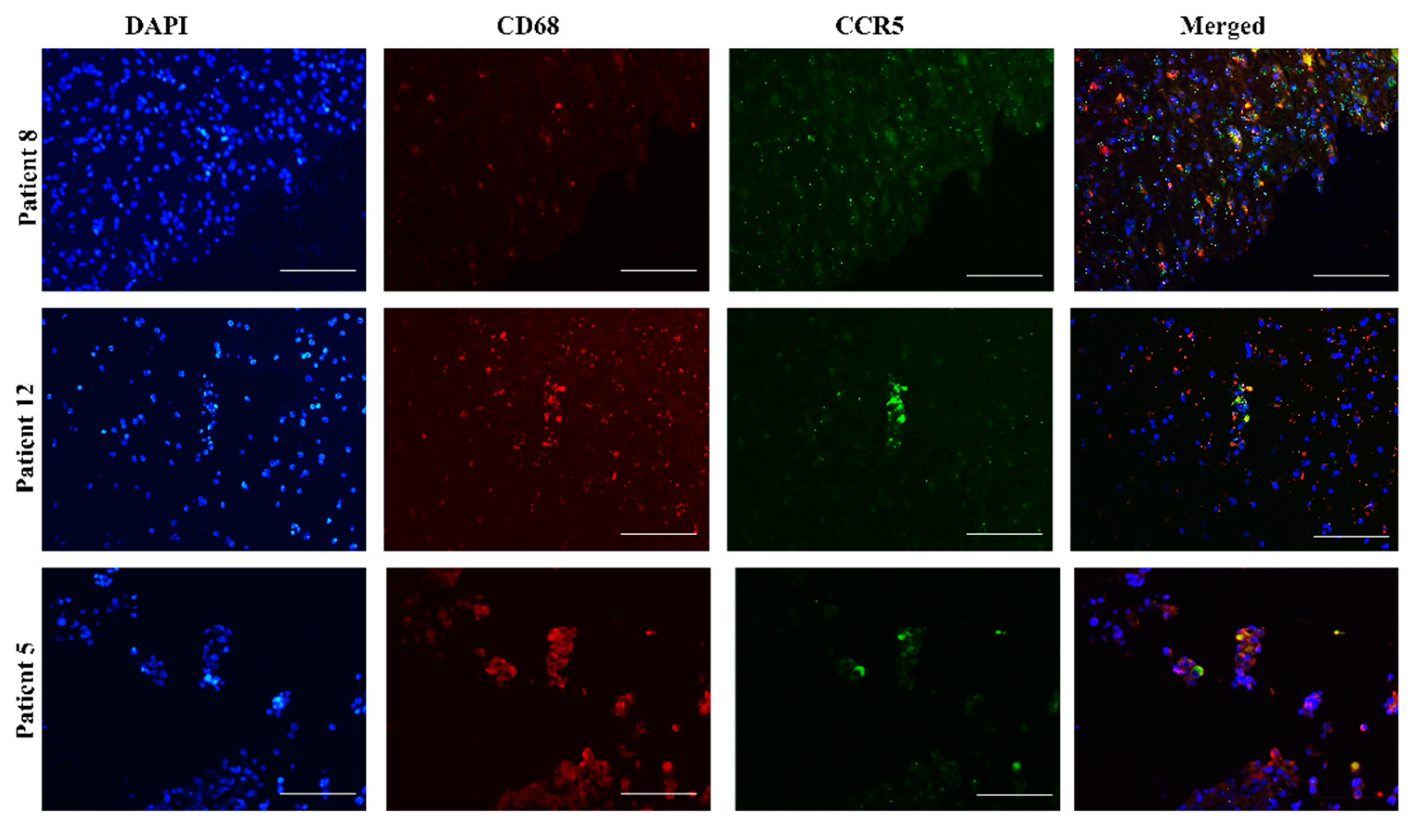
2.1. Heterogeneous Expression of CCL5/CCR5 Axis in Glioblastoma Tissue Sections
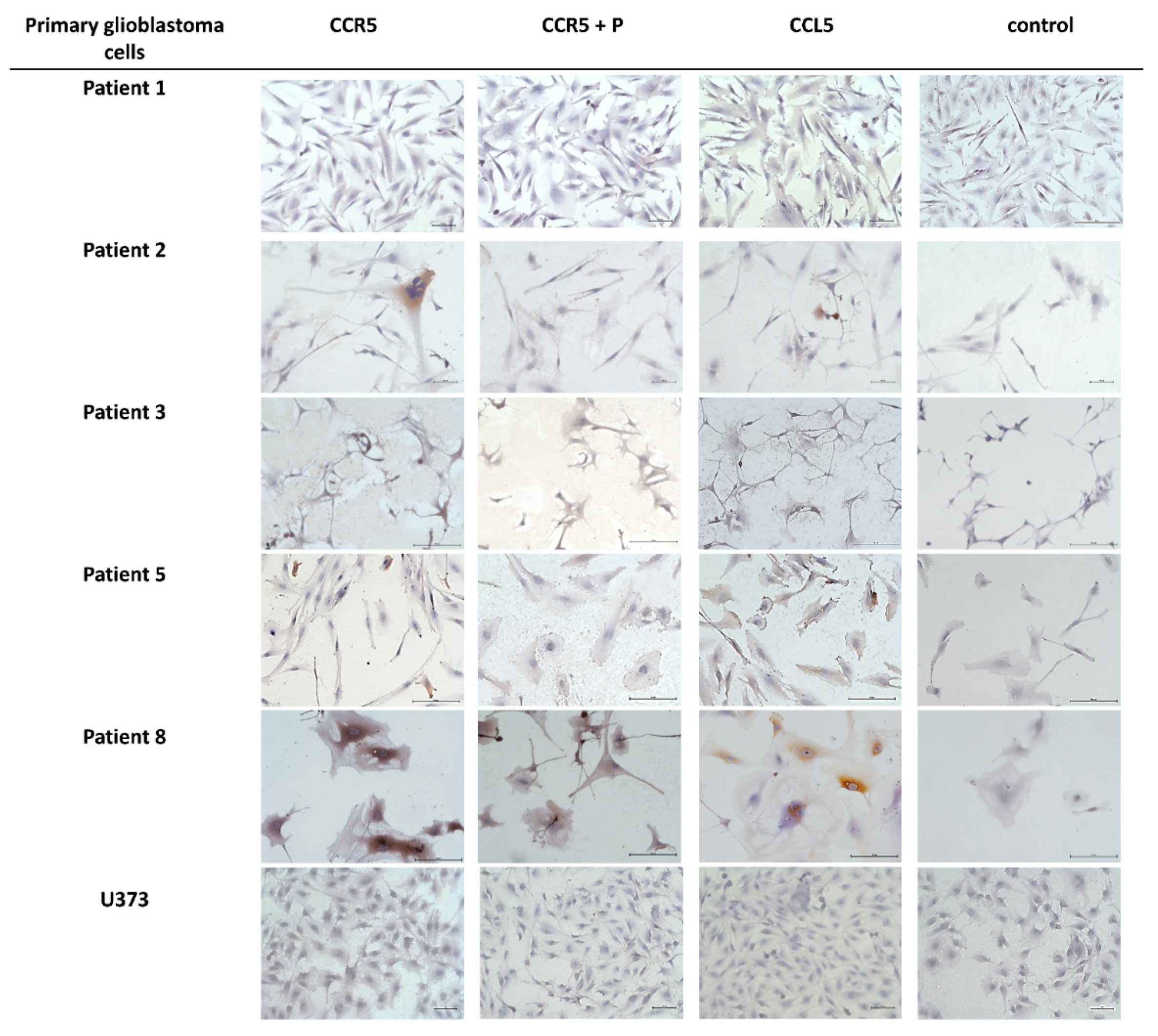
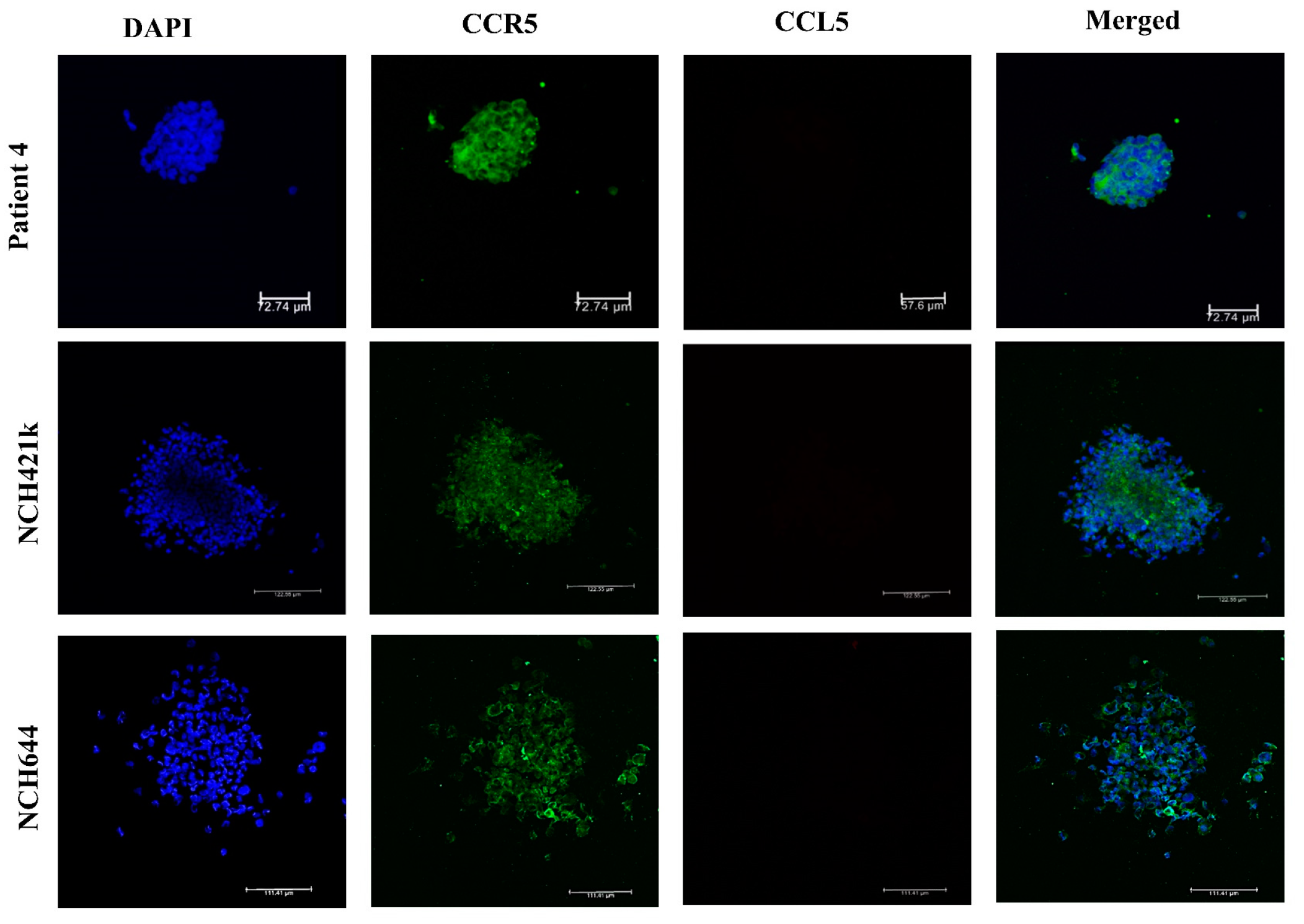
2.2. Expression of CCL5/CCR5 Axis in Primary Glioblastoma Cells and Glioblastoma Stem Cells
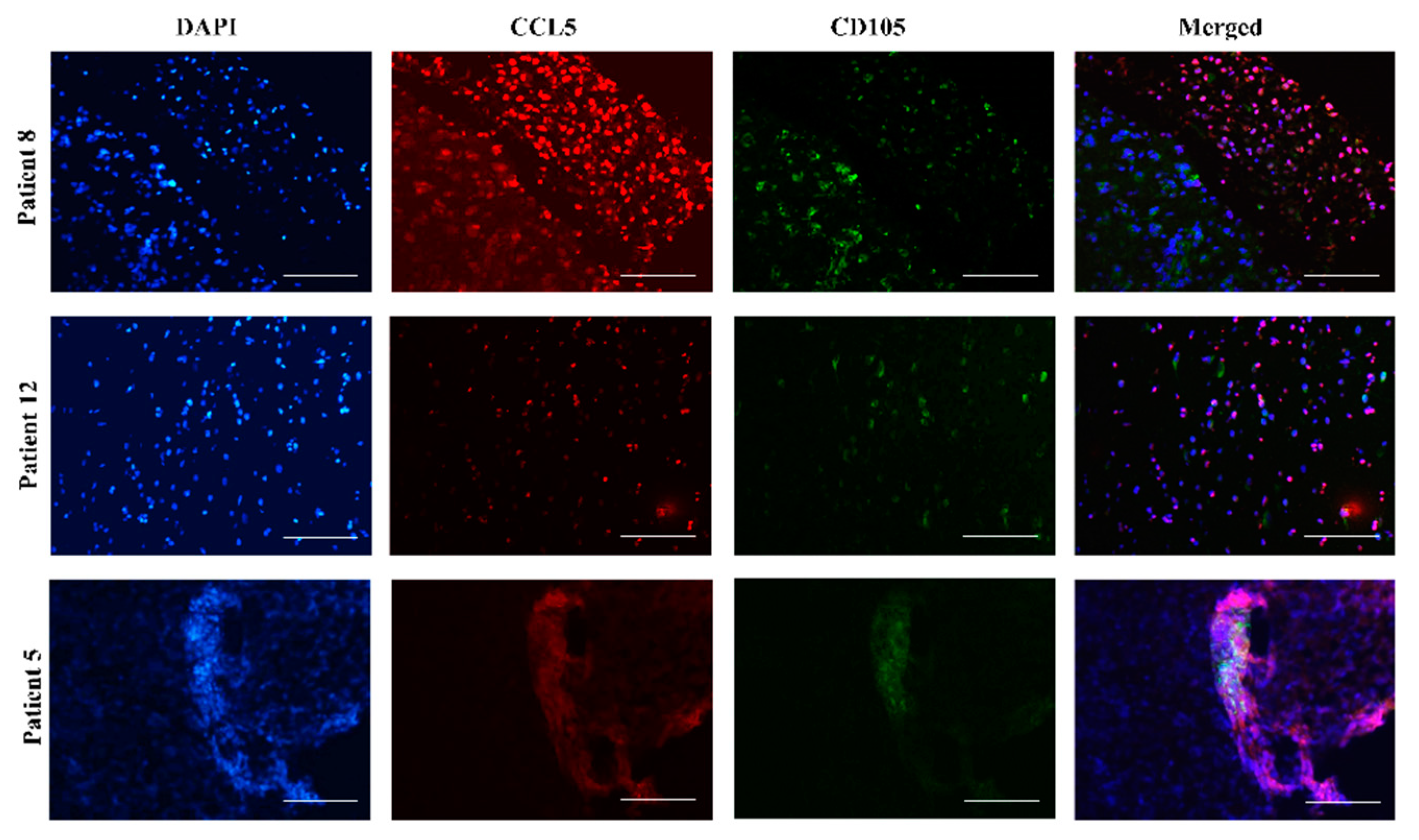
2.3. Mesenchymal Stem Cells In Vitro and In Vivo Tumor Sections Express CCL5
2.4. CCR5 Is Expressed in Glioblastoma-Associated Macrophages in Tumors
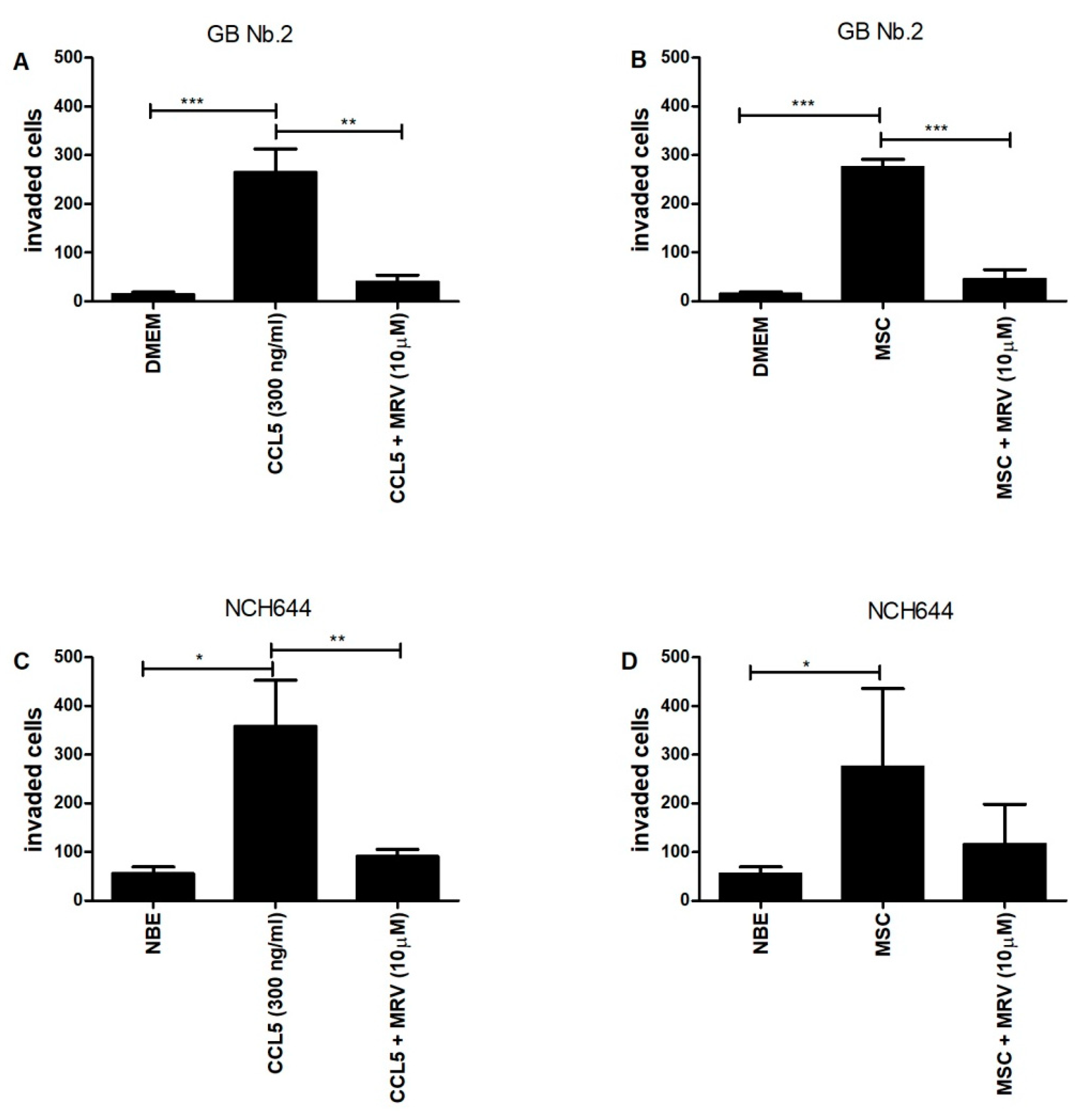
2.5. CCL5 and Mesenchymal Stem Cells Enhance the Invasion of Primary Glioblastoma Cells and Glioblastoma Stem Cells
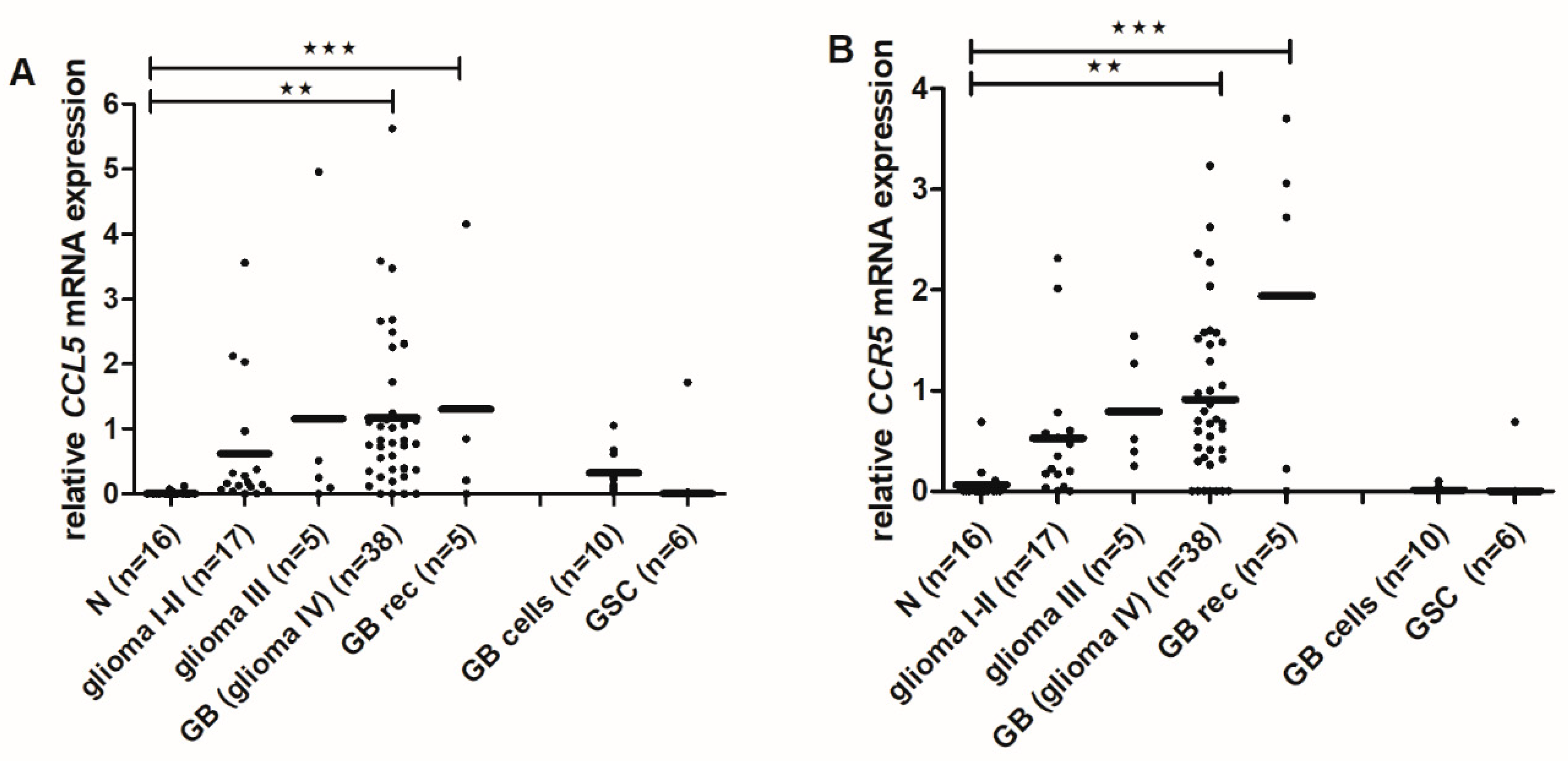
2.6. CCL5 and CCR5 mRNA Levels Are Increased in High-Grade Gliomas
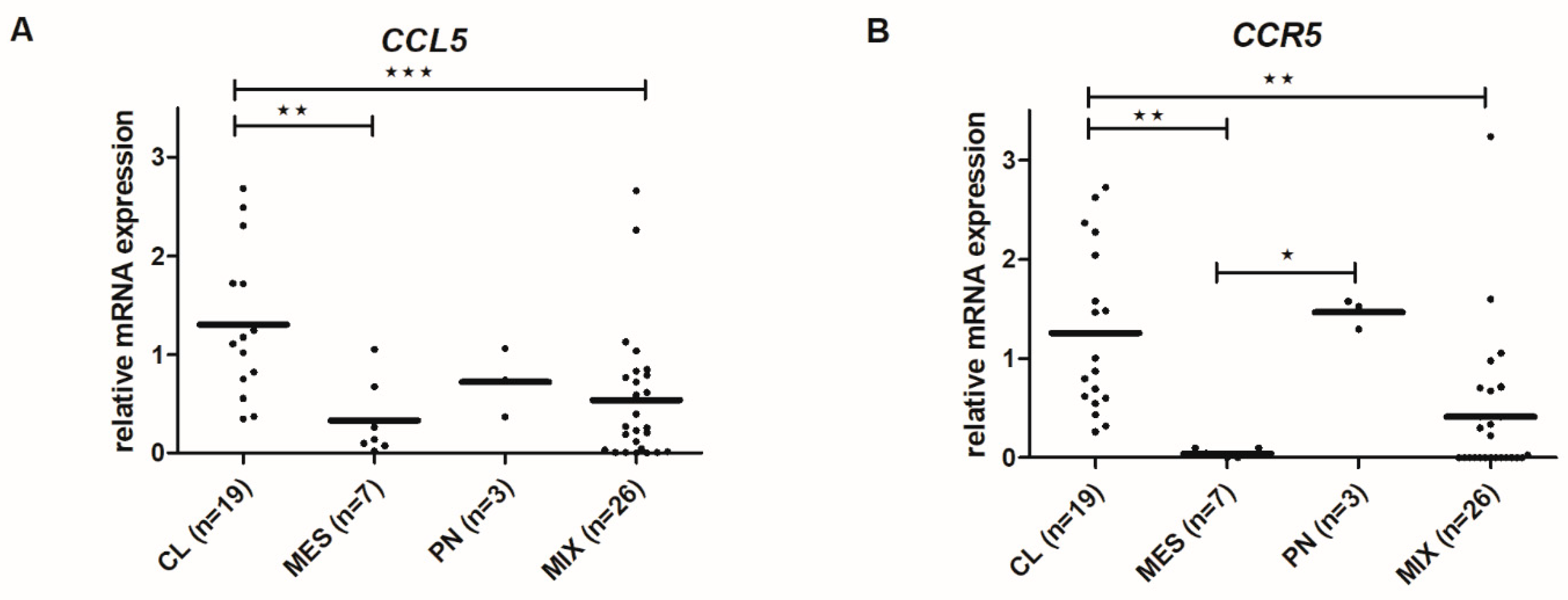
2.7. CCL5 and CCR5 mRNA Levels Differ among Glioblastoma Subtypes
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Glioblastoma Samples from Patients
4.3. Establishment of Primary Glioblastoma and Glioblastoma Stem Cell Lines
4.4. Immunohistochemistry and Immunocytochemistry
4.5. Immunofluorescence of Glioblastoma Stem Cell Spheroids
4.6. Immunofluorescence of Glioblastoma Tumor Tissue Sections
4.7. Invasion Assay
4.8. Cell Viability Assay
4.9. Gene Expression Analysis
4.10. Data Analysis
4.10.1. Glioblastoma Subtyping
4.10.2. Differentially Expressed Genes among Tissues and Glioblastoma Subtypes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Brennan, C.W.; DeAngelis, L.M.; Omuro, A.M. Emerging therapies for glioblastoma. JAMA Neurol. 2014, 71, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Noch, E.K.; Ramakrishna, R.; Magge, R. Challenges in the treatment of glioblastoma: Multisystem mechanisms of therapeutic resistance. World Neurosurg. 2018, 116, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Kranjc, M.K.; Novak, M.; Pestell, R.G.; Lah, T.T. Cytokine CCL5 and receptor CCR5 axis in glioblastoma multiforme. Radiol. Oncol. 2019, 53, 397–406. [Google Scholar] [CrossRef]
- Oliveira, M.N.; Breznik, B.; Pillat, M.M.; Pereira, R.L.; Ulrich, H.; Lah, T.T. Kinins in glioblastoma microenvironment. Cancer Microenviron. 2019, 12, 77–94. [Google Scholar] [CrossRef]
