Staying out or Going in? The Interplay between Type 3 and Type 5 Secretion Systems in Adhesion and Invasion of Enterobacterial Pathogens



Abstract
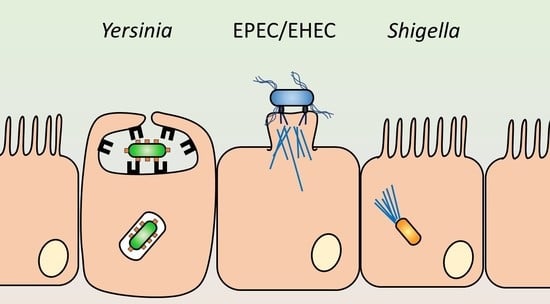
1. Introduction
2. Secretion Systems in Enterobacterial Pathogenesis
2.1. Type 3 Secretion Systems
2.1.1. Overview
2.1.2. Architecture and Assembly of the T3SS
2.1.3. Effector Proteins
2.2. Type 5 Secretion Systems
3. Staying out: The Attaching and Effacing Pathogens
3.1. The Esc-Esp T3SS
3.2. Intimin
3.3. Pedestal Formation
4. Going in: EIEC and Shigella
4.1. The Invasion Process
4.2. Actin-Based Motility
4.3. EIEC vs. Shigella
5. Maybe Going in? The Enteropathogenic Yersiniae
5.1. Invasin
5.2. YadA
5.3. The Ysc-Yop T3SS
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABM | Actin-based motility |
| A/E | Attaching and effacing |
| Ail | Attachment and invasion locus |
| C-ring | Cytoplasmic ring |
| FAK | Focal adhesion kinase |
| ECM | Extracellular matrix |
| EHEC | Enterohemorrhagic E. coli |
| EIEC | Enteroinvasive E. coli |
| EPEC | Enteropathogenic E. coli |
| Esp | E. coli secreted protein |
| Ics | Intercellular spread |
| IL | Interleukin |
| InvA | Invasin |
| Ipa | Invasion plasmid antigen |
| Ipg | Invasion plasmid gene |
| Lcr | Low calcium response |
| LEE | Locus of enterocyte effacement |
| M cell | Microfold cell |
| Mxi | Membrane expression of Ipa |
| Nle | Non-LEE effector |
| Sct | Secretion and cellular translocation |
| Sep | Secretion of E. coli proteins |
| Spa | Surface presentation of Ipa |
| SS | Secretion system |
| STEC | Shiga-toxigenic E. coli |
| T3SS | Type 3 secretion system |
| T5SS | Type 5 secretion system |
| TAA | Trimeric autotransporter adhesin |
| Tir | Translocated intimin receptor |
| N-WASP | Neural Wiskott–Aldrich syndrome protein |
| YadA | Yersinia adhesin A |
| Yop | Yersinia outer protein |
| Ysc | Yersinia secretion |
References
- Adeolu, M.; Alnajar, S.; Naushad, S.; Gupta, R.S. Genome-based phylogeny, and taxonomy of the ‘Enterobacteriales’: Proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.; Blacker, B.F.; Khalil, I.A.; Rao, P.C.; Cao, S.; Zimsen, S.R.; Albertson, S.B.; Stanaway, J.D.; Deshpande, A.; Abebe, Z.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1211–1228. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- Kotloff, K.; Nasrin, D.; Blackwelder, W.C.; Wu, Y.; Farag, T.; Panchalingham, S.; Sow, S.O.; Sur, D.; Zaidi, A.K.M.; Faruque, A.S.G.; et al. The incidence, aetiology, and adverse clinical consequences of less severe diarrhoeal episodes among infants and children residing in low-income and middle-income countries: A 12-month case-control study as a follow-on to the Global Enteric Multicenter Study (GEMS). Lancet Glob. Heal. 2019, 7, e568–e584. [Google Scholar] [CrossRef]
- Pettengill, E.A.; Hoffmann, M.; Binet, R.; Roberts, R.J.; Payne, J.; Allard, M.; Michelacci, V.; Minelli, F.; Morabito, S. Complete Genome Sequence of Enteroinvasive Escherichia coli O96:H19 Associated with a Severe Foodborne Outbreak. Genome Announc. 2015, 3, 883. [Google Scholar] [CrossRef]
- Sahl, J.W.; Morris, C.R.; Emberger, J.; Fraser, C.M.; Ochieng, J.B.; Juma, J.; Fields, B.; Breiman, R.F.; Gilmour, M.; Nataro, J.P.; et al. Defining the Phylogenomics of Shigella Species: A Pathway to Diagnostics. J. Clin. Microbiol. 2015, 53, 951–960. [Google Scholar] [CrossRef]
- Dhakal, R.; Wang, Q.; Lan, R.; Howard, P.; Sintchenko, V. Novel multiplex PCR assay for identification and subtyping of enteroinvasive Escherichia coli and differentiation from Shigella based on target genes selected by comparative genomics. J. Med. Microbiol. 2018, 67, 1257–1264. [Google Scholar] [CrossRef]
- Beld, M.J.C.V.D.; de Boer, R.F.; Reubsaet, F.A.G.; Rossen, J.W.A.; Zhou, K.; Kuiling, S.; Friedrich, A.W.; Kooistra-Smid, M.A.M.D. Evaluation of a Culture-Dependent Algorithm and a Molecular Algorithm for iIdentification of Shigella spp., Escherichia coli, and Enteroinvasive E. coli. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef]
- Baker, K.S.; Dallman, T.J.; Field, N.; Childs, T.; Mitchell, H.; Day, M.; Weill, F.-X.; Lefèvre, S.; Tourdjman, M.; Hughes, G.; et al. Genomic epidemiology of Shigella in the United Kingdom shows transmission of pathogen sublineages and determinants of antimicrobial resistance. Sci. Rep. 2018, 8, 7389. [Google Scholar] [CrossRef]
- Mitchell, H.D.; Mikhail, A.F.W.; Painset, A.; Dallman, T.J.; Jenkins, C.; Thomson, N.; Field, N.; Hughes, G. Use of whole-genome sequencing to identify clusters of Shigella flexneri associated with sexual transmission in men who have sex with men in England: A validation study using linked behavioural data. Microb. Genom. 2019, 5, e000311. [Google Scholar] [CrossRef]
- Sundström, K. Cost of iIllness for Five Major Foodborne Illnesses and Sequelae in Sweden. Appl. Heal. Econ. Heal. Policy 2018, 16, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.; Dabke, G.; Strakova, L.; Jenkins, C.; Saavedra-Campos, M.; McManus, O.; Paranthaman, K. Introduction of PCR testing reveals a previously unrecognized burden of yersiniosis in Hampshire, UK. J. Med. Microbiol. 2020, 69. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef]
- Bottichio, L.; Keaton, A.; Thomas, D.; Fulton, T.; Tiffany, A.; Frick, A.; Mattioli, M.; Kahler, A.; Murphy, J.; Otto, M.; et al. Shiga Toxin–Producing Escherichia coli Infections Associated With Romaine Lettuce—United States, 2018. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Christie, P.J. The Rich Tapestry of Bacterial Protein Translocation Systems. Protein J. 2019, 38, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Prehna, G.; Zhang, G.; Gong, X.; Duszyk, M.; Okon, M.; McIntosh, L.P.; Weiner, J.H.; Strynadka, N.C.J. A Protein Export Pathway Involving Escherichia coli Porins. Structure 2012, 20, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Rouse, S.L.; Hawthorne, W.; Berry, J.-L.; Chorev, D.S.; Ionescu, S.A.; Lambert, S.; Stylianou, F.; Ewert, W.; Mackie, U.; Morgan, R.M.L.; et al. A new class of hybrid secretion system is employed in Pseudomonas amyloid biogenesis. Nat. Commun. 2017, 8, 263. [Google Scholar] [CrossRef]
- Gómez-Santos, N.; Glatter, T.; Koebnik, R.; Świątek-Połatyńska, M.A.; Søgaard-Andersen, L. A TonB-dependent transporter is required for secretion of protease PopC across the bacterial outer membrane. Nat. Commun. 2019, 10, 1360. [Google Scholar] [CrossRef]
- Stones, D.H.; Krachler, A.M. Against the tide: The role of bacterial adhesion in host colonization. Biochem. Soc. Trans. 2016, 44, 1571–1580. [Google Scholar] [CrossRef]
- Colonne, P.M.; Winchell, C.G.; Voth, D.E. Hijacking Host Cell Highways: Manipulation of the Host Actin Cytoskeleton by Obligate Intracellular Bacterial Pathogens. Front. Microbiol. 2016, 6, 4609. [Google Scholar] [CrossRef]
- Diepold, A.; Armitage, J.P. Type III secretion systems: The bacterial flagellum and the injectisome. Philos. Trans. R. Soc. B Boil. Sci. 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Renault, T.T.; Guse, A.; Erhardt, M. Export Mechanisms and Energy Transduction in Type-III Secretion Machines. Curr. Top. Microbiol. Immunol. 2019, 1–17. [Google Scholar] [CrossRef]
- Schmidt, H.; Hensel, M. Pathogenicity Islands in Bacterial Pathogenesis. Clin. Microbiol. Rev. 2004, 17, 14–56. [Google Scholar] [CrossRef] [PubMed]
- Bancerz-Kisiel, A.; Pieczywek, M.; Łada, P.; Szweda, W. The Most Important Virulence Markers of Yersinia enterocolitica and Their Role during Infection. Genes 2018, 9, 235. [Google Scholar] [CrossRef] [PubMed]
