Abstract
Adipose tissue is an active endocrine and immune organ that controls systemic immunometabolism via multiple pathways. Diverse immune cell populations reside in adipose tissue, and their composition and immune responses vary with nutritional and environmental conditions. Adipose tissue dysfunction, characterized by sterile low-grade chronic inflammation and excessive immune cell infiltration, is a hallmark of obesity, as well as an important link to cardiometabolic diseases. Amongst the pro-inflammatory factors secreted by the dysfunctional adipose tissue, interleukin (IL)-1β, induced by the NLR family pyrin domain-containing 3 (NLRP3) inflammasome, not only impairs peripheral insulin sensitivity, but it also interferes with the endocrine and immune functions of adipose tissue in a paracrine manner. Human studies indicated that NLRP3 activity in adipose tissues positively correlates with obesity and its metabolic complications, and treatment with the IL-1β antibody improves glycaemia control in type 2 diabetic patients. In mouse models, genetic or pharmacological inhibition of NLRP3 activation pathways or IL-1β prevents adipose tissue dysfunction, including inflammation, fibrosis, defective lipid handling and adipogenesis, which in turn alleviates obesity and its related metabolic disorders. In this review, we summarize both the negative and positive regulators of NLRP3 inflammasome activation, and its pathophysiological consequences on immunometabolism. We also discuss the potential therapeutic approaches to targeting adipose tissue inflammasome for the treatment of obesity and its related metabolic disorders.
1. Introduction
Adipose tissue is an active endocrine organ, secreting a variety of hormones, peptides and metabolites (collectively called adipokines) that regulate systemic metabolism via cross-talk with multiple peripheral tissues and the central nervous system [1,2]. It is comprised of a heterogeneous collection of adipocytes, pre-adipocytes, endothelial cells, fibroblasts and other adipose tissue resident immune cells, such as adipose tissue macrophages (ATMs) and lymphocytes, which exert differential effects on metabolism [3,4]. Generally, adipose tissue can trifurcate into white (WAT), brown (BAT) and brown-like (beige) adipose tissues. WAT, dispersed throughout the body, primarily stores excess energy in the form of triglycerides, while BAT specializes in energy dissipation in the form of heat, via the uncoupling protein-1 (UCP-1) in the mitochondria [4,5]. WAT can be further divided into several regional depots, notably visceral WAT (vWAT), around internal organs, and subcutaneous WAT (sWAT) beneath the skin, which exhibit differences in their capacity for adipogenesis and lipid handling, insulin sensitivity, rate of lipolysis, cellular composition, adipokine profile, browning potential, etc. [6]. In general, subcutaneous fat is believed to protect against obesity and its cardiometabolic complications, whereas visceral fat exerts opposing metabolic effects.
The cellular heterogeneity and adipokine secretome of WAT are greatly altered in obesity and aging, given its indispensable roles in energy homeostasis and metabolic regulation, resulting in a systemic inflammation and cardiometabolic diseases. Responding to excessive calorie intake, WAT undergoes expansion via two pathways that are hyperplasia (increase in adipocyte number) and hypertrophy (increase in adipocytes size), as well as remodeling concurrent with hypoxia, endoplasmic reticulum stress, metabolic endotoxemia and adipocyte death [7]. These changes trigger the recruitment and activation of immune cells in a chronic and low-grade manner, unlike classical inflammation induced by infection [7]. Remarkably, ATMs, comprising around 10% of stromal vascular fraction (SVF), drastically increase in number and switch toward a pro-inflammatory M1 phenotype, secreting various pro-inflammatory cytokines, such as adipocyte fatty acid binding protein (FABP4), tumor necrosis factor-alpha (TNF-α), monocyte chemoattractant protein-1 (MCP-1), interleukin (IL)-1β and IL-6, which instigate insulin resistance, non-alcoholic fatty liver diseases and atherosclerosis [7,8,9,10,11,12,13]. Aside from energy overload, aging induces lipid redistribution from sWAT to vWAT, and the acquisition of the senescence-associated secretory phenotype (SASP) in senescent cells, which have also been linked to chronic low-grade inflammation, known as inflammaging, and it also shares similarities with obesity-induced inflammation, which provokes several lipid and glucose metabolic disorders [14,15,16].
IL-1 family cytokines, including IL-1β and IL-18, mediate both obesity- and aging-induced metabolic complications [17,18]. The production of these pro-inflammatory cytokines is conveyed by NLRP3 inflammasome. Among all inflammasome proteins, NLRP3 is most intensively studied in metabolic research, because of its crucial role in immune responses, glucose homeostasis, lipid metabolism and adipocyte functions. Multiple human and animal studies have indicated that NLRP3 is activated in adipose tissues with aging and obesity, and its inactivation significantly alleviates metabolic disorders [19,20]. Within adipose tissues, multiple cell types exhibit NLRP3 inflammasome activation induced by diverse stimuli, which in turn leads to the deterioration of metabolic control. In this review, we will discuss (1) the major factors that negatively and positively regulate the activation of NLRP3 inflammasome in adipose tissue; (2) the deleterious consequences of NLRP3 inflammasome activation in adipose tissues, in paracrine and endocrine aspects; and (3) the future prospect of targeting adipose tissue inflammasome for the treatment of metabolic diseases.
2. NLRP3 Inflammasome Activation in Adipose Tissue
2.1. General Overview of NLRP3 Inflammasome Activation
NLRP3 inflammasome is a group of intracellular multi-protein complexes consisting of a pattern recognition receptor (PRR), the apoptosis-associated speck-like protein containing a CARD (ASC/PYCARD), and caspase-1 (Figure 1) [21]. Upon activation by pathogen-(PAMPs) or danger (DAMPs)-associated molecular patterns, inflammasome initiates the proteolytic cleavage of dormant pro-caspase-1 into active caspase-1, which participates in gasdermin D (GSDMD)-dependent pyroptosis, and the processing of pro-IL-1β and pro-IL-18 into their biologically active forms [21,22,23]. A two-step activation model, in which priming and activation signals cooperatively activate inflammasome, has been well established over the past decade [24]. The first step is provided by microbial components [such as lipopolysaccharide (LPS)] or pro-inflammatory cytokines, to promote the expression of NLRP3 and pro-IL-1β at a transcriptional level, although post-translational regulation has also been shown [25,26,27]. The second step is initiated by a plethora of PAMPs and DAMPs which leads to inflammasome assembly, followed by caspase-1-driven IL-1β and IL-18 maturation [26,28,29]. Multiple intracellular signaling events, including ion fluxes, mitochondrial reactive oxygen species (ROS) production and DNA release, and lysosomal destabilization, have been implicated in relaying specific stimuli to NLRP3 sensor [26,28,29]. The NLRP3 inflammasome components are expressed in most of the WAT-resident cell types, including white adipocytes, ATMs, adipocyte progenitor cells, dendritic cells, B cells and T cells, and its expression is dynamically changed with adiposity, age, insulin sensitivity and other metabolic insults [30,31,32,33,34], highlighting its critical function in adipose tissues.
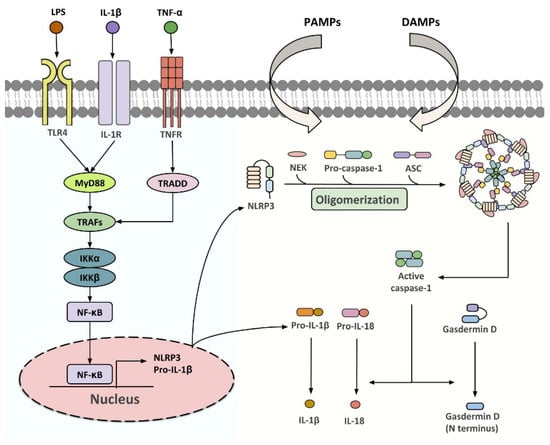
Figure 1.
Classical pathways for NLRP3 inflammasome activation. Upon stimulation of TLR4, IL-1R or TNFR, TNF receptor-associated factor 2 (TRAF2) and TNF receptor-associated factor 6 (TRAF6) recruit the inhibitor of nuclear factor-κB kinase α/β (IKKα/β) that drives the translocation of NF-κB subunits to the nucleus. This upregulates the transcription of NLRP3 and pro-IL-1β, which enables the following assembly of NLFPR3 inflammasome initiated by various PAMPs and DAMPs. Once activated, the dormant procaspase-1 is cleaved into active caspase-1, which initiates the processing of gasdermin D, pro-IL-1β and pro-IL-18 to their biologically active forms.
2.2. Association of NLRP3 Inflammasome Activation and Metabolic Disorders in Human
A recent systematic review revealed that increased expression of NLRP3 and IL-1β in the subcutaneous and visceral fat depots of obese individuals has been found in most of the previous studies [35]. For example, increased gene expressions of NLRP3, and its subsequent products IL-1β and IL-18, were observed in the visceral fat of metabolically unhealthy obese individuals, when compared to those isolated from lean healthy control or metabolically healthy obese individuals [36]. In addition, gene expressions of IL-1β, caspase-1 and NLRP3 are increased in obese individuals with a higher ratio of visceral fat over visceral fat plus subcutaneous fat [37]. In subcutaneous fat, expression of the inflammasome molecules is positively associated with ceramide levels. Increased expressions of NLRP3 and IL-1β were also observed in the adipocytes, but not the SVF, of subcutaneous fat isolated from obese females. A positive correlation between inflammasome expression and adiposity was also seen in the same cohort of subjects. In response to calorie restriction and exercise, gene expressions of IL-1β and NLRP3 are reduced in the subcutaneous fat of patients with obesity and type 2 diabetes, accompanied with improvement in insulin sensitivity [19]. Likewise, weight loss induced by bariatric surgery diminished IL-1β gene and IL-1β secretion in the adipose tissue of human and animal models [19,38,39,40]. Noticeably, inflammasome inducers (such as LPS) and inhibitors (such as adiponectin) are reduced and increased, respectively, after bariatric surgery, yet whether these changes directly contribute to the reduction of adipose tissue’s inflammasome activity remain elusive [41,42,43]. The expression of NLRP3 in sWAT is an independent predictor for atherosclerosis, and is positively associated with its severity [44]. Monocyte-derived macrophages from type 2 diabetic patients are more sensitive to inflammasome activation upon LPS stimulation, when compared to those isolated from healthy controls [45]. NLRP3 rs10754558 polymorphism was reported as associated with type 2 diabetes in the Chinese population [46]. Together, these findings indicate that inflammasome activity in adipose tissue and the circulating level of IL-1β are closely associated with metabolic functions in humans.