- Teng, J.; Da Hora, C.C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Chiocca, A.E.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol. 2017, 19, 820–832. [Google Scholar] [CrossRef]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal signaling between glioblastoma stem cells and differentiated tumor cells promotes malignant progression. Cell Stem Cell 2018, 22, 514–528. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Breznik, B.; Vittori, M.; de Jong, L.A.; Mlakar, J.; Oostra, R.-J.; Khurshed, M.; Molenaar, R.J.; Lah, T.; Van Noorden, C.J.F. Similarities between stem cell niches in glioblastoma and bone marrow: Rays of hope for novel treatment strategies. J. Histochem. Cytochem. 2019, 68, 33–57. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Aderetti, D.A.; van Noorden, C.J.F. Glioma stem cell niches in human glioblastoma are periarteriolar. J. Histochem. Cytochem. 2018, 66, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Breznik, B.; Motaln, H.; Turnšek, T.L. Proteases and cytokines as mediators of interactions between cancer and stromal cells in tumours. Biol. Chem. 2017, 398, 709–719. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Salanga, C.L.; Handel, T.M.; Allen, S.J. Chemokines and cancer: Migration, intracellular signalling and intercellular communication in the microenvironment. Biochem. J. 2008, 409, 635–649. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Sarvaiya, P.J.; Guo, D.; Ulasov, I.V.; Gabikian, P.; Lesniak, M.S.; Sarvaiya, P.J.; Guo, D.; Ulasov, I.V.; Gabikian, P.; Lesniak, M.S. Chemokines in tumor progression and metastasis. Oncotarget 2013, 4, 2171–2185. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.; Xolalpa, W.; Pestell, R.G. The potential to target CCL5/CCR5 in breast cancer. Expert Opin. Ther. Targets 2014, 18, 1265–1275. [Google Scholar] [CrossRef]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.A.; Bae, S. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1–2. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.; Jiao, X.; De La Fuente, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef]