- Pasqua, M.; Michelacci, V.; di Martino, M.L.; Tozzoli, R.; Grossi, M.; Colonna, B.; Morabito, S.; Prosseda, G. The Intriguing Evolutionary Journey of Enteroinvasive E. coli (EIEC) toward Pathogenicity. Front. Microbiol. 2017, 8, 2390. [Google Scholar] [CrossRef] [PubMed]
- Gaytán, M.O.; Martinez-Santos, V.; Soto, E.; González-Pedrajo, B. Type Three Secretion System in Attaching and Effacing Pathogens. Front. Microbiol. 2016, 6, 129. [Google Scholar] [CrossRef]
- Belotserkovsky, I.; Sansonetti, P. Shigella and Enteroinvasive Escherichia Coli. Mol. Asp. Myeloid Stem Cell Dev. 2018, 416, 1–26. [Google Scholar] [CrossRef]
- Mattock, E.; Blocker, A. How Do the Virulence Factors of Shigella Work Together to Cause Disease? Front. Microbiol. 2017, 7, 2457. [Google Scholar] [CrossRef]
- De Woody, R.S.; Merritt, P.M.; Marketon, M.M. Regulation of the Yersinia type III secretion system: Traffic control. Front. Microbiol. 2013, 3, 4. [Google Scholar] [CrossRef]
- Slater, S.L.; Sagfors, A.M.; Pollard, D.J.; Ruano-Gallego, D.; Frankel, G. The Type III Secretion System of Pathogenic Escherichia coli. Mol. Asp. Myeloid Stem Cell Dev. 2018, 416, 51–72. [Google Scholar] [CrossRef]
- Wagner, S.; Diepold, A. A Unified Nomenclature for Injectisome-Type Type III Secretion Systems. Mol. Asp. Myeloid Stem Cell Dev. 2020, 1–10. [Google Scholar] [CrossRef]
- Dohlich, K.; Zumsteg, A.B.; Goosmann, C.; Kolbe, M. A Substrate-Fusion Protein Is Trapped inside the Type III Secretion System Channel in Shigella flexneri. PLoS Pathog. 2014, 10, e1003881. [Google Scholar] [CrossRef] [PubMed]
- Radics, J.; Königsmaier, L.; Marlovits, T. Structure of a pathogenic type 3 secretion system in action. Nat. Struct. Mol. Boil. 2013, 21, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Edgren, T.; Forsberg, A.; Rosqvist, R.; Wolf-Watz, H. Type III Secretion in Yersinia: Injectisome or Not? PLoS Pathog. 2012, 8, e1002669. [Google Scholar] [CrossRef] [PubMed]
- Akopyan, K.; Edgren, T.; Wang-Edgren, H.; Rosqvist, R.; Fahlgren, A.; Wolf-Watz, H.; Fällman, M. Translocation of surface-localized effectors in type III secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Tejeda-Dominguez, F.; Huerta-Cantillo, J.; Chavez-Dueñas, L.; Navarro-Garcia, F. A Novel Mechanism for Protein Delivery by the Type 3 Secretion System for Extracellularly Secreted Proteins. mBio 2017, 8, 184. [Google Scholar] [CrossRef]
- Fujii, T.; Cheung, M.; Blanco, A.; Kato, T.; Blocker, A.J.; Namba, K. Structure of a type III secretion needle at 7-A resolution provides insights into its assembly and signaling mechanisms. Proc. Natl. Acad. Sci. USA 2012, 109, 4461–4466. [Google Scholar] [CrossRef]
- Espina, M.; Olive, A.J.; Kenjale, R.; Moore, D.S.; Ausar, S.F.; Kaminski, R.W.; Oaks, E.V.; Middaugh, C.R.; Picking, W.D.; Picking, W.L. IpaD Localizes to the Tip of the Type III Secretion System Needle of Shigella flexneri. Infect. Immun. 2006, 74, 4391–4400. [Google Scholar] [CrossRef]
- Veenendaal, A.K.J.; Hodgkinson, J.L.; Schwarzer, L.; Stabat, D.; Zenk, S.F.; Blocker, A.J. The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol. Microbiol. 2007, 63, 1719–1730. [Google Scholar] [CrossRef]
- Murillo, I.; Argudo, I.M.; Blocker, A. Genetic Dissection of the Signaling Cascade that Controls Activation of the Shigella Type III Secretion System from the Needle Tip. Sci. Rep. 2016, 6, 27649. [Google Scholar] [CrossRef]
- Roehrich, A.D.; Guillossou, E.; Blocker, A.; Argudo, I.M. Shigella IpaD has a dual role: Signal transduction from the type III secretion system needle tip and intracellular secretion regulation. Mol. Microbiol. 2013, 87, 690–706. [Google Scholar] [CrossRef] [PubMed]
- Blocker, A.; Gounon, P.; Larquet, E.; Niebuhr, K.; Cabiaux, V.; Parsot, C.; Sansonetti, P. The Tripartite Type III Secreton of Shigella flexneri Inserts Ipab and Ipac into Host Membranes. J. Cell Boil. 1999, 147, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Grin, I.; Malmsheimer, S.; Singh, N.; Torres-Vargas, C.E.; Westerhausen, S. Bacterial type III secretion systems: A complex device for the delivery of bacterial effector proteins into eukaryotic host cells. FEMS Microbiol. Lett. 2018, 365, 201. [Google Scholar] [CrossRef] [PubMed]
- Argudo, I.M.; Blocker, A.J. The Shigella T3SS needle transmits a signal for MxiC release, which controls secretion of effectors. Mol. Microbiol. 2010, 78, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Roe, A.J.; McAteer, S.; Shipston, M.J.; Gally, D.L. Hierarchal type III secretion of translocators and effectors from Escherichia coli O157:H7 requires the carboxy terminus of SepL that binds to Tir. Mol. Microbiol. 2008, 69, 1499–1512. [Google Scholar] [CrossRef]
- Cheng, L.W.; Kay, O.; Schneewind, O. Regulated Secretion of YopN by the Type III Machinery of Yersinia enterocolitica. J. Bacteriol. 2001, 183, 5293–5301. [Google Scholar] [CrossRef]
- Deane, J.E.; Roversi, P.; King, C.; Johnson, S.; Lea, S.M. Structures of the Shigella flexneri Type 3 Secretion System Protein MxiC Reveal Conformational Variability Amongst Homologues. J. Mol. Boil. 2008, 377, 985–992. [Google Scholar] [CrossRef][Green Version]
- Cepeda-Molero, M.; Berger, C.N.; Walsham, A.D.S.; Ellis, S.; Wemyss-Holden, S.; Schüller, S.; Frankel, G.; Fernández, L.A. Attaching and effacing (A/E) lesion formation by enteropathogenic E. coli on human intestinal mucosa is dependent on non-LEE effectors. PLoS Pathog. 2017, 13, e1006706. [Google Scholar] [CrossRef]
- Platenkamp, A.; Mellies, J.L. Environment Controls LEE Regulation in Enteropathogenic Escherichia coli. Front. Microbiol. 2018, 9, 1694. [Google Scholar] [CrossRef]
- Yoshida, S.; Katayama, E.; Kuwae, A.; Mimuro, H.; Suzuki, T.; Sasakawa, C. Shigella deliver an effector protein to trigger host microtubule destabilization, which promotes Rac1 activity and efficient bacterial internalization. EMBO J. 2002, 21, 2923–2935. [Google Scholar] [CrossRef]
- Ogawa, M.; Yoshimori, T.; Suzuki, T.; Sagara, H.; Mizushima, N.; Sasakawa, C. Escape of Intracellular Shigella from Autophagy. Science 2005, 307, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Van Nhieu, G.T.; Caron, E.; Hall, A.; Sansonetti, P.J. IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. 1999, 18, 3249–3262. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.S.; Finlay, B.B. Bringing down the host: Enteropathogenic and enterohaemorrhagic Escherichia coli effector-mediated subversion of host innate immune pathways. Cell. Microbiol. 2015, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Baumann, D.; Salia, H.; Greune, L.; Norkowski, S.; Körner, B.; Uckeley, Z.M.; Frankel, G.; Guenot, M.; Rüter, C.; Schmidt, M.A. Multitalented EspB of enteropathogenic Escherichia coli (EPEC) enters cells autonomously and induces programmed cell death in human monocytic THP-1 cells. Int. J. Med. Microbiol. 2018, 308, 387–404. [Google Scholar] [CrossRef]
- Ashida, H.; Mimuro, H.; Sasakawa, C. Shigella Manipulates Host Immune Responses by Delivering Effector Proteins with Specific Roles. Front. Immunol. 2015, 6, 219. [Google Scholar] [CrossRef]
- Ugalde-Silva, P.; Gonzalez-Lugo, O.; Navarro-Garcia, F. Tight Junction Disruption Induced by Type 3 Secretion System Effectors Injected by Enteropathogenic and Enterohemorrhagic Escherichia coli. Front. Microbiol. 2016, 6, 1162. [Google Scholar] [CrossRef]
- Grabowski, B.; Schmidt, M.A.; Rüter, C. Immunomodulatory Yersinia outer proteins (Yops)–useful tools for bacteria and humans alike. Virulence 2017, 8, 1124–1147. [Google Scholar] [CrossRef]