2.3. Key Regulators of NLRP3 Inflammasome in Adipose Tissues
With concerted efforts in deciphering inflammasome activation pathways, the cell types within obese or aged WAT that are responsible for inflammasome-mediated chronic inflammation and insulin resistance become apparent, each with distinct priming and activating stimuli, such as gut-derived endotoxin, adipocytokines and lipid metabolites, and mitochondrial dysfunction (Figure 2) [47,48,49,50,51,52].
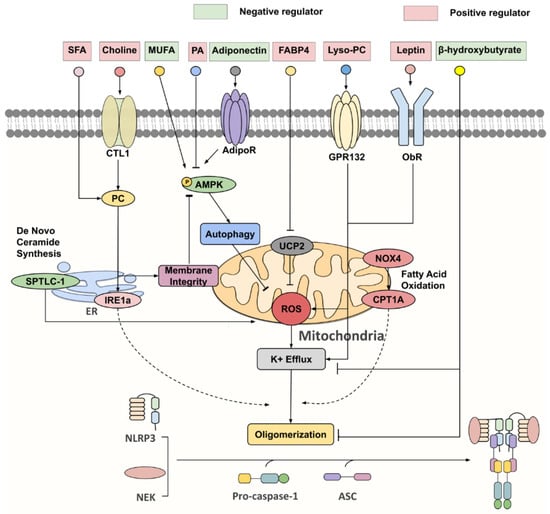
Figure 2.
Key negative and positive regulators for NLRP3 inflammasome. Under nutrient overload, SFAs [such as palmitic acid (PA)] and choline are extensively incorporated into phosphatidylcholine (PC), which activates inositol-requiring enzyme 1α (IRE1α), whose endonuclease activity promotes NLPR3 inflammasome activation via an undefined mechanism. Furthermore, PC synthesis through the choline pathway reciprocally regulates the AMP-activated protein kinase (AMPK)–autophagy–ROS signaling axis by maintaining mitochondrial membrane integrity. On the other hand, monounsaturated fatty acids (MUFA) and adiponectin were identified as initiators of AMPK-dependent autophagy, that attenuate ROS production and K+ efflux, thereby suppressing NLRP3 activation. FABP4, lyso-PC, leptin and serine palmitoyltransferase long chain base subunit 1 (SPTLC-1), a key enzyme involved in de novo ceramide synthesis, all partake in NLRP3 inflammasome activation via increasing ROS production. NADPH oxidase 4 (NOX4) enhances the protein expression of carnitine palmitoyl-transferase 1A (CPT1A), a rate-limiting fatty acid oxidation-related enzyme, which is responsible for heightening NLRP3 inflammasome response through a largely unknown pathway. β-hydroxybutyrate (BHB) was unveiled as a potent NLRP3 inflammasome inhibitor, targeting both K+ efflux and ASC oligomerization.
2.3.1. Lipopolysaccharide (LPS)
LPS, the endotoxin located on the outer membrane of Gram-negative bacteria, is one of the most potent PAMPs for the priming of the NLRP3 inflammasome. The circulating level of LPS is elevated in obese and diabetic states, due to increased gut permeability and/or a change in gut microbiota composition [53,54]. LPS-mediated chronic metabolic endotoxemia increase body weight and adiposity, adipose tissue inflammation, and systemic insulin resistance in rodent models [54,55]. A recent study found a higher number of Gram-negative Enterobacteriaceae in the mesenteric adipose tissue of individuals with type 2 diabetes than in individuals without diabetes [54,56]. LPS primes NLRP3 inflammasome activation via multiple pathways. At the transcriptional level, LPS induces mRNA expression of pro-IL-1β and NLRP3, via the toll-like receptor 4 (TLR4) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) dependent pathway [25]. At the post-translational level, LPS stimulates NLRP3 deubiquitination, which is required for priming and activation of NLRP3 [57]. Pharmacological inhibition of ROS abrogates the deubiquitinating effect of LPS. A subsequent study identified that the deubiquitinating enzyme, BRCC3, mediates the deubiquitination of NLRP3 [27]. Macrophages and adipocytes are able to absorb circulating LPS, that binds with the lipoproteins [58]. Apart from the canonical TLR4 pathway, the hexa-acyl lipid A component of LPS has also been shown to trigger IL-1β production via caspase 11 [59].
2.3.2. Lipids
Dysregulated fatty acid metabolism in the context of overnutrition modulates both NLRP3 inflammasome priming and activation steps. Dietary saturated fatty acids (SFAs), whose intake is strongly associated with an increased risk of obesity, have been identified as potent priming agents of the NLRP3 inflammasome via TLR4 in DCs, resulting in elevated expression of pro-IL-1β, caspase-1, TLR4 and NLRP3 [60]. The pathways that mediate the promoting effect of SFAs and LPS on NLRP3 can be distinct [61]. Palmitic acid, an abundant SFA usually elevated in obesity and diabetes, induces IL-1β production in macrophages and dendritic cells via multiple pathways [61,62]. The higher consumption of SFA is positively associated with insulin resistance and inflammatory status in humans [63]. Mice fed with a high fat diet (HFD) enriched with palmitic acid display increased mRNA levels of caspase-1, NLRP3 and IL-1β in their SVF of adipose tissues, accompanied with insulin resistance and glucose intolerance [63]. Mechanistically, palmitic acid attenuates AMPK activation, which diminishes autophagy and induces mitochondrial ROS accumulation, leading to inflammasome activation as well as IL-1β-mediated insulin resistance [62]. Secondly, excessive amounts of palmitic acid lead to the synthesis of ceramides, which enhances ROS generation and activates the NLRP3 inflammasome via upregulating serine palmitoyltransferase long chain (Sptlc)-2 [64]. Surprisingly, the myeloid cell-specific deletion of Sptlc-2 did not prevent HFD-induced adipose tissue inflammation and insulin resistance, suggesting its dispensable role in NLRP3 inflammasome activation in obesity [64]. Third, palmitic acid also elicits endoplasmic reticulum (ER) stress, and activates inositol-requiring enzyme 1α (IRE1α) via the flux to phosphatidylcholine, which in turn increases IL-1β production in macrophages [61].
Apart from palmitic acid, phosphatidylcholine derived from choline is also associated with the inflammasome-mediated IL-1β and IL-18 production in macrophages [65]. Impaired choline uptake, or incorporation into phosphatidylcholine, interferes with the NLRP3 inflammasome activity, accompanied by upregulated AMPK-mediated mitophagy (a specific form of autophagy targeting damaged mitochondria) and the reduced cytosolic release of mitochondrial ROS and oxidized mitochondrial DNA [65]. It is likely that phosphatidylcholine synthesis through the choline pathway is responsible for maintaining mitochondrial membrane integrity, which prevents excessive damage that leads to defective ATP synthase activities and the activation of AMPK-dependent mitochondrial clearance [65]. Aside from relaying metabolic signals to the inflammasome, several naturally-occurring phospholipids also act as the initiators of NLRP3 inflammasome-dependent IL-1β secretion, in which oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) and the platelet-activating factor (PAF) have recently come to light [66,67,68]. Of note, oxPAPC exerts differential effects on NLRP3 inflammasome activation, depending on adipose cell types. It interacts with caspase-11 and elicits IL-1β secretion, but not pyroptosis, in DCs, via an unknown mechanism, with no involvement of the K+ efflux that is required in the non-canonical inflammasome signaling pathway [66]; another study demonstrated that oxPAPC competes with LPS binding for caspase-11 and suppresses the downstream non-canonical NLRP3 inflammasome pathways in macrophages [68].
Lysophosphatidylcholine (lyso-PC; a pro-inflammatory lipid), released by adipocytes upon stimulation of homocysteine, serves as both the first and second signal activator of the NLRP3 inflammasome in adipocytes and ATMs [69]. It was speculated that lyso-PC promotes the full activation of the NLRP3 inflammasome via the interaction with G-protein coupled receptors 132 (GPR132), which triggers diverse intracellular signaling events, including Ca2+ signaling, K+ efflux, ROS generation and lysosomes damages [69,70,71,72].
Oleic acid, an unsaturated fatty acid known for its protective effects on coronary heart disease, favours neither priming nor activating steps, but promotes AMPK activation that negatively regulates the NLRP3 inflammasome [63]. Consistent with in vitro findings, mice fed with an oleic acid-enriched HFD exhibit lower adipose IL-1β levels and improved acute insulin responses [63]. In addition, omega-3 polyunsaturated fatty acids (such as docosahexaenoic acid) interfere with the NLRP3 inflammasome in the process, by reducing the cytosolic pool of NLRP3 [73,74].
2.3.3. Adipokines
Adiponectin is a fat-derived hormone with anti-inflammatory and insulin sensitizing effects [75,76]. The circulating level of adiponectin inversely correlates with IL-18 in human with type 2 diabetes [77]. The adiponectin or adiponectin receptor agonist AdipoRon inhibits inflammasome activation in diverse types of cells, including macrophages, endothelial cells, cardiomyocytes and hepatocytes [77,78,79,80]. The inhibitory effect of adiponectin on NLRP3 inflammasome activation is believed to be mediated by the AMPK, autophagy FoxO4 and/or NF-kB pathways. On the other hand, IL-1β was reported to reduce adiponectin expression and secretion in human and mouse mature adipocytes, forming a feedback loop [81]. These studies indicate that the reciprocal regulation of adiponectin and the NLRP3 inflammasome might be important for maintenance of metabolic health.