- Ignatov, A.; Robert, J.; Gregory-Evans, C.; Schaller, H.C. RANTES stimulates Ca 2+ mobilization and inositol trisphosphate (IP 3) formation in cells transfected with G protein-coupled receptor 75. Br. J. Pharmacol. 2006, 149, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. 2019, 79, 4801–4807. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, H.; Ichikura, T.; Kinoshita, M.; Ono, S.; Majima, T.; Tsujimoto, H.; Chochi, K.; Hiroi, S.; Takayama, E.; Saitoh, D.; et al. Gastric cancer cells exploit CD4+ cell-derived CCL5 for their growth and prevention of CD8+ cell-involved tumor elimination. Int. J. Cancer 2008, 122, 2535–2541. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Cambien, B.; Richard-Fiardo, P.; Karimdjee, B.F.; Martini, V.; Ferrua, B.; Pitard, B.; Schmid-Antomarchi, H.; Schmid-Alliana, A. CCL5 Neutralization restricts cancer growth and potentiates the targeting of PDGFRβ in colorectal carcinoma. PLoS ONE 2011, 6, e28842. [Google Scholar] [CrossRef]
- Chen, L.; Liu, X.; Zhang, H.-Y.; Du, W.; Qin, Z.; Yao, Y.; Mao, Y.; Zhou, L. Upregulation of chemokine receptor CCR10 is essential for glioma proliferation, invasion and patient survival. Oncotarget 2014, 5, 6576–6583. [Google Scholar] [CrossRef][Green Version]
- Kouno, J.; Nagai, H.; Nagahata, T.; Onda, M.; Yamaguchi, H.; Adachi, K.; Takahashi, H.; Teramoto, A.; Emi, M. Up-regulation of CC chemokine, CCL3L1, and receptors, CCR3, CCR5 in human glioblastoma that promotes cell growth. J. Neuro Oncol. 2004, 70, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Xue, Y.; Lv, W.; Zhang, Y.; He, S. Critical roles of chemokine receptor CCR5 in regulating glioblastoma proliferation and invasion. Acta Biochim. Biophys. Sin. 2015, 47, 890–898. [Google Scholar] [CrossRef]
- Moogooei, M.; Shamaei, M.; Khorramdelazad, H.; Fattahpour, S.; Seyedmehdi, S.M.; Moogooei, M.; Hassanshahi, G.; Kalantari Khandani, B. The intricate expression of CC chemokines in glial tumors: Evidence for involvement of CCL2 and CCL5 but Not CCL11. Acta Med. Iran. 2015, 53, 770–777. [Google Scholar]