- Pinaud, L.; Sansonetti, P.J.; Phalipon, A. Host Cell Targeting by Enteropathogenic Bacteria T3SS Effectors. Trends Microbiol. 2018, 26, 266–283. [Google Scholar] [CrossRef]
- Kenny, B.; Lai, L.-C.; Finlay, B.B.; Donnenberg, M. EspA, a protein secreted by enteropathogenic Escherichia coli, is required to induce signals in epithelial cells. Mol. Microbiol. 1996, 20, 313–323. [Google Scholar] [CrossRef]
- Hua, Y.; Yan, K.; Wan, C. Clever Cooperation: Interactions Between EspF and Host Proteins. Front. Microbiol. 2018, 9, 2831. [Google Scholar] [CrossRef]
- Garmendia, J.; Phillips, A.D.; Carlier, M.-F.; Chong, Y.; Schüller, S.; Marchès, O.; Dahan, S.; Oswald, E.; Shaw, R.K.; Knutton, S.; et al. TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell. Microbiol. 2004, 6, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Pollard, D.J.; Berger, C.N.; So, E.C.; Yu, L.; Hadavizadeh, K.; Jennings, P.; Tate, E.W.; Choudhary, J.S.; Frankel, G. Broad-Spectrum Regulation of Nonreceptor Tyrosine Kinases by the Bacterial ADP-Ribosyltransferase EspJ. mBio 2018, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.N.; Crepin, V.F.; Baruch, K.; Mousnier, A.; Rosenshine, I.; Frankel, G. EspZ of Enteropathogenic and Enterohemorrhagic Escherichia coli Regulates Type III Secretion System Protein Translocation. mBio 2012, 3, 317. [Google Scholar] [CrossRef]
- Campbell-Valois, F.-X.; Sachse, M.; Sansonetti, P.J.; Parsot, C. Escape of Actively Secreting Shigella flexneri from ATG8/LC3-Positive Vacuoles Formed during Cell-To-Cell Spread Is Facilitated by IcsB and VirA. mBio 2015, 6, 02567-14. [Google Scholar] [CrossRef] [PubMed]
- Izard, T.; van Nhieu, G.T.; Bois, P.R.J. Shigella applies molecular mimicry to subvert vinculin and invade host cells. J. Cell Biol. 2006, 175, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Hakansson, S.; Schesser, K.; Persson, C.; Galyov, E.E.; Rosqvist, R.; Homblé, F.; Wolf-Watz, H. The YopB protein of Yersinia pseudotuberculosis is essential for the translocation of Yop effector proteins across the target cell plasma membrane and displays a contact-dependent membrane disrupting activity. EMBO J. 1996, 15, 5812–5823. [Google Scholar] [CrossRef]
- Kresse, A.U.; Rohde, M.; Guzmán, C.A. The EspD Protein of Enterohemorrhagic Escherichia coli is Required for the Formation of Bacterial Surface Appendages and Is Incorporated in the Cytoplasmic Membranes of Target Cells. Infect. Immun. 1999, 67, 4834–4842. [Google Scholar] [CrossRef]
- Francis, M.; Wolf-Watz, H. YopD of Yersinia pseudotuberculosisis translocated into the cytosol of HeLa epithelial cells: Evidence of a structural domain necessary for translocation. Mol. Microbiol. 1998, 29, 799–813. [Google Scholar] [CrossRef]
- Luo, W.; Donnenberg, M. Analysis of the Function of Enteropathogenic Escherichia coli EspB by Random Mutagenesis. Infect. Immun. 2006, 74, 810–820. [Google Scholar] [CrossRef]
- Sarker, M.R.; Neyt, C.; Stainier, I.; Cornelis, G.R. The Yersinia Yop Virulon: LcrV Is Required for Extrusion of the Translocators YopB and YopD. J. Bacteriol. 1998, 180, 1207–1214. [Google Scholar] [CrossRef]
- Picking, W.L.; Nishioka, H.; Hearn, P.D.; Baxter, M.A.; Harrington, A.T.; Blocker, A.; Picking, W.D. IpaD of Shigella flexneri Is Independently Required for Regulation of Ipa Protein Secretion and Efficient Insertion of IpaB and IpaC into Host Membranes. Infect. Immun. 2005, 73, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Höfling, S.; Grabowski, B.; Norkowski, S.; Schmidt, M.A.; Rüter, C. Current activities of the Yersinia effector protein YopM. Int. J. Med Microbiol. 2015, 305, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Sasakawa, C. Shigella IpaH Family Effectors as a Versatile Model for Studying Pathogenic Bacteria. Front. Microbiol. 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Burnaevskiy, N.; Peng, T.; Reddick, L.E.; Hang, H.C.; Alto, N.M. Myristoylome profiling reveals a concerted mechanism of ARF GTPase deacylation by the bacterial protease IpaJ. Mol. Cell 2015, 58, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Alto, N.M.; Shao, F.; Lazar, C.S.; Brost, R.L.; Chua, G.; Mattoo, S.; McMahon, S.A.; Ghosh, P.; Hughes, T.R.; Boone, C.; et al. Identification of a Bacterial Type III Effector Family with G Protein Mimicry Functions. Cell 2006, 124, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Arbeloa, A.; Blanco, M.; Moreira, F.C.; Bulgin, R.; Lopez, C.; Dahbi, G.; Blanco, J.; Mora, A.; Alonso, M.P.; Mamani, R.C.; et al. Distribution of espM and espT among enteropathogenic and enterohaemorrhagic Escherichia coli. J. Med. Microbiol. 2009, 58, 988–995. [Google Scholar] [CrossRef]
- Garza-Mayers, A.C.; Miller, K.A.; Russo, B.C.; Nagda, D.V.; Goldberg, M.B. Shigella flexneri Regulation of ARF6 Activation during Bacterial Entry via an IpgD-Mediated Positive Feedback Loop. mBio 2015, 6, 02584. [Google Scholar] [CrossRef]
- Deng, W.; Yu, H.B.; de Hoog, C.L.; Stoynov, N.; Li, Y.; Foster, L.J.; Finlay, B.B. Quantitative proteomic analysis of type III secretome of enteropathogenic Escherichia coli reveals an expanded effector repertoire for attaching/effacing bacterial pathogens. Mol. Cell. Proteom. 2012, 11, 692–709. [Google Scholar] [CrossRef]
- Zurawski, D.V.; Mumy, K.L.; Faherty, C.S.; McCormick, B.A.; Maurelli, A.T. Shigella flexneri type III secretion system effectors OspB and OspF target the nucleus to downregulate the host inflammatory response via interactions with retinoblastoma protein. Mol. Microbiol. 2009, 71, 350–368. [Google Scholar] [CrossRef]
- Zurawski, D.V.; Mitsuhata, C.; Mumy, K.L.; McCormick, B.A.; Maurelli, A.T. OspF and OspC1 Are Shigella flexneri Type III Secretion System Effectors That Are Required for Postinvasion Aspects of Virulence. Infect. Immun. 2006, 74, 5964–5976. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ogawa, M.; Sanada, T.; Mimuro, H.; Kim, M.; Ashida, H.; Akakura, R.; Yoshida, M.; Kawalec, M.; Reichhart, J.-M.; et al. The Shigella OspC3 Effector Inhibits Caspase-4, Antagonizes Inflammatory Cell Death, and Promotes Epithelial Infection. Cell Host Microbe 2013, 13, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Parsot, C.; Ageron, E.; Penno, C.; Mavris, M.; Jamoussi, K.; D’Hauteville, H.; Sansonetti, P.; Demers, B. A secreted anti-activator, OspD1, and its chaperone, Spa15, are involved in the control of transcription by the type III secretion apparatus activity in Shigella flexneri. Mol. Microbiol. 2005, 56, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Mou, X.; Souter, S.; Du, J.; Reeves, A.Z.; Lesser, C.F. Synthetic bottom-up approach reveals the complex interplay of Shigella effectors in regulation of epithelial cell death. Proc. Natl. Acad. Sci. USA 2018, 115, 6452–6457. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.S.; Giogha, C.; Mühlen, S.; Nachbur, U.; Pham, C.L.L.; Zhang, Y.; Hildebrand, J.M.; Oates, C.V.; Lung, T.W.F.; Ingle, D.N.; et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat. Microbiol. 2017, 2, 16258. [Google Scholar] [CrossRef]
- Farfan, M.J.; Toro, C.S.; Barry, E.M.; Nataro, J.P. Shigella enterotoxin-2 is a type III effector that participates in Shigella-induced interleukin 8 secretion by epithelial cells. FEMS Immunol. Med. Microbiol. 2011, 61, 332–339. [Google Scholar] [CrossRef]