TNF-α was identified as a potent endogenous priming signal in the NLRP3 inflammasome, driving age-related inflammation [49]. Macrophage-intrinsic NLRP3 mRNA expression within adipose tissue and liver increases in response to a spontaneously elevated TNF-α level in aged mice [49]. Consistent with the fact that chronic inflammation in aging is independent of IL-1β secretion, the NLRP3 inflammasome in TNF-α-primed macrophages contributes to the maturation of caspase-1, without affecting IL-1β level [20,49].
Leptin, highly expressed in white adipocytes and mainly secreted by WAT serving as an activator of the hypothalamic anorexigenic pathway, has also been implicated in NLRP3 inflammasome activation [82,83,84]. Leptin level correlates with total fat mass, but different fat depots display distinct abilities in leptin secretion capacity. In humans, LEPTIN gene expression was significantly lower in the omental depot than the subcutaneous and mesenterial sites, while on the other hand, leptin is mainly expressed in the gonadal WAT of mice. [85,86,87]. Despite being often associated with obesity, aging confers independent effects on leptin in metabolically healthy aged rats that had received calorie restriction [88,89]. It was found to promote NLRP3 inflammasome-mediated IL-18 secretion in RAW264.7 cells, at least in part by augmenting ROS production and K+ efflux [84].
FABP4, an adipokine that is positively associated with obesity and metabolic syndrome, was demonstrated to positively control the NLRP3 inflammasome through downregulating UCP2 expression in a paracrine manner [90,91]. Given the ability of UCP2 to facilitate the re-entry of protons into the mitochondrial matrix, and to attenuate superoxide production, the activated FABP4–UCP2 signaling axis is concomitant with enhanced ROS production and mitochondrial UPR, targeting both priming and activating steps of the NLRP3 inflammasome [91,92].
2.3.4. Defective Autophagy and Mitochondrial Dysfunction
Mitochondria plays a central role in the regulation of energy metabolism, but its function deteriorates in obesity and aging. Mitochondrial dysfunction, characterized by an accumulation of damaged mitochondria, an increased production of ROS and mitochondrial DNA release to cytosol, has been recently linked to NLRP3 activation in macrophages [93,94,95,96]. Autophagy, a highly conserved cellular process that delivers dysfunctional components to lysosome for degradation, was also demonstrated to mitigate NLRP3 inflammasome activity in two ways; either through the destruction of ubiquitinated inflammasomes, or the removal of multiple mitochondrial-derived DAMPs, such as mitochondrial ROS [94,97]. As mentioned above, palmitic acid activates the NLRP3 inflammasome via inhibition of autophagy [62]. Autophagy also suppresses IL-1β secretion through the degradation of pro-IL-1β, which limits the availability of substrate for IL-1β maturation by inflammasome [98]. Myeloid cell-specific deletion of Atg7, a key gene involve in autophagy, exacerbates obesity-induced glucose intolerance, accompanied with adipose inflammasome activation [99]. Likewise, defective mitophagy (mitochondrial autophagy), induced by deletion of the mitophagy receptor FUNDC1, also accelerates adipose tissue inflammation and systemic insulin resistance under the obese condition [100]. On the other hand, inflammasome activation has been shown to inhibit mitophagy and exacerbate mitochondrial damage [101]. Therefore, inflammasome activation and defective mitophagy may form a vicious cycle that worsens adipose tissue function in obesity.
2.3.5. Glucose Metabolism
Hyperglycaemia can stimulate NLRP3 inflammasome activation in multiple cell types, including THP-1-derived macrophages, 3T3-L1 mature adipocytes and human adipose tissue [102,103,104,105,106,107]. Both caspase-1 and thioredoxin-interacting protein (TXNIP) expression are increased in ob/ob mice. High glucose upregulates the expression of TXNIP, accompanied by an increased caspase-1 level in human adipose tissue. The siRNA-mediated knockdown expression of TXNIP abolishes the high-glucose-induced activation of capase-1 in human primary adipocytes [107]. In addition, inhibition of glycolysis by inactivating either mTORC1 or hexkinase-1 attenuates the maturation of IL-1β in macrophages treated with LPS and ATP [108].
3. Detrimental Consequences of Adipose NLRP3 Inflammasome Activation
The activation of NLRP3 exerts diverse detrimental effects on multiple tissues, in both paracrine and endocrine manners (Figure 3). In this section, we discuss the major consequences of inflammasome activation on metabolism and immune responses.
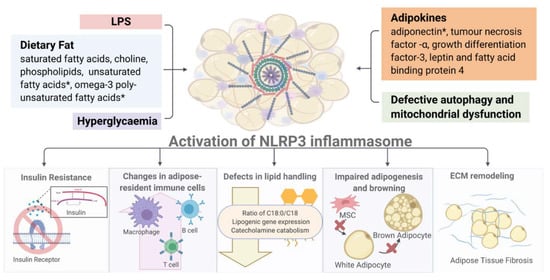
Figure 3.
Overview of NLRP3 inflammasome-associated metabolic consequences. (Image created with BioRender.com). * Activation of NLRP3 inflammasome by diverse metabolic stimuli (such as LPS, adipokines, hyperglycemia and mitochondrial dysfunction) leads to multiple metabolic and immune dysregulations including insulin resistance, altered immune cell composition, defective lipid handing and adipogenesis and increased fibrosis in white and brown fat depots. Detailed description and explanation for each consequence can be found in Section 3.
3.1. Insulin Resistance in Type 2 Diabetes
The NLRP3 inflammasome impairs insulin sensitivity in dietary-induced obesity via the disruption of phosphatidylinositol 3-kinase-protein kinase B (PI3K-Akt) signaling, a major pathway orchestrating the metabolic effects of insulin in peripheral tissue [19,32,109]. In addition, IL-1β was found to alter the protein abundance and phosphorylation of the insulin receptor substrate (IRS)-1, the p85α regulatory subunit of PI3K, and Akt in adipocytes [110]. Genetic ablation of NLRP3 activates Akt, paralleled by reduced phosphorylation of IRS-1 within liver and adipose tissue, resulting in improved insulin signaling and a lower fasting glucose level [19]. Improvement in insulin signaling was also evident in the skeletal muscle of NLRP3-deficient mice, while the knockdown of NLRP3 expression partially reversed perilipin 2-induced insulin resistance in C2C12 myoblast via upregulation of IRS-1 mRNA [19,62,111]. Additionally, the plasma levels of leptin and resistin, known to impair glucose tolerance, were shown to be positively associated with the NLRP3 inflammasome [107]. However, it is worth noting that IL-18 was found to promote insulin sensitivity in skeletal muscle by activating AMPK [112].
3.2. Changes in Adipose-Resident Immune Cells
It is widely accepted that a preponderance of CD11c+ macrophages in obese vWAT originates from newly recruited CCR2+ monocytes [113,114,115], though recent findings also highlight the importance of local proliferation in sustaining ATMs’ accumulation in vWAT [115,116,117]. The mechanism of how the NLRP3 inflammasome promotes macrophage recruitment becomes apparent. Upon activation of TLR4-MyD88 signalling, ATMs promote myelopoiesis in bone marrow in an NLRP3 inflammasome/IL-1β-dependent fashion, which increases the circulating level of monocytes that subsequently infiltrate vWAT and aggravate chronic local inflammation [118]. More evidence has arisen from the finding that HFD-fed mice with caspase-1 deletion displayed a reduction in the number of ATMs as well as in monocyte chemoattractant protein-1 (MCP-1) expression, a key chemokine that facilitates the macrophage influx to WAT [107]. Conversely, the inflammasome-independent protective effect of caspase-1 on adipose tissue macrophage recruitment was also reported [119]. IL-1β is also positively associated with the gene expression of MCP-1 and chemokine (C-C motif) ligand 5 (CCL5) [33,107]. Surprisingly, the absence of ASC does not attenuate HFD-induced macrophage localization, which can be attributed to the direct transcriptional regulation of cytokine genes by ASC [107]. In addition, the NLRP3 inflammasome amplifies M1-like macrophage polarization within obese vWAT [19]. Genetic ablation of NLRP3 is associated with downregulation of the M1 macrophage-specific genes TNF-α and chemokine (C-C motif) ligand 20 (CXC20) in vWAT-derived ATMs, accompanied by increased numbers of M2 macrophages (F4/80+CD11c−CD206+) in sWAT without changing the M1 population [19].
Decreases in T regulatory (Treg) cells are accompanied by increased NLRP3 inflammasome activity in the vWAT of metabolically unhealthy subjects [36]. Absence of NLRP3 was shown to specifically reduce both CD4+ and CD8+ effector/memory T cell subsets in the vWAT of diet-induced obese mice, without affecting these cell populations in sWAT, while CD4+/CD8+ naïve T cells and Treg cells remained unchanged in vWAT [19]. This may be accomplished by the reduced ATMs expression of chemokines, which promotes T cell infiltration and IL-1β/IL-18-induced naïve T cell differentiation [120,121]. IL-18 secretion, driven by the NLRP3 inflammasome, also activates T helper 1 (Th1) response, which leads to increased IFN-γ expression in both vWAT and sWAT, whereas NLRP3 inflammasome-related gene expression is positively correlated with the expression of the markers of Th1 cells, Th17 cells, pan T cells and Treg cells [19,33]. Collectively, these suggest the inducing role of the NLRP3 inflammasome in the pro-inflammatory shift of adipose tissue-resident T cells, and the expansion of Treg cells is thought to be a negative feedback in response to the inflammation induced by Th1 cells and Th17 cells [33].
IL-1β from macrophages promotes IL-17 and IL-22 secretion from adipose tissue CD4+ T cells. Activation of the c-Jun pathway in adipose tissue macrophages, by IL-17 and IL-22, subsequently increases pro-IL-1β in a feed forward manner, to propagate inflammation [39]. The age-related expansion of adipose B cells in vWAT requires the NLRP3 inflammasome [34]. Multiple NLRP3-related ligands and receptors on macrophages appear to interact with adipose B cells, contributing to their expansion during aging. Of these, IL-1 signalling is crucial for the adipose B cell proliferative capability in aged vWAT, as a blockade of IL-1R reduced B cell expansion and restored lipolysis in aged vWAT [34].