- Podergajs, N.; Motaln, H.; Rajčević, U.; Verbovšek, U.; Koršič, M.; Obad, N.; Espedal, H.; Vittori, M.; Herold-Mende, C.; Miletic, H.; et al. Transmembrane protein CD9 is glioblastoma biomarker, relevant for maintenance of glioblastoma stem cells. Oncotarget 2015, 7, 593–609. [Google Scholar] [CrossRef]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.G.; Joyce, C.; et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef]
- Appaix, F. Brain mesenchymal stem cells: The other stem cells of the brain? World J. Stem Cells 2014, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Motaln, H.; Koren, A.; Gruden, K.; Ramšak, Ž.; Schichor, C.; Lah, T.T. Heterogeneous glioblastoma cell cross-talk promotes phenotype alterations and enhanced drug resistance. Oncotarget 2015, 6, 40998–41017. [Google Scholar] [CrossRef] [PubMed]
- Motaln, H.; Turnsek, T.L. Cytokines play a key role in communication between mesenchymal stem cells and brain cancer cells. Protein Pept. Lett. 2015, 22, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.N.; Pillat, M.M.; Motaln, H.; Ulrich, H.; Lah, T.T. Kinin-B1 receptor stimulation promotes invasion and is involved in cell-cell interaction of co-cultured glioblastoma and mesenchymal stem cells. Sci. Rep. 2018, 8, 1299. [Google Scholar] [CrossRef] [PubMed]
- Breznik, B.; Motaln, H.; Vittori, M.; Rotter, A.; Turnšek, L.T. Mesenchymal stem cells differentially affect the invasion of distinct glioblastoma cell lines. Oncotarget 2017, 8, 25482–25499. [Google Scholar] [CrossRef]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/astrocytes-glioblastoma crosstalk: Crucial molecular mechanisms and microenvironmental factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef]
- Morantz, R.A.; Wood, G.W.; Foster, M.; Clark, M.; Gollahon, K. Macrophages in experimental and human brain tumors. Part 2: Studies of the macrophage content of human brain tumors. J. Neurosurg. 1979, 50, 305–311. [Google Scholar] [CrossRef]
- Laudati, E.; Currò, D.; Navarra, P.; Lisi, L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem. Int. 2017, 108, 100–108. [Google Scholar] [CrossRef]