- Tobe, T. Cytoskeleton-modulating effectors of enteropathogenic and enterohemorrhagic Escherichia coli: Role of EspL2 in adherence and an alternative pathway for modulating cytoskeleton through Annexin A2 function. FEBS J. 2010, 277, 2403–2408. [Google Scholar] [CrossRef]
- Faherty, C.S.; Redman, J.C.; Rasko, D.A.; Barry, E.M.; Nataro, J.P. Shigella flexneri effectors OspE1 and OspE2 mediate induced adherence to the colonic epithelium following bile salts exposure. Mol. Microbiol. 2012, 85, 107–121. [Google Scholar] [CrossRef]
- Morita-Ishihara, T.; Miura, M.; Iyoda, S.; Izumiya, H.; Watanabe, H.; Ohnishi, M.; Terajima, J. EspO1-2 Regulates EspM2-Mediated RhoA Activity to Stabilize Formation of Focal Adhesions in Enterohemorrhagic Escherichia coli-Infected Host Cells. PLoS ONE 2013, 8, e55960. [Google Scholar] [CrossRef]
- Zhou, Y.; Dong, N.; Hu, L.; Shao, F. The Shigella Type Three Secretion System Effector OspG Directly and Specifically Binds to Host Ubiquitin for Activation. PLoS ONE 2013, 8, e57558. [Google Scholar] [CrossRef]
- Sanada, T.; Kim, M.; Mimuro, H.; Suzuki, M.; Ogawa, M.; Oyama, A.; Ashida, H.; Kobayashi, T.; Koyama, T.; Nagai, S.; et al. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 2012, 483, 623–626. [Google Scholar] [CrossRef]
- Yao, Q.; Cui, J.; Zhu, Y.; Wang, G.; Hu, L.; Long, C.; Cao, R.; Liu, X.; Huang, N.; Chen, S.; et al. A bacterial type III effector family uses the papain-like hydrolytic activity to arrest the host cell cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 3716–3721. [Google Scholar] [CrossRef] [PubMed]
- Zurawski, D.V.; Mumy, K.L.; Badea, L.; Prentice, J.A.; Hartland, E.L.; McCormick, B.A.; Maurelli, A.T. The NleE/OspZ Family of Effector Proteins Is Required for Polymorphonuclear Transepithelial Migration, a Characteristic Shared by Enteropathogenic Escherichia coli and Shigella flexneri Infections. Infect. Immun. 2007, 76, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Kenny, B.; de Vinney, R.; Stein, M.; Reinscheid, D.J.; Frey, E.A.; Finlay, B.B. Enteropathogenic E. coli (EPEC) Transfers Its Receptor for Intimate Adherence into Mammalian Cells. Cell 1997, 91, 511–520. [Google Scholar] [CrossRef]
- Black, D.S.; Bliska, J.B. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol. Microbiol. 2002, 37, 515–527. [Google Scholar] [CrossRef]
- Persson, C.; Carballeira, N.; Wolf-Watz, H.; Fällman, M. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Casand FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO J. 1997, 16, 2307–2318. [Google Scholar] [CrossRef]
- Orth, K. Disruption of Signaling by Yersinia Effector YopJ, a Ubiquitin-Like Protein Protease. Science 2000, 290, 1594–1597. [Google Scholar] [CrossRef]
- Lee, W.L.; Grimes, J.M.; Robinson, R.C. Yersinia effector YopO uses actin as bait to phosphorylate proteins that regulate actin polymerization. Nat. Struct. Mol. Boil. 2015, 22, 248–255. [Google Scholar] [CrossRef]
- Shao, F.; Vacratsis, P.O.; Bao, Z.; Bowers, K.E.; Fierke, C.A.; Dixon, J.E. Biochemical characterization of the Yersinia YopT protease: Cleavage site and recognition elements in Rho GTPases. Proc. Natl. Acad. Sci. USA 2003, 100, 904–909. [Google Scholar] [CrossRef]
- Drobnak, I.; Braselmann, E.; Chaney, J.L.; Leyton, D.L.; Bernstein, H.D.; Lithgow, T.; Luirink, J.; Nataro, J.P.; Clark, P.L. Of linkers and autochaperones: An unambiguous nomenclature to identify common and uncommon themes for autotransporter secretion. Mol. Microbiol. 2014, 95, 1–16. [Google Scholar] [CrossRef]
- Leo, J.; Grin, I.; Linke, D. Type V secretion: Mechanism(s) of autotransport through the bacterial outer membrane. Philos. Trans. R. Soc. B Boil. Sci. 2012, 367, 1088–1101. [Google Scholar] [CrossRef]
- Bernstein, H.D. Type V Secretion in Gram-Negative Bacteria. EcoSal Plus 2019, 8, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.; Chauhan, N.; Udatha, D.G.; Leo, J.; Linke, D. Type V Secretion Systems in Bacteria. Microbiol. Spectr. 2016, 4, 305–335. [Google Scholar] [CrossRef] [PubMed]
- Meuskens, I.; Saragliadis, A.; Leo, J.; Linke, D. Type V Secretion Systems: An Overview of Passenger Domain Functions. Front. Microbiol. 2019, 10, 1163. [Google Scholar] [CrossRef] [PubMed]
- Leo, J.; Oberhettinger, P.; Schütz, M.; Linke, D. The inverse autotransporter family: Intimin, invasin and related proteins. Int. J. Med Microbiol. 2015, 305, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Łyskowski, A.; Leo, J.; Goldman, A. Structure and Biology of Trimeric Autotransporter Adhesins. Adv. Exp. Med. Biol. 2011, 715, 143–158. [Google Scholar] [CrossRef]
- Bassler, J.; Alvarez, B.H.; Hartmann, M.D.; Lupas, A.N. A domain dictionary of trimeric autotransporter adhesins. Int. J. Med Microbiol. 2015, 305, 265–275. [Google Scholar] [CrossRef]
- Carter, M.Q.; Brandl, M.T.; Kudva, I.T.; Katani, R.; Moreau, M.R.; Kapur, V. Conditional Function of Autoaggregative Protein Cah and Common cah Mutations in Shiga Toxin-Producing Escherichia coli. Appl. Environ. Microbiol. 2017, 84, e01739-17. [Google Scholar] [CrossRef]
- Wells, T.; Sherlock, O.; Rivas, L.; Mahajan, A.; Beatson, S.A.; Torpdahl, M.; Webb, R.I.; Allsopp, L.; Gobius, K.; Gally, D.L.; et al. EhaA is a novel autotransporter protein of enterohemorrhagic Escherichia coli O157:H7 that contributes to adhesion and biofilm formation. Environ. Microbiol. 2008, 10, 589–604. [Google Scholar] [CrossRef]
- Wells, T.; McNeilly, T.N.; Totsika, M.; Mahajan, A.; Gally, D.L.; Schembri, M.A. The Escherichia coli O157:H7 EhaB autotransporter protein binds to laminin and collagen I and induces a serum IgA response in O157:H7 challenged cattle. Environ. Microbiol. 2009, 11, 1803–1814. [Google Scholar] [CrossRef]
- Totsika, M.; Wells, T.; Beloin, C.; Valle, J.; Allsopp, L.; King, N.; Ghigo, J.-M.; Schembri, M.A. Molecular Characterization of the EhaG and UpaG Trimeric Autotransporter Proteins from Pathogenic Escherichia coli. Appl. Environ. Microbiol. 2012, 78, 2179–2189. [Google Scholar] [CrossRef]
- Easton, D.M.; Totsika, M.; Allsopp, L.; Phan, M.-D.; Idris, A.; Wurpel, D.J.; Sherlock, O.; Zhang, B.; Venturini, C.; Beatson, S.A.; et al. Characterization of EhaJ, a New Autotransporter Protein from Enterohemorrhagic and Enteropathogenic Escherichia coli. Front. Microbiol. 2011, 2, 120. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, P.; Habouria, H.; Bessaiah, H.; Dozois, C.M. Serine Protease Autotransporters of the Enterobacteriaceae (SPATEs): Out and About and Chopping It Up. Microorganisms 2019, 7, 594. [Google Scholar] [CrossRef] [PubMed]
- Nesta, B.; Spraggon, G.; Alteri, C.J.; Moriel, D.G.; Rosini, R.; Veggi, D.; Smith, S.; Bertoldi, I.; Pastorello, I.; Ferlenghi, I.; et al. FdeC, a Novel Broadly Conserved Escherichia coli Adhesin Eliciting Protection against Urinary Tract Infections. mBio 2012, 3, e00010-12. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.G.K.; Moriel, D.G.; Hancock, S.J.; Phan, M.-D.; Schembri, M.A. Bioinformatic and Molecular Analysis of Inverse Autotransporters from Escherichia coli. mSphere 2019, 4, 572. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, B.D.; Torres, A.G. Enterohemorrhagic Escherichia coli Adhesins. Microbiol. Spectr. 2014, 2, 157–174. [Google Scholar] [CrossRef]
- Martinez-Gil, M.; Goh, K.; Rackaityte, E.; Sakamoto, C.; Audrain, B.; Moriel, D.G.; Totsika, M.; Ghigo, J.-M.; Schembri, M.A.; Beloin, C. YeeJ is an inverse autotransporter from Escherichia coli that binds to peptidoglycan and promotes biofilm formation. Sci. Rep. 2017, 7, 11326. [Google Scholar] [CrossRef]