In addition, SFA-primed DCs and DCs isolated from HFD-fed mice secreted elevated level of IL-1β. A co-culture of DCs derived from HFD-fed mice with adipocytes potentiated caspase-1 maturation and IL-1β secretion in adipocytes, resulting in impairment of insulin sensitivity. Therefore, DCs-derived IL-1β might mediate the development of insulin resistance in obese adipose tissue [60]. Adipose tissue CD11b+ DCs were found to express high IL-1β, which partake in Th17 maturation. While the CD103+ DCs subset express IL-18, which promotes Th1 polarization [122].
3.3. Defects in Lipid Handling
Prior work has shown the deleterious effects of NLRP3 inflammasome on lipid synthesis and utilization in mature adipocytes. Fat oxidation rate and mitochondrial energy dissipation markedly decrease upon NLRP3 activation, contributing to elevated diurnal caloric expenditure and adiposity [32]. The absence of capase-1 alters the fatty acid composition of WAT, with an increase in the amount of palmitic acid (C16:0) and stearic acid (C18:0), accompanied by decreased oleic acid (C18:1) [32]. The lower ratio of C18:0/C18:1 is likely to be accomplished by the attenuation of stearoyl-CoA desaturase-1 activity, a key lipogenic mediator, and this implies NLRP3 inflammasome positively regulates lipogenesis [32].
As discussed earlier, senescent ATMs with activated NLRP3 inflammasome diminish the lipolysis in adipocytes by downregulating the expression of genes implicated in catecholamine catabolism, such as the growth differentiation factor-3 (GDF3) and monoamine oxidase A (MAOA). This leads to the reduction of glycerol and free fatty acids (FFAs) released from vWAT in response to fasting in aging [34]. Deletion of either NLRP3 or GDF3 is sufficient to reverse age-related catecholamine degradation, and to restore the proper expression of two major lipolytic enzymes: hormone-sensitive lipase (HSL) and adipose triglyceride lipase (ATGL) [34]. During aging, the B cell population, including adipose B cells, considerably expands in vWAT in an NLRP3 inflammasome-dependent manner, which perturbs lipolytic signaling, whereas both B cell depletion and NLRP3 ablation restores lipolysis with normal levels of the lipases [34].
3.4. Adipose Tissue Remodelling
Adipocytes undergo regeneration and death in response to different nutritional statuses and environment factors. The differentiation of progenitor cells into mature adipocytes is known as adipogenesis. Expressions of caspase 1 and IL-1β dynamically change during adipocyte differentiation [32]. Inhibition of caspase-1 by Pralnacasan increases the expression of genes related to adipogenesis, which include adiponectin and PPARγ [32]. In vitro, treatment with IL-1β but not IL-18 inhibits adipocyte differentiation. Genetic abrogation of caspase 1 or NLRP3 promotes adipogenesis, thereby improving adipose tissue function and insulin sensitivity in animal models [32]. On the contrary, an in vitro study recently indicated that activation of NLRP3 inflammation by LPS and palmitic acid promotes adipogenesis, but represses osteogenesis in mesenchymal stem cells [123]. The discrepancy may be due to the use of different NLRP3 activators and/or cells. As mentioned above, the NLRP3 inflammasome is associated with the downregulation of adipogenesis in abdominal SAT from obese adolescents [37]. Defects in adipogenesis are likely to impair the recruitment of new adipocytes and contribute to adipocyte enlargement. Indeed, genetic ablation of NLRP3 or caspase-1 prevents obesity-induced adipocyte hypertrophy [19,107], yet whether this is due to changes in adipogenesis, lipolysis and/or energy metabolism remains to be further clarified.
Inflammation in BAT during obesity is less studied, but was demonstrated to impair thermogenic capacity and browning [124]. The IL-1β antibody and IL-1 receptor antagonist restores isoproterenol-induced UCP1 mRNA expression in C3H10T1/2 adipocytes, treated with a conditioned medium from LPS-stimulated macrophages [125]. In vivo, treatment with LPS abolishes CL316243 (a β3 adrenergic receptor agonist)-induced browning of sWAT, accompanied with lower core body temperature. The negative effect of LPS on browning is mediated by TLR4. IL-1β indeed impairs mitochondrial function and browning in adipocytes via the upregulation of oxidative stress [126]. The adipose triglyceride lipase knockout mice display a whitening of BAT, accompanied with a strong induction of the NLRP3 inflammasome [127].
There is a positive correlation between the expression of NLRP3 inflammasome components and extracellular matrix (ECM) remodeling genes, including matrix metallopeptidase 2 (MMP2), MMP9, and transforming growth factor β (TGF-β), in both vWAT and the liver [128]. Paradoxically, NLRP3 inflammasome was revealed to aggravate adipose tissue fibrosis during the progression of obesity, which limits healthy adipocyte expansion and elevates circulating levels of FFAs [128,129]. Consistently, genetic ablation of TLR4, the upstream regulator of the NLRP3 inflammasome, in immune cells has also been shown to prevent adipose tissue fibrosis in mice fed with HFD [130].
3.5. Others
The NLRP3 inflammasome in adventitial macrophages plays pivotal roles in vascular fibrosis and inflammation, contributing to the progression of abdominal aortic aneurysm [131]. Dysfunctions of perivascular adipose tissue (PVAT), a special type of adipose tissue surrounding blood vessels, are observed during obesity, with excessive vascular injury-induced adventitia fibroblast proliferation and differentiation, while the NLRP3 inflammasome and IL-1β signaling in obese PVAT are drastically upregulated, in order to aggravate this adventitial remodeling [132].
Apart from its deleterious effects on the peripheral tissues, activation of the NLRP3 inflammasome in vWAT has been recently report to impair the central nervous system in cases of obesity [133]. Genetic abrogation of NLRP3 prevents HFD-induced hippocampus dysfunction in mice. In addition, wild-type mice transplanted with obese visceral fat display deficits in memory and synaptic plasticity, whereas wild-type mice transplanted with obese visceral fat lacking NLRP3 show normal brain function. Further analysis indicated that NLRP3 inflammasome activation in adipose tissues induces microglial activation via the interleukin-1 receptor [133]. Treatment with the natural flavonoid quercetin has been shown to reduce hypothalamic inflammation, by downregulating NF-kB and NLRP3 inflammasome activities, accompanied with improvement in insulin sensitivity in the hypothalamus of rats fed with high fructose [134].
4. Potential Approaches Targeting Inflammasome Activation
4.1. Pharmacological Inhibition of NLRP3 Inflammasome
With the identification of inflammasome stimuli within obese and aged adipose tissue, several endogenous molecules and chemical compounds have been identified to directly suppress the NLRP3 inflammasome, targeting either priming, activation or ASC oligomerization, some of which are being translated from bench-side theory into promising bedside applications (Table 1).

Table 1.
NLRP3 inflammasome deactivators.
4.1.1. Inhibitors of NOX4/CPT1A and FASN
In line with the positive association between (NADPH oxidase 4) NOX4/carnitine palmitoyltransferase 1A (CPT1A)-dependent fatty acid oxidation and the NLRP3 inflammasome, treatments of GKT137831 and VAS-2870, two chemically distinct NOX4 inhibitors, demonstrated therapeutic potential against NLRP3 inflammasome activation in vitro and in vivo, by abolishing palmitic acid-induced caspase-1 activation, and IL-1β and IL-18 production [135]. Furthermore, etomoxir, an inhibitor of CPT1A that was clinically used for tackling type 2 diabetes and heart failure, yet was withdrawn owing to severe hepatotoxicity, has been identified as reducing the activation of caspase-1, and the secretion of IL-1β and IL-18, in vitro, when encountering nigericin and ATP [135,153,154]. Other than the NOX4/CPT1A signaling axis, the inhibition of fatty acid synthase (FASN), a key enzyme of fatty acid synthesis, is also likely to be a valuable therapeutic strategy for ameliorating NLRP3 inflammasome-mediated inflammation. Chemical inhibitors C75 and cerulenin are all capable of diminishing caspase-1 activation, as well as the protein expression of NLRP3 and pro-IL-1β in peritoneal macrophages [136], whereas C75 administration can markedly decrease the circulating levels of IL-1β and IL-18, and inhibit hepatic lipid accumulation in wild-type mice injected with LPS [136].
4.1.2. Direct Inhibitors of NLRP3 Protein
At present, a plethora of chemical compounds have been reported to impede ATPase activity in the NACHT domain of NLRP3, which is necessary for inflammasome assembly, including CY-09, MNS, OLT1177, BOT-4-one, INF39 and MCC950 [29,137,138,139,140,141,155]. Parthenolide, a naturally occurring sesquiterpene lactone from Feverfew, and Bay 11-7082, a phenyl vinyl sulfone compound, disrupt the ATPase activity of NLRP3, concurrent with suppressed IκB kinase, NF-κB and caspase-1 activation [142,143,144]. Tranilast, an analogue of tryptophan metabolite, can interfere with the NLRP3–NLRP3 and NLRP3–ASC interaction, while oridonin, a diterpenoid isolated from the medicinal herb Rabdosia rubescens, alters the NLPR3–NEK7 interaction, thereby obstructing inflammasome oligomerization in an ATPase-independent fashion [139,145]. Of note, the therapeutic effects of CY-09, OLT11771, BOT-4-one, INF39, parthenolide, tranilast and oridonin have been confirmed in vivo, while the others warrant more pre-clinical studies for their translational values. In addition, their direct effects on adipose tissue inflammation and metabolism also warrant further investigation.