- Baebler, Š.; Svalina, M.; Petek, M.; Stare, K.; Rotter, A.; Pompe-Novak, M.; Gruden, K. quantGenius: Implementation of a decision support system for qPCR-based gene quantification. BMC Bioinform. 2017, 18, 276. [Google Scholar] [CrossRef]
- Behnan, J.; Stangeland, B.; Hosainey, S.A.M.; Joel, M.; Olsen, T.K.; Micci, F.; Glover, J.C.; Isakson, P.; Brinchmann, J.E. Differential propagation of stroma and cancer stem cells dictates tumorigenesis and multipotency. Oncogene 2017, 36, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Velasco-Velazquez, M.A.; Wang, M.; Li, Z.; Rui, H.; Peck, A.R.; Korkola, J.E.; Chen, X.; Xu, S.; Du Hadaway, J.B.; et al. CCR5 governs DNA damage repair and breast cancer stem cell expansion. Cancer Res. 2018, 78, 1657–1671. [Google Scholar] [CrossRef] [PubMed]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 4, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Smithson, L.J.; Ma, Y.; Hambardzumyan, D.; Gutmann, D.H. Ccl5 establishes an autocrine high-grade glioma growth regulatory circuit critical for mesenchymal glioblastoma survival. Oncotarget 2017, 8, 32977–32989. [Google Scholar] [CrossRef] [PubMed]
- Pham, K.; Luo, D.; Liu, C.; Harrison, J.K. CCL5, CCR1 and CCR5 in murine glioblastoma: Immune cell infiltration and survival rates are not dependent on individual expression of either CCR1 or CCR5. J. Neuroimmunol. 2012, 246, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Watabe, K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci. 2017, 22, 1805–1829. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Roscic-Mrkic, B.; Fischer, M.; Leemann, C.; Manrique, A.; Gordon, C.J.; Moore, J.P.; Proudfoot, A.E.I.; Trkola, A. RANTES (CCL5) uses the proteoglycan CD44 as an auxiliary receptor to mediate cellular activation signals and HIV-1 enhancement. Blood 2003, 102, 1169–1177. [Google Scholar] [CrossRef]