- Zumsteg, A.B.; Goosmann, C.; Brinkmann, V.; Morona, R.; Zychlinsky, A. IcsA Is a Shigella flexneri Adhesin Regulated by the Type III Secretion System and Required for Pathogenesis. Cell Host Microbe 2014, 15, 435–445. [Google Scholar] [CrossRef]
- Grassl, G.A.; Bohn, E.; Müller, Y.; Bühler, O.T.; Autenrieth, I.B. Interaction of Yersinia enterocolitica with epithelial cells: Invasin beyond invasion. Int. J. Med. Microbiol. 2003, 293, 41–54. [Google Scholar] [CrossRef]
- Strong, P.C.; Hinchliffe, S.J.; Patrick, H.; Atkinson, S.; Champion, O.L.; Wren, B. Identification and characterisation of a novel adhesin Ifp in Yersinia pseudotuberculosis. BMC Microbiol. 2011, 11, 85. [Google Scholar] [CrossRef]
- Pisano, F.; Kochut, A.; Uliczka, F.; Geyer, R.; Stolz, T.; Thiermann, T.; Rohde, M.; Dersch, P. In Vivo-Induced InvA-Like Autotransporters Ifp and InvC of Yersinia pseudotuberculosis Promote Interactions with Intestinal Epithelial Cells and Contribute to Virulence. Infect. Immun. 2011, 80, 1050–1064. [Google Scholar] [CrossRef]
- Sadana, P.; Geyer, R.; Pezoldt, J.; Helmsing, S.; Huehn, J.; Hust, M.; Dersch, P.; Scrima, A. The invasin D protein from Yersinia pseudotuberculosis selectively binds the Fab region of host antibodies and affects colonization of the intestine. J. Boil. Chem. 2018, 293, 8672–8690. [Google Scholar] [CrossRef] [PubMed]
- Mühlenkamp, M.; Oberhettinger, P.; Leo, J.; Linke, D.; Schütz, M.S. Yersinia adhesin A (YadA) – Beauty & beast. Int. J. Med. Microbiol. 2015, 305, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Felek, S.; Lawrenz, M.B.; Krukonis, E.S. The Yersinia pestis autotransporter YapC mediates host cell binding, autoaggregation and biofilm formation. Microbiology 2008, 154, 1802–1812. [Google Scholar] [CrossRef]
- Lawrenz, M.B.; Lenz, J.D.; Miller, V.L. A Novel Autotransporter Adhesin Is Required for Efficient Colonization during Bubonic Plague. Infect. Immun. 2008, 77, 317–326. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nair, M.K.M.; de Masi, L.; Yue, M.; Galván, E.M.; Chen, H.; Wang, F.; Schifferli, D.M. Adhesive Properties of YapV and Paralogous Autotransporter Proteins of Yersinia pestis. Infect. Immun. 2015, 83, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.A. LEEways: Tales of EPEC, ATEC and EHEC. Cell Microbiol. 2010, 12, 1544–1552. [Google Scholar] [CrossRef]
- Lai, Y.; Rosenshine, I.; Leong, J.M.; Frankel, G. Intimate host attachment: Enteropathogenic and enterohaemorrhagic Escherichia coli. Cell Microbiol. 2013, 15, 1796–1808. [Google Scholar] [CrossRef]
- Velle, K.B.; Campellone, K.G. Extracellular motility and cell-to-cell transmission of enterohemorrhagic E. coli is driven by EspFU-mediated actin assembly. PLoS Pathog. 2017, 13, e1006501. [Google Scholar] [CrossRef]
- Klapproth, J.-M.A. The Role of Lymphostatin/EHEC Factor for Adherence-1 in the Pathogenesis of Gram-Negative Infection. Toxins 2010, 2, 954–962. [Google Scholar] [CrossRef]
- Franzin, F.M.; Sircili, M.P. Locus of Enterocyte Effacement: A Pathogenicity Island Involved in the Virulence of Enteropathogenic and Enterohemorragic Escherichia coli Subjected to a Complex Network of Gene Regulation. BioMed Res. Int. 2015, 1–10. [Google Scholar] [CrossRef]
- Furniss, R.C.D.; Clements, A. Regulation of the Locus of Enterocyte Effacement in Attaching and Effacing Pathogens. J. Bacteriol. 2017, 200, e00336-17. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Puente, J.L.; Gruenheid, S.; Li, Y.; Vallance, B.A.; Vázquez, A.; Barba, J.; Ibarra, J.A.; O’Donnell, P.; Metalnikov, P.; et al. Dissecting virulence: Systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. USA 2004, 101, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Padavannil, A.; Jobichen, C.; Mills, E.; Velázquez-Campoy, A.; Li, M.; Leung, K.Y.; Mok, Y.K.; Rosenshine, I.; Sivaraman, J. Structure of GrlR–GrlA complex that prevents GrlA activation of virulence genes. Nat. Commun. 2013, 4, 2546. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.P.; Frankel, G.M. The Locus of Enterocyte Effacement and Associated Virulence Factors of Enterohemorrhagic Escherichia coli. Microbiol. Spectr. 2014, 2, 131–155. [Google Scholar] [CrossRef]
- Liu, H.; Magoun, L.; Luperchio, S.; Schauer, D.B.; Leong, J.M. The Tir-binding region of enterohaemorrhagic Escherichia coli intimin is sufficient to trigger actin condensation after bacterial-induced host cell signalling. Mol. Microbiol. 1999, 34, 67–81. [Google Scholar] [CrossRef]
- Vidal, J.E.; Navarro-García, F. EspC translocation into epithelial cells by enteropathogenic Escherichia coli requires a concerted participation of type V and III secretion systems. Cell Microbiol. 2008, 10, 1975–1986. [Google Scholar] [CrossRef]
- Guignot, J.; Segura, A.; van Nhieu, G.T. The Serine Protease EspC from Enteropathogenic Escherichia coli Regulates Pore Formation and Cytotoxicity Mediated by the Type III Secretion System. PLoS Pathog. 2015, 11, e1005013. [Google Scholar] [CrossRef]
- Cameron, E.A.; Curtis, M.M.; Kumar, A.; Dunny, G.M.; Sperandio, V.; Skaar, E.; Trent, M.S. Microbiota and Pathogen Proteases Modulate Type III Secretion Activity in Enterohemorrhagic Escherichia coli. mBio 2018, 9, e02204-18. [Google Scholar] [CrossRef]
- Xicohtencatl-Cortes, J.; Saldaña, Z.; Deng, W.; Castañeda, E.; Freer, E.; Tarr, P.I.; Finlay, B.B.; Puente, J.L.; Girón, J.A. Bacterial Macroscopic Rope-like Fibers with Cytopathic and Adhesive Properties. J. Boil. Chem. 2010, 285, 32336–32342. [Google Scholar] [CrossRef]
- Drago-Serrano, M.E.; Parra, S.G.; Manjarrez-Hernã¡ndez, H.A. EspC, an autotransporter protein secreted by enteropathogenic Escherichia coli (EPEC), displays protease activity on human hemoglobin. FEMS Microbiol. Lett. 2006, 265, 35–40. [Google Scholar] [CrossRef]
- Navarro-Garcia, F.; Serapio-Palacios, A.; Vidal, J.E.; Salazar, M.I.; Tapia-Pastrana, G. EspC Promotes Epithelial Cell Detachment by Enteropathogenic Escherichia coli via Sequential Cleavages of a Cytoskeletal Protein and then Focal Adhesion Proteins. Infect. Immun. 2014, 82, 2255–2265. [Google Scholar] [CrossRef] [PubMed]
- Serapio-Palacios, A.; Navarro-Garcia, F. EspC, an Autotransporter Protein Secreted by Enteropathogenic Escherichia coli, Causes Apoptosis and Necrosis through Caspase and Calpain Activation, Including Direct Procaspase-3 Cleavage. mBio 2016, 7, 479. [Google Scholar] [CrossRef] [PubMed]
- Leibiger, K.; Schweers, J.M.; Schütz, M.S. Biogenesis and function of the autotransporter adhesins YadA, intimin and invasin. Int. J. Med. Microbiol. 2019, 309, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Frey, E.A.; Pfuetzner, R.A.; Creagh, A.L.; Knoechel, D.G.; Haynes, C.A.; Finlay, B.B.; Strynadka, N.C.J. Crystal structure of enteropathogenic Escherichia coli intimin–receptor complex. Nature 2000, 405, 1073–1077. [Google Scholar] [CrossRef]
- Fairman, J.W.; Dautin, N.; Wojtowicz, D.; Liu, W.; Noinaj, N.; Barnard, T.J.; Udho, E.; Przytycka, T.M.; Cherezov, V.; Buchanan, S.K. Crystal structures of the outer membrane domain of intimin and invasin from enterohemorrhagic E. coli and enteropathogenic Y. pseudotuberculosis. Structure 2012, 20, 1233–1243. [Google Scholar] [CrossRef]
- Leo, J.; Oberhettinger, P.; Chaubey, M.; Schütz, M.; Kühner, D.; Bertsche, U.; Schwarz, H.; Götz, F.; Autenrieth, I.B.; Coles, M.; et al. The Intimin periplasmic domain mediates dimerisation and binding to peptidoglycan. Mol. Microbiol. 2014, 95, 80–100. [Google Scholar] [CrossRef]