4.1.3. AMPK Activators
AMPK is a key regulator of metabolic balance and NLRP3 inflammasome activity [145,156]. As mentioned above, the activity of AMPK is reduced by inflammasome stimuli, such as palmitic acid, and its activation is able to antagonize NLRP3 activation in macrophages. Metformin, the first line anti-diabetic drug, exhibits inhibitory activity on the NLRP3 inflammasome in multiple cell types. Treatment with metformin for two months dramatically suppresses the production of IL-1β and IL-18 in monocyte-derived macrophages isolated from type 2 diabetic subjects [45]. These suppressive effects are abolished by the knockdown of AMPK expression. Resveratrol is a polyphenolic compound with anti-diabetic activity, and shows similar actions in modulating AMPK to metformin [157,158]. It was reported that both are capable of attenuating ER stress and mitochondrial fission in adipose tissue, with reduced IRE1α and eIF2α phosphorylation, in an AMPK-dependent manner, which in turn blunts the activity of the NLRP3 inflammasome [146]. Oral administration of metformin or resveratrol can effectively ameliorate inflammation and adipose dysfunction in diabetic mice [146]. Berberine, a natural alkaloid compound isolated from various medicinal herbs, augments AMPK-dependent autophagy, with increased autophagic protein expression and autophagosome formation, which eliminates ROS and blunts NLRP3 inflammasome activity [148,159]. To be noteworthy, HFD-fed mice with oral administration of berberine displayed reduced adipose tissue mass, and improved insulin sensitivity and glucose tolerance [148].
4.1.4. Others
Glycyrrhizin (GL) and Isoliquiritigenin (ILG), the active compounds in the Glycyrrhiza plant, have been implicated in the blockade of TLR4 signaling, which leads to reduced downstream NF-κB and mitogen-activated protein kinase (MAPK) activation, resulting in the repression of NLRP3 transcription [160,161,162]. Moreover, their inhibitory effects are not only confined to the priming step, as both GL and ILG diminish ASC oligomerization in response to ATP, dampening the NLRP3 inflammasome activation signal [147]. Treatment with ILG suppresses dietary-induced IL-1β production and adipose tissue inflammation in mice, as expected [147].
Melatonin, a hormone synthesized by the pineal gland engaging in the circadian rhythm, abolishes NF-κB signaling via reducing the protein levels of NF-κB and p65, in cytoplasm and nucleus, respectively [149,163,164]. Melatonin injection in HFD-fed mice thus exhibited decreased protein expression of the NLRP3 inflammasome components and the serum level of IL-1β [149]. Notably, HFD-induced pyrotopsis in adipose tissue was also markedly suppressed upon melatonin treatment, through downregulation of caspase-1, GSDMD and interferon regulatory factor 7 (IRF7) [149].
Eplerenone is a selective aldosterone antagonist approved by The Food and Drug Administration for treatment of hypertension and heart failure [165]. Its potent anti-inflammatory effects have been well documented, which suppress ATM accumulation and inflammasome activation in epididymal WAT and the liver, thereby improving glucose homeostasis [150]. Mechanistically, these compelling biological functions are largely attributed to its inhibitory roles in the NLRP3 inflammasome’s priming and activation steps [150]. Transcription of the NLRP3 inflammasome’s components, phosphorylation of NF-κB and ROS production are all attenuated by eplerenone in epididymal WAT in mice [150].
β-hydroxybutyrate, a ketone body serving as an alternative source of ATP during an energy deficit, has been shown to abrogate the activating effects of ATP, monosodium urate and ceramide on NLRP3 inflammasome, through diminishing K+ efflux and ASC oligomerization [151]. β-hydroxybutyrate enclosed with nanolipogels conferred protection against NLRP3 inflammasome-induced inflammatory diseases, such as Muckle–Wells syndrome and familial cold autoinflammatory syndrome [151]. Furthermore, a ketogenic diet, that elevates serum β-hydroxybutyrate level, considerably suppresses caspase-1 activation, and attenuates neutrophilia and hyperglycaemia in the mouse model [151]. However, the effect of β-hydroxybutyrate on adipose tissue inflammasome remains to be determined.
4.2. Genetic Approach
Other than the pharmacological approach, direct deletion of NLRP3 at the genomic level is confined to molecular studies, and still rarely applied in clinic, largely owing to safety concerns. Promisingly, CRISPR/Cas9, the third-generation gene editing tool, with an in vivo delivery system of cationic lipid-assisted nanoparticles encapsulating mCas9 and gRNA, was first utilized to disrupt NLRP3 in peritoneal macrophages [166]. The strategy is effective in combatting multiple inflammatory diseases, as evidenced by the mitigation of HFD-induced type 2 diabetes and LPS-induced septic shock in NLRP3 knockout mice [166]. Nevertheless, further studies need to address the immune-related side effects, considering the critical role of the NLRP3 inflammasome in innate immunity.
5. Conclusions and Remarks
Adipose tissue inflammation is a key pathogenic link between obesity and cardiometabolic diseases. It is likely that adipose tissue also adopts a two-signal model for NLRP3 inflammasome activation. Metabolic insults, including SFAs, pro-inflammatory adipokines, hyperglycemia and endotoxemia, represent major stimuli of NLRP3 inflammasome activation, and subsequent IL-1β production, in adipose tissue. Noticeably, some of the metabolic insults, such as palmitic acid, can act as both priming and activation signals. NLRP3 inflammasome exacerbates dietary-induced insulin resistance, immune dysregulation, plaque growth and vascular remodeling, whereas a series of aging-associated metabolic dysregulations in adipose tissue, such as impaired glycaemic control, increased visceral adiposity and reduced lipolysis, are also exacerbated by the NLRP3 inflammasome. A positive correlation between adipose NLRP3 inflammation activity and cardiometabolic disorders is observed in different human populations, although a causative relationship remains to be shown. In addition, the underlying mechanism by which NLRP3 is activated in obesity and aging has recently be revealed. For instance, deacetylation of NLRP3 by SIRT2 reduces IL-1β production in macrophages, and hence improves aging-related inflammation and insulin resistance in rodents [167]. With these insights into molecular control, an NLPR3 inflammasome-targeting strategy might hold a promise for combating metabolic disorders in obesity and aging, by improving adipose tissue inflammation. In addition, it is worth further exploring the beneficial effects of NLRP3 deactivators (Table 1) on adipose tissue inflammation and metabolic health during aging and obesity, yet their potential side effects on immunosuppression should also be considered.
Author Contributions
Writing, review and editing by K.K.-L.W., S.W.-M.C. and K.K.-Y.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (NSFC) (grant number: 91857119), Research grant council Hong Kong (general research grant 17101815 and 17100717), Health Medical Research Fund (05161286) and PolyU internal funding to Kenneth King Yip Cheng.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Francisco, V.; Pino, J.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gomez, R.; Mobasheri, A.; Gualillo, O. Adipokines and inflammation: Is it a question of weight? Br. J. Pharmacol. 2018, 175, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, A.W., Jr. The immune cells in adipose tissue. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 34–38. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol. Metab. 2018, 29, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Thomou, T.; Zhu, Y.; Karagiannides, I.; Pothoulakis, C.; Jensen, M.D.; Kirkland, J.L. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013, 17, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol. Metab. 2008, 19, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Kwok, K.H.M.; Cheng, K.K.Y.; Hoo, R.L.C.; Ye, D.; Xu, A.; Lam, K.S.L. Adipose-specific inactivation of JNK alleviates atherosclerosis in apoE-deficient mice. Clin. Sci. (Lond.) 2016, 130, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, K.K.L.; Jiang, X.; Xu, A.; Cheng, K.K.Y. The role of adipose tissue senescence in obesity- and ageing-related metabolic disorders. Clin. Sci. (Lond.) 2020, 134, 315–330. [Google Scholar] [CrossRef]
- Liu, Z.H.; Jin, L.G.; Yang, J.K.; Wang, B.L.; Wu, K.K.L.; Hallenborg, P.; Xu, A.M.; Cheng, K.K.Y. The Dysfunctional MDM2-p53 Axis in Adipocytes Contributes to Aging-Related Metabolic Complications by Induction of Lipodystrophy. Diabetes 2018, 67, 2397–2409. [Google Scholar] [CrossRef]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N. Targeting senescent cells enhances adipogenesis and metabolic function in old age. elife 2015, 4, e12997. [Google Scholar] [CrossRef]