- Mentlein, R.; Hattermann, K.; Held-Feindt, J. Lost in disruption: Role of proteases in glioma invasion and progression. Biochim. Biophys. Acta 2012, 1825, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Fong, Y.C.; Lee, C.Y.; Chen, M.Y.; Tsai, H.C.; Hsu, H.C.; Tang, C.H. CCL5 increases lung cancer migration via PI3K, Akt and NF-κB pathways. Biochem. Pharmacol. 2009, 77, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.-J.; Chen, C.-H.; Lin, C.-Y.; Feng, L.-Y.; Lin, Y.-C.; Wei, K.-C.; Huang, C.-Y.; Fang, J.-Y.; Chen, P.-Y. CCL5 of glioma-associated microglia/macrophages regulates glioma migration and invasion via calcium-dependent matrix metalloproteinase-2. Neuro Oncol. 2019, 1–14. [Google Scholar]
- Winer, A.; Adams, S.; Mignatti, P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Sicoli, D.; Jiao, X.; Ju, X.; Velasco-Velazquez, M.; Ertel, A.; Addya, S.; Li, Z.; Andò, S.; Fatatis, A.; Paudyal, B.; et al. CCR5 receptor antagonists block metastasis to bone of v-Src oncogene-transformed metastatic prostate cancer cell lines. Cancer Res. 2014, 74, 7103–7114. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Ardebili, S.; Zajc, I.; Gole, B.; Campos, B.; Herold-Mende, C.; Drmota, S.; Lah, T. CD133/prominin1 is prognostic for GBM patient’s survival, but inversely correlated with cysteine cathepsins’ expression in glioblastoma derived spheroids. Radiol. Oncol. 2011, 45, 1–13. [Google Scholar] [CrossRef]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef]
- Hira, V.V.V.; Verbovšek, U.; Breznik, B.; Srdič, M.; Novinec, M.; Kakar, H.; Wormer, J.; der Swaan, B.V.; Lenarčič, B.; Juliano, L.; et al. Cathepsin K cleavage of SDF-1α inhibits its chemotactic activity towards glioblastoma stem-like cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 594–603. [Google Scholar] [CrossRef]
- Kast, R.E. Glioblastoma: Synergy of growth promotion between CCL5 and NK-1R can be thwarted by blocking CCL5 with miraviroc, an FDA approved anti-HIV drug and blocking NK-1R with aprepitant, an FDA approved anti-nausea drug. J. Clin. Pharm. Ther. 2010, 35, 657–663. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Mazzucco, M.; Mellai, M. The microenvironment in gliomas: Phenotypic expressions. Cancers 2015, 7, 2352–2359. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L.; Wolf, M.J.; Roblek, M.; Lorentzen, A.; Heikenwalder, M. Inflammatory chemokines and metastasis—Tracing the accessory. Oncogene 2014, 33, 3217–3224. [Google Scholar] [CrossRef] [PubMed]