- Touzé, T.; Hayward, R.D.; Eswaran, J.; Leong, J.M.; Koronakis, V. Self-association of EPEC intimin mediated by the β-barrel-containing anchor domain: A role in clustering of the Tir receptor. Mol. Microbiol. 2003, 51, 73–87. [Google Scholar] [CrossRef]
- Phillips, A.D.; Frankel, G. Intimin-Mediated Tissue Specificity in Enteropathogenic Escherichia coli Interaction with Human Intestinal Organ Cultures. J. Infect. Dis. 2000, 181, 1496–1500. [Google Scholar] [CrossRef]
- Mundy, R.; Schüller, S.; Girard, F.; Fairbrother, J.M.; Phillips, A.D.; Frankel, G. Functional studies of intimin in vivo and ex vivo: Implications for host specificity and tissue tropism. Microbiology 2007, 153, 959–967. [Google Scholar] [CrossRef]
- Mallick, E.M.; Brady, M.J.P.; Luperchio, S.A.; Vanguri, V.K.; Magoun, L.; Liu, H.P.; Sheppard, B.J.; Mukherjee, J.; Donohue-Rolfe, A.; Tzipori, S.; et al. Allele- and Tir-Independent Functions of Intimin in Diverse Animal Infection Models. Front. Microbiol. 2012, 3, 11. [Google Scholar] [CrossRef]
- Hartland, E.L.; Batchelor, M.; Delahay, R.M.; Hale, C.; Matthews, S.J.; Dougan, G.; Knutton, S.; Connerton, I.F.; Frankel, G. Binding of intimin from enteropathogenic Escherichia coli to Tir and to host cells. Mol. Microbiol. 1999, 32, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Kenny, B. Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol. Microbiol. 1999, 31, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.; Hayward, R.D.; Koronakis, V. Phosphorylation of the enteropathogenic E. coli receptor by the Src-family kinase c-Fyn triggers actin pedestal formation. Nature 2004, 6, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Swimm, A.; Bommarius, B.; Li, Y.; Cheng, D.; Reeves, P.; Sherman, M.; Veach, D.; Bornmann, W.; Kalman, D. Enteropathogenic Escherichia coli Use Redundant Tyrosine Kinases to Form Actin Pedestals. Mol. Boil. Cell 2004, 15, 3520–3529. [Google Scholar] [CrossRef] [PubMed]
- Bommarius, B.; Maxwell, D.; Swimm, A.; Leung, S.; Corbett, A.; Bornmann, W.; Kalman, D. Enteropathogenic Escherichia coli Tir is an SH2/3 ligand that recruits and activates tyrosine kinases required for pedestal formation. Mol. Microbiol. 2007, 63, 1748–1768. [Google Scholar] [CrossRef]
- Hayward, R.D.; Hume, P.J.; Humphreys, D.; Phillips, N.; Smith, K.; Koronakis, V. Clustering transfers the translocated Escherichia coli receptor into lipid rafts to stimulate reversible activation of c-Fyn. Cell. Microbiol. 2009, 11, 433–441. [Google Scholar] [CrossRef]
- Gruenheid, S.; de Vinney, R.; Bladt, F.; Goosney, D.; Gelkop, S.; Gish, G.D.; Pawson, T.; Finlay, B.B. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nature 2001, 3, 856–859. [Google Scholar] [CrossRef]
- Velle, K.B.; Campellone, K.G. Enteropathogenic E. coli relies on collaboration between the formin mDia1 and the Arp2/3 complex for actin pedestal biogenesis and maintenance. PLoS Pathog. 2018, 14, e1007485. [Google Scholar] [CrossRef]
- De Vinney, R.; Stein, M.; Reinscheid, D.; Abe, A.; Ruschkowski, S.; Finlay, B.B. Enterohemorrhagic Escherichia coli O157:H7 Produces Tir, Which Is Translocated to the Host Cell Membrane but Is Not Tyrosine Phosphorylated. Infect. Immun. 1999, 67, 2389–2398. [Google Scholar] [CrossRef]
- Weiss, S.M.; Ladwein, M.; Schmidt, D.; Ehinger, J.; Lommel, S.; Städing, K.; Beutling, U.; Disanza, A.; Frank, R.; Jänsch, L.; et al. IRSp53 Links the Enterohemorrhagic E. coli Effectors Tir and EspFU for Actin Pedestal Formation. Cell Host Microbe 2009, 5, 244–258. [Google Scholar] [CrossRef]
- Vingadassalom, D.; Kazlauskas, A.; Skehan, B.; Cheng, H.-C.; Magoun, L.; Robbins, D.; Rosen, M.K.; Saksela, K.; Leong, J.M. Insulin receptor tyrosine kinase substrate links the E. coli O157:H7 actin assembly effectors Tir and EspFU during pedestal formation. Proc. Natl. Acad. Sci. USA 2009, 106, 6754–6759. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.H.; Kumar, A.; Abe, C.M.; Carvalho, E.; Nishiyama-Jr, M.; Xing, C.; Sperandio, V.; Elias, W.P. EspFU-Mediated Actin Assembly Enhances Enteropathogenic Escherichia coli Adherence and Activates Host Cell Inflammatory Signaling Pathways. mBio 2020, 11, 617. [Google Scholar] [CrossRef] [PubMed]
- Frankel, G.; Lider, O.; Hershkoviz, R.; Mould, P.; Kachalsky, S.G.; Candy, D.C.A.; Cahalon, L.; Humphries, M.J.; Dougan, G. The Cell-binding Domain of Intimin from Enteropathogenic Escherichia coli Binds to β1 Integrins. J. Boil. Chem. 1996, 271, 20359–20364. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, J.F.; O’Brien, A.D. Cell Surface-localized Nucleolin Is a Eukaryotic Receptor for the Adhesin Intimin-γ of Enterohemorrhagic Escherichia coli O157:H7. J. Boil. Chem. 2001, 277, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Du, M.; Zhu, M.-J. Quercetin Prevents Escherichia coli O157:H7 Adhesion to Epithelial Cells via Suppressing Focal Adhesions. Front. Microbiol. 2019, 9, 3278. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Magoun, L.; Leong, J.M. β1-Chain Integrins Are Not Essential for Intimin-Mediated Host Cell Attachment and Enteropathogenic Escherichia coli-Induced Actin Condensation. Infect. Immun. 1999, 67, 2045–2049. [Google Scholar] [CrossRef]
- Oberhettinger, P.; Schütz, M.; Leo, J.C.; Heinz, N.; Berger, J.; Autenrieth, I.B.; Linke, D. Intimin and invasin export their C-terminus to the bacterial cell surface using an inverse mechanism compared to classical autotransport. PLoS ONE 2012, 7, e47069. [Google Scholar] [CrossRef]
- Cleary, J.; Lai, L.-C.; Shaw, R.K.; Straatman-Iwanowska, A.; Donnenberg, M.; Frankel, G.; Knutton, S. Enteropathogenic Escherichia coli (EPEC) adhesion to intestinal epithelial cells: Role of bundle-forming pili (BFP), EspA filaments and intimin. Microbiology 2004, 150, 527–538. [Google Scholar] [CrossRef]
- Saldaña, Z.; Erdem, A.L.; Schüller, S.; Okeke, I.N.; Lucas, M.; Sivananthan, A.; Phillips, A.D.; Kaper, J.; Puente, J.L.; Girón, J.A. The Escherichia coli Common Pilus and the Bundle-Forming Pilus Act in Concert during the Formation of Localized Adherence by Enteropathogenic E. coli. J. Bacteriol. 2009, 191, 3451–3461. [Google Scholar] [CrossRef]
- Santos, F.F.; Yamamoto, D.; Abe, C.M.; Bryant, J.A.; Hernandes, R.T.; Kitamura, F.C.; Castro, F.S.; Valiatti, T.B.; Piazza, R.M.F.; Elias, W.P.; et al. The Type III Secretion System (T3SS)-Translocon of Atypical Enteropathogenic Escherichia coli (aEPEC) Can Mediate Adherence. Front. Microbiol. 2019, 10, 1527. [Google Scholar] [CrossRef]
- Rey, C.; Chang, Y.-Y.; Latour-Lambert, P.; Varet, H.; Proux, C.; Legendre, R.; Coppée, J.-Y.; Enninga, J. Transcytosis subversion by M cell-to-enterocyte spread promotes Shigella flexneri and Listeria monocytogenes intracellular bacterial dissemination. PLoS Pathog. 2020, 16, e1008446. [Google Scholar] [CrossRef] [PubMed]
- Ud-Din, A.; Wahid, S. Relationship among Shigella spp. and enteroinvasive Escherichia coli (EIEC) and their differentiation. Braz. J. Microbiol. 2015, 45, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J.; Kopecko, D.J.; Formal, S.B. Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect. Immun. 1982, 35, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Pilla, G.; Tang, C.M. Going around in circles: Virulence plasmids in enteric pathogens. Nat. Rev. Genet. 2018, 16, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Tobe, T.; Nagai, S.; Okada, N.; Adter, B.; Yoshikawa, M.; Sasakawa, C. Temperature-regulated expression of invasion genes in Shigella flexneriis controlled through the transcriptional activation of the virB gene on the large plasmid. Mol. Microbiol. 1991, 5, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Marteyn, B.; West, N.P.; Browning, D.F.; Cole, J.A.; Shaw, J.G.; Palm, F.; Mounier, J.; Prévost, M.-C.; Sansonetti, P.; Tang, C.M. Modulation of Shigella virulence in response to available oxygen in vivo. Nature 2010, 465, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Killackey, S.A.; Sorbara, M.T.; Girardin, S.E. Cellular Aspects of Shigella Pathogenesis: Focus on the Manipulation of Host Cell Processes. Front. Microbiol. 2016, 6, 530. [Google Scholar] [CrossRef]