- Ballak, D.B.; Stienstra, R.; Tack, C.J.; Dinarello, C.A.; van Diepen, J.A. IL-1 family members in the pathogenesis and treatment of metabolic disease: Focus on adipose tissue inflammation and insulin resistance. Cytokine 2015, 75, 280–290. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 2006, 83, 447S–455S. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.L.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 2011, 32, 110–116. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Harder, J.; Franchi, L.; Muñoz-Planillo, R.; Park, J.-H.; Reimer, T.; Núñez, G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-κB activation but proceeds independently of TLR signaling and P2X7 receptor. J. Immunol. 2009, 183, 5823–5829. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, H. T cells in adipose tissue: Critical players in immunometabolism. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Sundara Rajan, S.; Longhi, M.P. Dendritic cells and adipose tissue. Immunology 2016, 149, 353–361. [Google Scholar] [CrossRef]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; Van Tits, B.; Van Diepen, J.A.; Van Den Berg, S.A.; Rensen, P.C.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Goossens, G.H.; Blaak, E.E.; Theunissen, R.; Duijvestijn, A.M.; Clément, K.; Tervaert, J.-W.C.; Thewissen, M.M. Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Mol. Immunol. 2012, 50, 142–149. [Google Scholar] [CrossRef]
- Camell, C.D.; Sander, J.; Spadaro, O.; Lee, A.; Nguyen, K.Y.; Wing, A.; Goldberg, E.L.; Youm, Y.-H.; Brown, C.W.; Elsworth, J. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 2017, 550, 119. [Google Scholar] [CrossRef] [PubMed]
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism 2017, 74, 1–9. [Google Scholar] [CrossRef]
- Esser, N.; L’homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef]
- Kursawe, R.; Dixit, V.D.; Scherer, P.E.; Santoro, N.; Narayan, D.; Gordillo, R.; Giannini, C.; Lopez, X.; Pierpont, B.; Nouws, J. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes 2016, 65, 610–618. [Google Scholar] [CrossRef]
- Moschen, A.R.; Molnar, C.; Enrich, B.; Geiger, S.; Ebenbichler, C.F.; Tilg, H. Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol. Med. 2011, 17, 840–845. [Google Scholar] [CrossRef]
- Dalmas, E.; Venteclef, N.; Caer, C.; Poitou, C.; Cremer, I.; Aron-Wisnewsky, J.; Lacroix-Desmazes, S.; Bayry, J.; Kaveri, S.V.; Clement, K.; et al. T cell-derived IL-22 amplifies IL-1beta-driven inflammation in human adipose tissue: Relevance to obesity and type 2 diabetes. Diabetes 2014, 63, 1966–1977. [Google Scholar] [CrossRef]
- Mocanu, A.O.; Mulya, A.; Huang, H.; Dan, O.; Shimizu, H.; Batayyah, E.; Brethauer, S.A.; Dinischiotu, A.; Kirwan, J.P. Effect of Roux-en-Y Gastric Bypass on the NLRP3 Inflammasome in Adipose Tissue from Obese Rats. PLoS ONE 2015, 10, e0139764. [Google Scholar] [CrossRef]
- Hoffstedt, J.; Andersson, D.P.; Eriksson Hogling, D.; Theorell, J.; Naslund, E.; Thorell, A.; Ehrlund, A.; Ryden, M.; Arner, P. Long-term Protective Changes in Adipose Tissue After Gastric Bypass. Diabetes Care 2017, 40, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guan, W.; Ma, S.; Lin, S.; Yang, N.; Liu, R.; Liang, H.; Zhou, H. Lipopolysaccharide and inflammatory cytokines levels decreased after sleeve gastrectomy in Chinese adults with obesity. Endocr. J. 2019, 66, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Frikke-Schmidt, H.; O’Rourke, R.W.; Lumeng, C.N.; Sandoval, D.A.; Seeley, R.J. Does bariatric surgery improve adipose tissue function? Obes. Rev. 2016, 17, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Bando, S.; Fukuda, D.; Soeki, T.; Nishimoto, S.; Uematsu, E.; Matsuura, T.; Ise, T.; Tobiume, T.; Yamaguchi, K.; Yagi, S.; et al. Expression of NLRP3 in subcutaneous adipose tissue is associated with coronary atherosclerosis. Atherosclerosis 2015, 242, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, D.; Zhang, L.; Fu, M.; Zeng, Y.; Russell, R. Variants of NLRP3 gene are associated with insulin resistance in Chinese Han population with type-2 diabetes. Gene 2013, 530, 151–154. [Google Scholar] [CrossRef]
- Camell, C.; Goldberg, E.; Dixit, V.D. Regulation of Nlrp3 inflammasome by dietary metabolites. In Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2015; pp. 334–342. [Google Scholar]
- Xia, M.; Boini, K.M.; Abais, J.M.; Xu, M.; Zhang, Y.; Li, P.-L. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am. J. Pathol. 2014, 184, 1617–1628. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Niepmann, S.; Knolle, P.A.; Hornung, V. Aging-associated TNF production primes inflammasome activation and NLRP3-related metabolic disturbances. J. Immunol. 2016, 197, 2900–2908. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaraj, S.; Jialal, I. High glucose induces IL-1beta expression in human monocytes: Mechanistic insights. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E337–E346. [Google Scholar] [CrossRef]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thevenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Luche, E.; Cousin, B.; Garidou, L.; Serino, M.; Waget, A.; Barreau, C.; Andre, M.; Valet, P.; Courtney, M.; Casteilla, L.; et al. Metabolic endotoxemia directly increases the proliferation of adipocyte precursors at the onset of metabolic diseases through a CD14-dependent mechanism. Mol. Metab. 2013, 2, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Jensen, B.A.H.; Varin, T.V.; Servant, F.; Van Blerk, S.; Richard, D.; Marceau, S.; Surette, M.; Biertho, L.; Lelouvier, B.; et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat. Metab. 2020, 2, 233–242. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef]
- Hersoug, L.G.; Moller, P.; Loft, S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: Implications for inflammation and obesity. Obes. Rev. 2016, 17, 297–312. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.; Roche, H.M. Dietary saturated fatty acids prime the NLRP 3 inflammasome via TLR 4 in dendritic cells—implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 56, 1212–1222. [Google Scholar] [CrossRef]
- Robblee, M.M.; Kim, C.C.; Abate, J.P.; Valdearcos, M.; Sandlund, K.L.; Shenoy, M.K.; Volmer, R.; Iwawaki, T.; Koliwad, S.K. Saturated fatty acids engage an IRE1α-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep. 2016, 14, 2611–2623. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408. [Google Scholar] [CrossRef] [PubMed]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N. Monounsaturated fatty acid–enriched high-fat diets impede adipose NLRP3 inflammasome–mediated IL-1β secretion and insulin resistance despite obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Nguyen, K.Y.; Jurczak, M.J.; Christian, B.E.; Shulman, G.I.; Shadel, G.S.; Dixit, V.D. Macrophage-specific de novo synthesis of ceramide is dispensable for inflammasome-driven inflammation and insulin resistance in obesity. J. Biol. Chem. 2015, 290, 29402–29413. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Zhong, Z.; Stubelius, A.; Sweeney, S.R.; Booshehri, L.M.; Antonucci, L.; Liu-Bryan, R.; Lodi, A.; Terkeltaub, R.; Lacal, J.C. Choline Uptake and Metabolism Modulate Macrophage IL-1β and IL-18 Production. Cell Metab. 2019, 29, 1350–1362.e1357. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef]
- Foley, J.F. PAF and the inflammasome. Sci. Signal. 2019, 12, eaaz7370. [Google Scholar] [CrossRef]
- Chu, L.H.; Indramohan, M.; Ratsimandresy, R.A.; Gangopadhyay, A.; Morris, E.P.; Monack, D.M.; Dorfleutner, A.; Stehlik, C. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat. Commun. 2018, 9, 996. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Dong, Y.-Q.; Wang, P.; Zhang, X.; Yan, Y.; Sun, L.; Liu, B.; Zhang, D.; Zhang, H.; Liu, H. Adipocyte-derived lysophosphatidylcholine activates adipocyte and adipose tissue macrophage nod-like receptor protein 3 inflammasomes mediating homocysteine-induced insulin resistance. EBioMedicine 2018, 31, 202–216. [Google Scholar] [CrossRef]
- Kabarowski, J.H. G2A and LPC: Regulatory functions in immunity. Prostaglandins Other Lipid Mediat. 2009, 89, 73–81. [Google Scholar] [CrossRef]
- Khan, S.Y.; McLaughlin, N.J.; Kelher, M.R.; Eckels, P.; Gamboni-Robertson, F.; Banerjee, A.; Silliman, C.C. Lysophosphatidylcholines activate G2A inducing Gαi-1-/Gαq/11-Ca2+ flux, Gβγ-Hck activation and clathrin/β-arrestin-1/GRK6 recruitment in PMNs. Biochem. J. 2010, 432, 35–45. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Sodium dependence of lysophosphatidylcholine-induced caspase-1 activity and reactive oxygen species generation. Immunobiology 2011, 216, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Micaelo, N.; González-Abuín, N.; Pinent, M.; Ardévol, A.; Blay, M. Dietary fatty acid composition is sensed by the NLRP3 inflammasome: Omega-3 fatty acid (DHA) prevents NLRP3 activation in human macrophages. Food Funct. 2016, 7, 3480–3487. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.-H.; Luo, W.-J.; Xu, Q.-Y.; Hua, J. Dietary saturated fatty acid and polyunsaturated fatty acid oppositely affect hepatic NOD-like receptor protein 3 inflammasome through regulating nuclear factor-kappa B activation. World J. Gastroenterol. 2016, 22, 2533. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, A.; Monaco, M.L.; Capasso, M.; Forestieri, P.; Pilone, V.; Nardelli, C.; Buono, P.; Daniele, A. Adiponectin oligomers as potential indicators of adipose tissue improvement in obese subjects. Eur. J. Endocrinol. 2013, 169, 37–43. [Google Scholar] [CrossRef]
- Cheng, K.K.; Lam, K.S.; Wang, B.; Xu, A. Signaling mechanisms underlying the insulin-sensitizing effects of adiponectin. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 3–13. [Google Scholar] [CrossRef]