- Kološa, K.; Motaln, H.; Herold-Mende, C.; Koršič, M.; Lah, T.T. Paracrine effects of mesenchymal stem cells induce senescence and differentiation of glioblastoma stem-like cells. Cell Transplant. 2015, 24, 631–644. [Google Scholar] [CrossRef]
- Motaln, H.; Gruden, K.; Hren, M.; Schichor, C.; Primon, M.; Rotter, A.; Lah, T.T. Human mesenchymal stem cells exploit the immune response mediating chemokines to impact the phenotype of glioblastoma. Cell Transplant. 2012, 21, 1529–1545. [Google Scholar] [CrossRef]
- Choi, S.A.; Lee, J.Y.; Kwon, S.E.; Wang, K.C.; Phi, J.H.; Choi, J.W.; Jin, X.; Lim, J.Y.; Kim, H.; Kim, S.K. Human adipose tissue-derived mesenchymal stem cells target brain tumor-initiating cells. PLoS ONE 2015, 10, 1–15. [Google Scholar]
- Ramšak, Ž.; Coll, A.; Stare, T.; Tzfadia, O.; Baebler, Š.; Van de Peer, Y.; Gruden, K. Network modeling unravels mechanisms of crosstalk between ethylene and salicylate signaling in potato. Plant Physiol. 2018, 178, 488–499. [Google Scholar] [CrossRef] [PubMed]
- FPC: Flexible Procedures for Clustering. Rpackage Version 2.2-3. Available online: https://CRAN.R-project.org/package=fpc. (accessed on November 2019).
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster: Cluster Analysis Basics and Extensions. R Package Version 2.1.0.; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Dvinge, H.; Bertone, P. HTqPCR: High-throughput analysis and visualization of quantitative real-time PCR data in R. Bioinformatics 2009, 25, 3325–3326. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCR5 | CCL5 | |||||
|---|---|---|---|---|---|---|
| Tumor | Intensity | % Positive Cells | Localization | Intensity | % Positive Cells | Localization |
| Patient 2 | + | 1 | c | − | 0 | / |
| Patient 5 | − | 0 | / | + | 1 | c, e |
| Patient 8 | + | 1 | c | ++ | 3 | c |
| Patient 9 | − | 0 | / | − | 0 | / |
| NB 1 | − | 0 | / | ++ | 1 | c |
| NB 2 | + | 2 | c | ++ | 2 | c |
| Patients | Age (Years) | Gender | Survival * (Months) | Diagnosis | Necrosis ** | Angiogenesis ** | Karnofsky Score *** | Subtype (MES, PN, CL, MIX) **** | IDH ***** |