- Tinevez, J.-Y.; Arena, E.T.; Anderson, M.; Nigro, G.; Injarabian, L.; André, A.; Ferrari, M.; Campbell-Valois, F.-X.; Devin, A.; Shorte, S.L.; et al. Shigella-mediated oxygen depletion is essential for intestinal mucosa colonization. Nat. Microbiol. 2019, 4, 2001–2009. [Google Scholar] [CrossRef]
- Russo, B.C.; Stamm, L.M.; Raaben, M.; Kim, C.M.; Kahoud, E.; Robinson, L.R.; Bose, S.; Queiroz, A.L.; Herrera, B.B.; Baxt, L.A.; et al. Intermediate filaments enable pathogen docking to trigger type 3 effector translocation. Nat. Microbiol. 2016, 1, 16025. [Google Scholar] [CrossRef]
- Russo, B.C.; Duncan, J.K.; Wiscovitch, A.L.; Hachey, A.C.; Goldberg, M.B. Activation of Shigella flexneri type 3 secretion requires a host-induced conformational change to the translocon pore. PL0S Pathog. 2019, 15, e1007928. [Google Scholar] [CrossRef]
- Arizmendi, O.; Picking, W.D.; Picking, W.L. Macrophage Apoptosis Triggered by IpaD from Shigella flexneri. Infect. Immun. 2016, 84, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Skoudy, A.; Mounier, J.; Aruffo, A.; Ohayon, H.; Gounon, P.; Sansonetti, P.; Van Nhieu, G.T. CD44 binds to the Shigella IpaB protein and participates in bacterial invasion of epithelial cells. Cell. Microbiol. 2000, 2, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J.; Phalipon, A.; Arondel, J.; Thirumalai, K.; Banerjee, S.; Akira, S.; Takeda, K.; Zychlinsky, A. Caspase-1 Activation of IL-1β and IL-18 Are Essential for Shigella flexneri–Induced Inflammation. Immunity 2000, 12, 581–590. [Google Scholar] [CrossRef]
- Goldberg, M.B.; Bârzu, O.; Parsot, C.; Sansonetti, P.J. Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. J. Bacteriol. 1993, 175, 2189–2196. [Google Scholar] [CrossRef] [PubMed]
- Agaisse, H. Molecular and Cellular Mechanisms of Shigella flexneri Dissemination. Front. Microbiol. 2016, 6, 1605. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, M.; Mounier, J.; D’Hauteville, H.; Coquis-Rondon, M.; Sansonetti, P.J. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. USA 1989, 86, 3867–3871. [Google Scholar] [CrossRef]
- Goldberg, M.B.; Theriot, J.A. Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc. Natl. Acad. Sci. USA 1995, 92, 6572–6576. [Google Scholar] [CrossRef]
- Egile, C.; Loisel, T.P.; Laurent, V.; Li, R.; Pantaloni, D.; Sansonetti, P.J.; Carlier, M.-F. Activation of the Cdc42 Effector N-Wasp by the Shigella flexneri Icsa Protein Promotes Actin Nucleation by Arp2/3 Complex and Bacterial Actin-Based Motility. J. Cell Boil. 1999, 146, 1319–1332. [Google Scholar] [CrossRef]
- Suzuki, T.; Miki, H.; Takenawa, T.; Sasakawa, C. Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 1998, 17, 2767–2776. [Google Scholar] [CrossRef]
- Pope, L.M.; Reed, E.K.; Payne, S.M. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect. Immun. 1995, 63, 3642–3648. [Google Scholar] [CrossRef]
- Qin, J.; Doyle, M.T.; Tran, E.N.H.; Morona, R. The virulence domain of Shigella IcsA contains a subregion with specific host cell adhesion function. PLoS ONE 2020, 15, e0227425. [Google Scholar] [CrossRef] [PubMed]
- Köseoğlu, V.K.; Hall, C.P.; Rodríguez-López, E.M.; Agaisse, H. The Autotransporter IcsA Promotes Shigella flexneri Biofilm Formation in the Presence of Bile Salts. Infect. Immun. 2019, 87, 861. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.C.R.; Ferreira, L.G.; Martinez, M.B. Enteroinvasive Escherichia coli vs. Shigella flexneri: How different patterns of gene expression affect virulence. FEMS Microbiol. Lett. 2009, 301, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Bando, S.Y.; Moreno, A.C.R.; Albuquerque, J.A.T.; Amhaz, J.M.K.; Moreira-Filho, A.C.; Martinez, M.B. Expression of bacterial virulence factors and cytokines during in vitro macrophage infection by enteroinvasive Escherichia coli and Shigella flexneri: A comparative study. Mem. Inst. Oswaldo Cruz 2010, 105, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Parsot, C. Shigella spp. and enteroinvasive Escherichia coli pathogenicity factors. FEMS Microbiol. Lett. 2005, 252, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.; Darboe, N.; Kenjale, R.; Picking, W.L.; Middaugh, C.R.; Birket, S.; Picking, W.D. Characterization of the Interaction of Single Tryptophan Containing Mutants of IpaC from Shigella flexneri with Phospholipid Membranes. Biochemistry 2006, 45, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.; Hilbi, H. Molecular Pathogenesis of Shigella spp.: Controlling Host Cell Signaling, Invasion, and Death by Type III Secretion. Clin. Microbiol. Rev. 2008, 21, 134–156. [Google Scholar] [CrossRef]
- Jaumouillé, V.; Francetic, O.; Sansonetti, P.J.; van Nhieu, G.T. Cytoplasmic targeting of IpaC to the bacterial pole directs polar type III secretion in Shigella. EMBO J. 2008, 27, 447–457. [Google Scholar] [CrossRef]
- Di Martino, M.L.; Romilly, C.; Wagner, E.G.H.; Colonna, B.; Prosseda, G. One Gene and Two Proteins: A Leaderless mRNA Supports the Translation of a Shorter Form of the Shigella VirF Regulator. mBio 2016, 7. [Google Scholar] [CrossRef]
- Galindo, C.L.; Rosenzweig, J.A.; Kirtley, M.L.; Chopra, A.K. Pathogenesis of Y. enterocolitica and Y. pseudotuberculosis in Human Yersiniosis. J. Pathog. 2011, 2011, 1–16. [Google Scholar] [CrossRef]
- Barnes, P.D.; Bergman, M.A.; Mecsas, J.; Isberg, R.R. Yersinia pseudotuberculosis disseminates directly from a replicating bacterial pool in the intestine. J. Exp. Med. 2006, 203, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Drechsler-Hake, D.; Alamir, H.; Hahn, J.; Günter, M.; Wagner, S.; Schütz, M.S.; Bohn, E.; Schenke-Layland, K.; Pisano, F.; Dersch, P.; et al. Mononuclear phagocytes contribute to intestinal invasion and dissemination of Yersinia enterocolitica. Int. J. Med. Microbiol. 2016, 306, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Bottone, E.J. Yersinia enterocolitica: Revisitation of an Enduring Human Pathogen. Clin. Microbiol. Newsl. 2015, 37, 1–8. [Google Scholar] [CrossRef]
- Pepe, J.C.; Wachtel, M.R.; Wagar, E.; Miller, V.L. Pathogenesis of defined invasion mutants of Yersinia enterocolitica in a BALB/c mouse model of infection. Infect. Immun. 1995, 63, 4837–4848. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, G.R.; Boland, A.; Boyd, A.; Geuijen, C.; Iriarte, M.; Neyt, C.; Sory, M.-P.; Stainier, I. The Virulence Plasmid of Yersinia, an Antihost Genome. Microbiol. Mol. Boil. Rev. 1998, 62, 1315–1352. [Google Scholar] [CrossRef]
- Han, Y.W.; Miller, V.L. Reevaluation of the virulence phenotype of the inv yadA double mutants of Yersinia pseudotuberculosis. Infect. Immun. 1997, 65, 327–330. [Google Scholar] [CrossRef]
- Atkinson, S.; Williams, P. Yersinia virulence factors—A sophisticated arsenal for combating host defences. F1000 Res. 2016, 5, 1370. [Google Scholar] [CrossRef]
- Pujol, C.; Bliska, J.B. Turning Yersinia pathogenesis outside in: Subversion of macrophage function by intracellular yersiniae. Clin. Immunol. 2005, 114, 216–226. [Google Scholar] [CrossRef]