- Wang, F.; Liu, Y.; Yang, W.; Yuan, J.; Mo, Z. Adiponectin inhibits NLRP3 inflammasome by modulating the AMPK-ROS pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 3338–3347. [Google Scholar]
- Zhang, L.; Yuan, M.; Zhang, L.; Wu, B.; Sun, X. Adiponectin alleviates NLRP3-inflammasome-mediated pyroptosis of aortic endothelial cells by inhibiting FoxO4 in arteriosclerosis. Biochem. Biophys. Res. Commun. 2019, 514, 266–272. [Google Scholar] [CrossRef]
- Kim, E.H.; Park, P.H. Globular adiponectin protects rat hepatocytes against acetaminophen-induced cell death via modulation of the inflammasome activation and ER stress: Critical role of autophagy induction. Biochem. Pharmacol. 2018, 154, 278–292. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, E.H.; Pun, N.T.; Chang, J.-H.; Kim, J.; Jeong, J.-H.; Choi, D.Y.; Kim, S.-H.; Park, P.-H. Globular adiponectin inhibits lipopolysaccharide-primed inflammasomes activation in macrophages via autophagy induction: The critical role of AMPK signaling. Int. J. Mol. Sci. 2017, 18, 1275. [Google Scholar] [CrossRef]
- Lagathu, C.; Yvan-Charvet, L.; Bastard, J.-P.; Maachi, M.; Quignard-Boulange, A.; Capeau, J.; Caron, M. Long-term treatment with interleukin-1β induces insulin resistance in murine and human adipocytes. Diabetologia 2006, 49, 2162–2173. [Google Scholar] [CrossRef]
- Naylor, C.; Petri, W.A., Jr. Leptin Regulation of Immune Responses. Trends Mol. Med. 2016, 22, 88–98. [Google Scholar] [CrossRef]
- Abella, V.; Scotece, M.; Conde, J.; Pino, J.; Gonzalez-Gay, M.A.; Gomez-Reino, J.J.; Mera, A.; Lago, F.; Gomez, R.; Gualillo, O. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017, 13, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Liu, L.; Han, L.; Yu, Y. Leptin promotes IL-18 secretion by activating the NLRP3 inflammasome in RAW 264.7 cells. Mol. Med. Rep. 2017, 16, 9770–9776. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schoof, E.; Stuppy, A.; Harig, F.; Carbon, R.; Horbach, T.; Stohr, W.; Rascher, W.; Dotsch, J. Comparison of leptin gene expression in different adipose tissues in children and adults. Eur. J. Endocrinol. 2004, 150, 579–584. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shimizu, H.; Shimomura, Y.; Hayashi, R.; Ohtani, K.; Sato, N.; Futawatari, T.; Mori, M. Serum leptin concentration is associated with total body fat mass, but not abdominal fat distribution. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 536–541. [Google Scholar] [CrossRef]
- Zhao, S.; Zhu, Y.; Schultz, R.D.; Li, N.; He, Z.; Zhang, Z.; Caron, A.; Zhu, Q.; Sun, K.; Xiong, W.; et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab. 2019, 30, 706–719. [Google Scholar] [CrossRef]
- Gabriely, I.; Ma, X.H.; Yang, X.M.; Rossetti, L.; Barzilai, N. Leptin resistance during aging is independent of fat mass. Diabetes 2002, 51, 1016–1021. [Google Scholar] [CrossRef]
- Moller, N.; O’Brien, P.; Nair, K.S. Disruption of the relationship between fat content and leptin levels with aging in humans. J. Clin. Endocrinol. Metab. 1998, 83, 931–934. [Google Scholar] [CrossRef]
- Wu, L.E.; Samocha-Bonet, D.; Whitworth, P.T.; Fazakerley, D.J.; Turner, N.; Biden, T.J.; James, D.E.; Cantley, J. Identification of fatty acid binding protein 4 as an adipokine that regulates insulin secretion during obesity. Mol. Metab. 2014, 3, 465–473. [Google Scholar] [CrossRef]
- Steen, K.A.; Xu, H.; Bernlohr, D.A. FABP4/aP2 regulates macrophage redox signaling and inflammasome activation via control of UCP2. Mol. Cell. Biol. 2017, 37, e00282-16. [Google Scholar] [CrossRef]
- Dando, I.; Fiorini, C.; Dalla Pozza, E.; Padroni, C.; Costanzo, C.; Palmieri, M.; Donadelli, M. UCP2 inhibition triggers ROS-dependent nuclear translocation of GAPDH and autophagic cell death in pancreatic adenocarcinoma cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Yabal, M.; Calleja, D.J.; Simpson, D.S.; Lawlor, K.E. Stressing out the mitochondria: Mechanistic insights into NLRP3 inflammasome activation. J. Leukoc. Biol. 2019, 105, 377–399. [Google Scholar] [CrossRef]
- Shi, C.-S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, J.; Quan, W.; Lee, J.C.; Kim, M.S.; Kim, S.H.; Bae, J.W.; Hur, K.Y.; Lee, M.S. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy 2016, 12, 1390–1403. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Y.; Li, W.; Chen, H.; Du, L.; Liu, D.; Wang, X.; Xu, T.; Liu, L.; Chen, Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019, 15, 1882–1898. [Google Scholar] [CrossRef]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef]
- Zhang, X.; Dai, J.; Li, L.; Chen, H.; Chai, Y. NLRP3 inflammasome expression and signaling in human diabetic wounds and in high glucose induced macrophages. J. Diabetes Res. 2017, 2017, 5281358. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Gu, J.; Gou, F.; Huang, W.; Gao, C.; Chen, G.; Long, Y.; Zhou, X.; Yang, M.; Liu, S. High glucose and lipopolysaccharide prime NLRP3 inflammasome via ROS/TXNIP pathway in mesangial cells. J. Diabetes Res. 2016, 2016, 6973175. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhang, Z.; Wang, X.; Li, R.; Hou, W.; Bi, W.; Zhang, X. Inhibition of autophagy induces IL-1β release from ARPE-19 cells via ROS mediated NLRP3 inflammasome activation under high glucose stress. Biochem. Biophys. Res. Commun. 2015, 463, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.; Ergul, A. Relationship of endothelin-1 and NLRP3 inflammasome activation in HT22 hippocampal cells in diabetes. Life Sci. 2016, 159, 97–103. [Google Scholar] [CrossRef]
- Hu, T.X.; Zhang, N.N.; Ruan, Y.; Tan, Q.Y.; Wang, J. Hydrogen sulfide modulates high glucose-induced NLRP3 inflammasome activation in 3T3-L1 adipocytes. Exp. Ther. Med. 2020, 19, 771–776. [Google Scholar] [CrossRef]
- Koenen, T.B.; Stienstra, R.; van Tits, L.J.; de Graaf, J.; Stalenhoef, A.F.; Joosten, L.A.; Tack, C.J.; Netea, M.G. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes 2011, 60, 517–524. [Google Scholar] [CrossRef]
- Moon, J.S.; Hisata, S.; Park, M.A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M.K. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Pessin, J.E. Insulin signaling pathways in time and space. Trends Cell Biol. 2002, 12, 65–71. [Google Scholar] [CrossRef]
- Gao, D.; Madi, M.; Ding, C.; Fok, M.; Steele, T.; Ford, C.; Hunter, L.; Bing, C. Interleukin-1β mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E289–E304. [Google Scholar] [CrossRef]
- Cho, K.A.; Kang, P.B. PLIN2 inhibits insulin-induced glucose uptake in myoblasts through the activation of the NLRP3 inflammasome. Int. J. Mol. Med. 2015, 36, 839–844. [Google Scholar] [CrossRef]
- Lindegaard, B.; Matthews, V.B.; Brandt, C.; Hojman, P.; Allen, T.L.; Estevez, E.; Watt, M.J.; Bruce, C.R.; Mortensen, O.H.; Syberg, S.; et al. Interleukin-18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes 2013, 62, 3064–3074. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, H.; Talukdar, S.; Bae, E.J.; Olefsky, J.M. Increased macrophage migration into adipose tissue in obese mice. Diabetes 2012, 61, 346–354. [Google Scholar]
- Wouters, K.; Gaens, K.; Bijnen, M.; Verboven, K.; Jocken, J.; Wetzels, S.; Wijnands, E.; Hansen, D.; Van Greevenbroek, M.; Duijvestijn, A. Circulating classical monocytes are associated with CD11c+ macrophages in human visceral adipose tissue. Sci. Rep. 2017, 7, 42665. [Google Scholar] [CrossRef] [PubMed]
- Amano, S.U.; Cohen, J.L.; Vangala, P.; Tencerova, M.; Nicoloro, S.M.; Yawe, J.C.; Shen, Y.; Czech, M.P.; Aouadi, M. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014, 19, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Luche, E.; Robert, V.; Cuminetti, V.; Pomie, C.; Sastourne-Arrey, Q.; Waget, A.; Arnaud, E.; Varin, A.; Labit, E.; Laharrague, P.; et al. Corrupted adipose tissue endogenous myelopoiesis initiates diet-induced metabolic disease. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Zamarron, B.F.; Mergian, T.A.; Cho, K.W.; Martinez-Santibanez, G.; Luan, D.; Singer, K.; DelProposto, J.L.; Geletka, L.M.; Muir, L.A.; Lumeng, C.N. Macrophage Proliferation Sustains Adipose Tissue Inflammation in Formerly Obese Mice. Diabetes 2017, 66, 392–406. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Kraakman, M.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.S.; Abdel-Latif, A.; Smyth, S.S. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014, 19, 821–835. [Google Scholar] [CrossRef]
- Kimura, H.; Karasawa, T.; Usui, F.; Kawashima, A.; Endo, Y.; Kobayashi, M.; Sadatomo, A.; Nakamura, J.; Iwasaki, Y.; Yada, T.; et al. Caspase-1 deficiency promotes high-fat diet-induced adipose tissue inflammation and the development of obesity. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E881–E890. [Google Scholar] [CrossRef]
- Ben-Sasson, S.Z.; Hu-Li, J.; Quiel, J.; Cauchetaux, S.; Ratner, M.; Shapira, I.; Dinarello, C.A.; Paul, W.E. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 7119–7124. [Google Scholar] [CrossRef]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev. 2001, 12, 53–72. [Google Scholar] [CrossRef]