|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | 51 | F | 3 | GB | yes | yes | 90 | ND | wt |
| Patient 2 | 54 | M | 10 | GB | yes | yes | 80 | MIX | wt |
| Patient 3 | 61 | M | 8 | GB | yes | yes | 70 | PN | wt |
| Patient 4 | 76 | F | 16 | GB | no | yes | 50 | ND | n.a. |
| Patient 5 | 61 | M | 8 | GB | no | yes | 80 | MIX | wt |
| Patient 8 | 51 | M | 6 | GB | no | yes | 100 | CL | wt |
| Patient 9 | 67 | M | 9 | GB | no | no | 80 | ND | wt |
| Patient 12 | 63 | F | 9 | GB | no | no | 90 | CL | wt |
| Cell Sample Name | CCR5 * | CCL5 * |
|---|---|---|
| Patient 1 | − | + |
| Patient 2 | + | + |
| Patient 3 | − | − |
| Patient 5 | + | ++ |
| Patient 8 | ++ | +++ |
| U373 | − | − |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novak, M.; Koprivnikar Krajnc, M.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porčnik, A.; Mlakar, J.; Stare, K.; et al. CCR5-Mediated Signaling is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. Int. J. Mol. Sci. 2020, 21, 4199. https://doi.org/10.3390/ijms21124199
Novak M, Koprivnikar Krajnc M, Hrastar B, Breznik B, Majc B, Mlinar M, Rotter A, Porčnik A, Mlakar J, Stare K, et al. CCR5-Mediated Signaling is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. International Journal of Molecular Sciences. 2020; 21(12):4199. https://doi.org/10.3390/ijms21124199
Chicago/Turabian StyleNovak, Metka, Miha Koprivnikar Krajnc, Barbara Hrastar, Barbara Breznik, Bernarda Majc, Mateja Mlinar, Ana Rotter, Andrej Porčnik, Jernej Mlakar, Katja Stare, and et al. 2020. "CCR5-Mediated Signaling is Involved in Invasion of Glioblastoma Cells in Its Microenvironment" International Journal of Molecular Sciences 21, no. 12: 4199. https://doi.org/10.3390/ijms21124199
APA StyleNovak, M., Koprivnikar Krajnc, M., Hrastar, B., Breznik, B., Majc, B., Mlinar, M., Rotter, A., Porčnik, A., Mlakar, J., Stare, K., Pestell, R. G., & Lah Turnšek, T. (2020). CCR5-Mediated Signaling is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. International Journal of Molecular Sciences, 21(12), 4199. https://doi.org/10.3390/ijms21124199