- Ivanov, S.; Merlin, J.; Lee, M.K.S.; Murphy, A.J.; Guinamard, R.R. Biology and function of adipose tissue macrophages, dendritic cells and B cells. Atherosclerosis 2018, 271, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, K.; Wan, X.; Wang, F.; Guo, Z.; Mo, Z. NLRP3 inflammasome activation in mesenchymal stem cells inhibits osteogenic differentiation and enhances adipogenic differentiation. Biochem. Biophys. Res. Commun. 2017, 484, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, F.; Cereijo, R.; Gavalda-Navarro, A.; Villarroya, J.; Giralt, M. Inflammation of brown/beige adipose tissues in obesity and metabolic disease. J. Intern. Med. 2018, 284, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Naknukool, S.; Yoshitake, R.; Hanafusa, Y.; Tokiwa, S.; Li, Y.; Sakamoto, T.; Nitta, T.; Kim, M.; Takahashi, N.; et al. Proinflammatory cytokine interleukin-1beta suppresses cold-induced thermogenesis in adipocytes. Cytokine 2016, 77, 107–114. [Google Scholar] [CrossRef]
- Okla, M.; Zaher, W.; Alfayez, M.; Chung, S. Inhibitory effects of toll-like receptor 4, NLRP3 inflammasome, and interleukin-1β on white adipocyte browning. Inflammation 2018, 41, 626–642. [Google Scholar] [CrossRef]
- Kotzbeck, P.; Giordano, A.; Mondini, E.; Murano, I.; Severi, I.; Venema, W.; Cecchini, M.P.; Kershaw, E.E.; Barbatelli, G.; Haemmerle, G.; et al. Brown adipose tissue whitening leads to brown adipocyte death and adipose tissue inflammation. J. Lipid Res. 2018, 59, 784–794. [Google Scholar] [CrossRef]
- Unamuno, X.; Gómez-Ambrosi, J.; Ramírez, B.; Rodríguez, A.; Becerril, S.; Valentí, V.; Moncada, R.; Silva, C.; Salvador, J.; Frühbeck, G. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell. Mol. Immunol. 2019. [Google Scholar] [CrossRef]
- Khan, T.; Muise, E.S.; Iyengar, P.; Wang, Z.V.; Chandalia, M.; Abate, N.; Zhang, B.B.; Bonaldo, P.; Chua, S.; Scherer, P.E. Metabolic dysregulation and adipose tissue fibrosis: Role of collagen VI. Mol. Cell. Biol. 2009, 29, 1575–1591. [Google Scholar] [CrossRef]
- Vila, I.K.; Badin, P.M.; Marques, M.A.; Monbrun, L.; Lefort, C.; Mir, L.; Louche, K.; Bourlier, V.; Roussel, B.; Gui, P.; et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014, 7, 1116–1129. [Google Scholar] [CrossRef]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Yoshimura, K.; Aoki, H.; Tsutsui, H.; Noda, T. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II–induced aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 127–136. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, H.-W.; Chen, H.-N.; Deng, X.-J.; Tu, Y.-X.; Jackson, A.O.; Qing, J.-N.; Wang, A.-P.; Patel, V.; Yin, K. Perivascular adipose tissue dysfunction aggravates adventitial remodeling in obese mini pigs via NLRP3 inflammasome/IL-1 signaling pathway. Acta Pharmacol. Sin. 2019, 40, 46–54. [Google Scholar] [CrossRef]
- Guo, D.H.; Yamamoto, M.; Hernandez, C.M.; Khodadadi, H.; Baban, B.; Stranahan, A.M. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J. Clin. Investig. 2020, 130, 1961–1976. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Pan, Y.; Wang, R.; Kang, L.-L.; Xue, Q.-C.; Wang, X.-N.; Kong, L.-D. Quercetin inhibits AMPK/TXNIP activation and reduces inflammatory lesions to improve insulin signaling defect in the hypothalamus of high fructose-fed rats. J. Nutr. Biochem. 2014, 25, 420–428. [Google Scholar] [CrossRef]
- Moon, J.-S.; Nakahira, K.; Chung, K.-P.; DeNicola, G.M.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.-H.; Ryter, S.W.; Stout-Delgado, H. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002. [Google Scholar] [CrossRef]
- Moon, J.-S.; Lee, S.; Park, M.-A.; Siempos, I.I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J. Clin. Investig. 2015, 125, 665–680. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- He, Y.; Varadarajan, S.; Muñoz-Planillo, R.; Burberry, A.; Nakamura, Y.; Núñez, G. 3, 4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem. 2014, 289, 1142–1150. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef]
- Shim, D.-W.; Shin, W.-Y.; Yu, S.-H.; Kim, B.-H.; Ye, S.-K.; Koppula, S.; Won, H.-S.; Kang, T.-B.; Lee, K.-H. BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Cocco, M.; Pellegrini, C.; Martínez-Banaclocha, H.; Giorgis, M.; Marini, E.; Costale, A.; Miglio, G.; Fornai, M.; Antonioli, L.; Lopez-Castejon, G. Development of an acrylate derivative targeting the NLRP3 inflammasome for the treatment of inflammatory bowel disease. J. Med. Chem. 2017, 60, 3656–3671. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef]
- Hehner, S.P.; Hofmann, T.G.; Dröge, W.; Schmitz, M.L. The antiinflammatory sesquiterpene lactone parthenolide inhibits NF-κB by targeting the IκB kinase complex. J. Immunol. 1999, 163, 5617–5623. [Google Scholar]
- Saadane, A.; Masters, S.; DiDonato, J.; Li, J.; Berger, M. Parthenolide inhibits IκB kinase, NF-κB activation, and inflammatory response in cystic fibrosis cells and mice. Am. J. Respir. Cell Mol. Biol. 2007, 36, 728–736. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mole. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef]
- Honda, H.; Nagai, Y.; Matsunaga, T.; Okamoto, N.; Watanabe, Y.; Tsuneyama, K.; Hayashi, H.; Fujii, I.; Ikutani, M.; Hirai, Y. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 2014, 96, 1087–1100. [Google Scholar] [CrossRef]
- Zhou, H.; Feng, L.; Xu, F.; Sun, Y.; Ma, Y.; Zhang, X.; Liu, H.; Xu, G.; Wu, X.; Shen, Y. Berberine inhibits palmitate-induced NLRP3 inflammasome activation by triggering autophagy in macrophages: A new mechanism linking berberine to insulin resistance improvement. Biomed. Pharmacother. 2017, 89, 864–874. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Xu, Y.; Luo, D.; Ren, Q.; Wu, S.; Sun, C. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose tissue. J. Pineal Res. 2017, 63, e12414. [Google Scholar] [CrossRef]
- Wada, T.; Ishikawa, A.; Watanabe, E.; Nakamura, Y.; Aruga, Y.; Hasegawa, H.; Onogi, Y.; Honda, H.; Nagai, Y.; Takatsu, K. Eplerenone prevented obesity-induced inflammasome activation and glucose intolerance. J. Endocrinol. 2017, 235, 179–191. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263. [Google Scholar] [CrossRef]
- Ip, W.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M. Etomoxir: A new approach to treatment of chronic heart failure. Lancet 2000, 356, 1621–1622. [Google Scholar] [CrossRef]
- Ratheiser, K.; Schneeweiss, B.; Waldhäusl, W.; Fasching, P.; Korn, A.; Nowotny, P.; Rohac, M.; Wolf, H. Inhibition by etomoxir of carnitine palmitoyltransferase I reduces hepatic glucose production and plasma lipids in non-insulin-dependent diabetes mellitus. Metabolism 1991, 40, 1185–1190. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Cordero, M.D.; Williams, M.R.; Ryffel, B. AMP-Activated Protein Kinase Regulation of the NLRP3 Inflammasome during Aging. Trends Endocrinol. Metab. 2018, 29, 8–17. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Xiao, N.; Wang, M.; Kou, J.; Qi, L.; Huang, F.; Liu, B.; Liu, K. Pharmacological activation of AMPK ameliorates perivascular adipose/endothelial dysfunction in a manner interdependent on AMPK and SIRT1. Pharmacol. Res. 2014, 89, 19–28. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, H.; Kim, S.; Ha, T. Resveratrol inhibits TNF-α-induced changes of adipokines in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2007, 364, 972–977. [Google Scholar] [CrossRef]
- Neag, M.A.; Mocan, A.; Echeverría, J.; Pop, R.M.; Bocsan, C.I.; Crişan, G.; Buzoianu, A.D. Berberine: Botanical occurrence, traditional uses, extraction methods, and relevance in cardiovascular, metabolic, hepatic, and renal disorders. Front. Pharmacol. 2018, 9, 557. [Google Scholar] [CrossRef]
- Takahashi, T.; Takasuka, N.; Iigo, M.; Baba, M.; Nishino, H.; Tsuda, H.; Okuyama, T. Isoliquiritigenin, a flavonoid from licorice, reduces prostaglandin E2 and nitric oxide, causes apoptosis, and suppresses aberrant crypt foci development. Cancer Sci. 2004, 95, 448–453. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Park, S.J.; Yun, K.-J.; Cho, Y.-W.; Park, H.-J.; Lee, K.-T. Isoliquiritigenin isolated from the roots of Glycyrrhiza uralensis inhibits LPS-induced iNOS and COX-2 expression via the attenuation of NF-κB in RAW 264.7 macrophages. Eur. J. Pharmacol. 2008, 584, 175–184. [Google Scholar] [CrossRef]
- Takei, H.; Baba, Y.; Hisatsune, A.; Katsuki, H.; Miyata, T.; Yokomizo, K.; Isohama, Y. Glycyrrhizin Inhibits Interleukin-8 Production and Nuclear Factor–κB Activity in Lung Epithelial Cells, but Not Through Glucocorticoid Receptors. J. Pharmacol. Sci. 2008, 106, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Hariharasubramanian, N.; Pilapil, C. Circadian rhythm of plasma melatonin in endogenous depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1984, 8, 715–718. [Google Scholar] [CrossRef]
- Reiter, R.J. Pineal melatonin: Cell biology of its synthesis and of its physiological interactions. Endocr. Rev. 1991, 12, 151–180. [Google Scholar] [CrossRef] [PubMed]
- Craft, J. Eplerenone (Inspra), a new aldosterone antagonist for the treatment of systemic hypertension and heart failure. In Baylor University Medical Center Proceedings; Taylor & Francis: Milton, UK, 2004; pp. 217–220. [Google Scholar]
- Xu, C.; Lu, Z.; Luo, Y.; Liu, Y.; Cao, Z.; Shen, S.; Li, H.; Liu, J.; Chen, K.; Chen, Z. Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Chiang, H.-H.; Luo, H.; Zheng, Z.; Qiao, Q.; Wang, L.; Tan, M.; Ohkubo, R.; Mu, W.-C.; Zhao, S. An Acetylation Switch of the NLRP3 Inflammasome Regulates Aging-Associated Chronic Inflammation and Insulin Resistance. Cell Metab. 2020, 31, 580–591. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).