- Pujol, C.; Klein, K.A.; Romanov, G.A.; Palmer, L.E.; Cirota, C.; Zhao, Z.; Bliska, J.B. Yersinia pestis Can Reside in Autophagosomes and Avoid Xenophagy in Murine Macrophages by Preventing Vacuole Acidification. Infect. Immun. 2009, 77, 2251–2261. [Google Scholar] [CrossRef]
- Connor, M.; Pulsifer, A.R.; Price, C.T.; Abu Kwaik, Y.; Lawrenz, M.B. Yersinia pestis Requires Host Rab1b for Survival in Macrophages. PLoS Pathog. 2015, 11, e1005241. [Google Scholar] [CrossRef]
- Connor, M.; Pulsifer, A.R.; Chung, D.; Rouchka, E.C.; Ceresa, B.K.; Lawrenz, M.B. Yersinia pestis Targets the Host Endosome Recycling Pathway during the Biogenesis of the Yersinia-Containing Vacuole to Avoid Killing by Macrophages. mBio 2018, 9, e01800-17. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.J.V.; Schimmeck, H.; Gropengießer, J.; Middendorf, L.; Quitmann, M.; Schneider, C.; Holstermann, B.; Wacker, R.; Heussler, V.; Reimer, R.; et al. Activation of the macroautophagy pathway by Yersinia enterocolitica promotes intracellular multiplication and egress of yersiniae from epithelial cells. Cell. Microbiol. 2019, 21, e13046. [Google Scholar] [CrossRef]
- Besingi, R.N.; Chaney, J.L.; Clark, P.L. An alternative outer membrane secretion mechanism for an autotransporter protein lacking a C-terminal stable core. Mol. Microbiol. 2013, 90, 1028–1045. [Google Scholar] [CrossRef] [PubMed]
- Revell, P.A.; Miller, V.L. A chromosomally encoded regulator is required for expression of the Yersinia enterocolitica inv gene and for virulence. Mol. Microbiol. 2002, 35, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Avican, K.; Fahlgren, A.; Huss, M.; Heroven, A.K.; Beckstette, M.; Dersch, P.; Fällman, M. Reprogramming of Yersinia from Virulent to Persistent Mode Revealed by Complex In Vivo RNA-seq Analysis. PLoS Pathog. 2015, 11, e1004600. [Google Scholar] [CrossRef]
- Hamburger, Z.A.; Brown, M.S.; Isberg, R.R.; Bjorkman, P.J. Crystal Structure of Invasin: A Bacterial Integrin-Binding Protein. Science 1999, 286, 291–295. [Google Scholar] [CrossRef]
- Isberg, R.R.; Leong, J.M. Multiple β1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell 1990, 60, 861–871. [Google Scholar] [CrossRef]
- Clark, M.A.; Hirst, B.H.; Jepson, M.A. M-Cell Surface β1 Integrin Expression and Invasin-Mediated Targeting of Yersinia pseudotuberculosis to Mouse Peyer’s Patch M Cells. Infect. Immun. 1998, 66, 1237–1243. [Google Scholar] [CrossRef]
- Schulte, R.; Kerneis, S.; Klinke, S.; Bartels, H.; Preger, S.; Kraehenbuhl, J.; Pringault, E.; Autenrieth, I.B. Translocation of Yersinia enterocolitica across reconstituted intestinal epithelial monolayers is triggered by Yersinia invasin binding to β1 integrins apically expressed on M-like cells. Cell. Microbiol. 2000, 2, 173–185. [Google Scholar] [CrossRef]
- Dersch, P.; Isberg, R.R. A region of the Yersinia pseudotuberculosis invasin protein enhances integrin-mediated uptake into mammalian cells and promotes self-association. EMBO J. 1999, 18, 1199–1213. [Google Scholar] [CrossRef]
- Dersch, P.; Isberg, R.R. An Immunoglobulin Superfamily-Like Domain Unique to the Yersinia pseudotuberculosis Invasin Protein Is Required for Stimulation of Bacterial Uptake via Integrin Receptors. Infect. Immun. 2000, 68, 2930–2938. [Google Scholar] [CrossRef] [PubMed]
- Alrutz, M.A.; Isberg, R.R. Involvement of focal adhesion kinase in invasin-mediated uptake. Proc. Natl. Acad. Sci. USA 1998, 95, 13658–13663. [Google Scholar] [CrossRef] [PubMed]
- Alrutz, M.A.; Srivastava, A.; Wong, K.-W.; D’Souza-Schorey, C.; Tang, M.; Ch’Ng, L.-E.; Snapper, S.B.; Isberg, R.R. Efficient uptake of Yersinia pseudotuberculosis via integrin receptors involves a Rac1-Arp 2/3 pathway that bypasses N-WASP function. Mol. Microbiol. 2008, 42, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Weidow, C.L.; Black, D.S.; Bliska, J.B.; Bouton, A.H. CAS/Crk signalling mediates uptake of Yersinia into human epithelial cells. Cell. Microbiol. 2000, 2, 549–560. [Google Scholar] [CrossRef] [PubMed]
- McGee, K.; Zettl, M.; Way, M.; Fällman, M. A role for N-WASP in invasin-promoted internalisation. FEBS Lett. 2001, 509, 59–65. [Google Scholar] [CrossRef]
- Chauhan, N.; Wróbel, A.; Skurnik, M.; Leo, J. Yersinia adhesins: An arsenal for infection. Proteom. Clin. Appl. 2016, 10, 949–963. [Google Scholar] [CrossRef]
- Freund, S.; Czech, B.; Trülzsch, K.; Ackermann, N.; Heesemann, J. Unusual, Virulence Plasmid-Dependent Growth Behavior of Yersinia enterocolitica in Three-Dimensional Collagen Gels. J. Bacteriol. 2008, 190, 4111–4120. [Google Scholar] [CrossRef]
- Trunk, T.; Khalil, H.S.; Leo, J. Bacterial autoaggregation. AIMS Microbiol. 2018, 4, 140–164. [Google Scholar] [CrossRef]
- Keller, B.; Mühlenkamp, M.; Deuschle, E.; Siegfried, A.; Mössner, S.; Schade, J.; Griesinger, T.; Katava, N.; Lemberg, C.; Fehrenbacher, B.; et al. Yersinia enterocolitica exploits different pathways to accomplish adhesion and toxin injection into host cells. Cell. Microbiol. 2015, 17, 1179–1204. [Google Scholar] [CrossRef]
- Maldonado-Arocho, F.J.; Green, C.; Fisher, M.L.; Paczosa, M.K.; Mecsas, J. Adhesins and Host Serum Factors Drive Yop Translocation by Yersinia into Professional Phagocytes during Animal Infection. PLoS Pathog. 2013, 9, e1003415. [Google Scholar] [CrossRef]
- Mota, L.J.; Journet, L.; Sorg, I.; Agrain, C.; Cornelis, G.R. Bacterial Injectisomes: Needle Length Does Matter. Science 2005, 307, 1278. [Google Scholar] [CrossRef] [PubMed]
- Deuschle, E.; Keller, B.; Siegfried, A.; Manncke, B.; Spaeth, T.; Köberle, M.; Drechsler-Hake, D.; Reber, J.; Böttcher, R.T.; Autenrieth, S.E.; et al. Role of β1 integrins and bacterial adhesins for Yop injection into leukocytes in Yersinia enterocolitica systemic mouse infection. Int. J. Med. Microbiol. 2016, 306, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Eitel, J.; Dersch, P. The YadA Protein of Yersinia pseudotuberculosis Mediates High-Efficiency Uptake into Human Cells under Environmental Conditions in Which Invasin Is Repressed. Infect. Immun. 2002, 70, 4880–4891. [Google Scholar] [CrossRef] [PubMed]
- Eitel, J.; Heise, T.; Thiesen, U.; Dersch, P. Cell invasion and IL-8 production pathways initiated by YadA of Yersinia pseudotuberculosis require common signalling molecules (FAK, c-Src, Ras) and distinct cell factors. Cell. Microbiol. 2004, 7, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Felek, S.; Krukonis, E.S. The Yersinia pestis Ail Protein Mediates Binding and Yop Delivery to Host Cells Required for Plague Virulence. Infect. Immun. 2008, 77, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.M.; Felek, S.; Krukonis, E.S. Ail Binding to Fibronectin Facilitates Yersinia pestis Binding to Host Cells and Yop Delivery. Infect. Immun. 2010, 78, 3358–3368. [Google Scholar] [CrossRef]
- Bohn, E.; Sonnabend, M.; Klein, K.; Autenrieth, I.B. Bacterial adhesion and host cell factors leading to effector protein injection by type III secretion system. Int. J. Med. Microbiol. 2019, 309, 344–350. [Google Scholar] [CrossRef]
- Chen, S.; Thompson, K.; Francis, M. Environmental Regulation of Yersinia Pathophysiology. Front. Microbiol. 2016, 6, 1424. [Google Scholar] [CrossRef]
- Köberle, M.; Klein-Günther, A.; Schütz, M.; Fritz, M.; Berchtold, S.; Tolosa, E.; Autenrieth, I.B.; Bohn, E. Yersinia enterocolitica Targets Cells of the Innate and Adaptive Immune System by Injection of Yops in a Mouse Infection Model. PLoS Pathog. 2009, 5, e1000551. [Google Scholar] [CrossRef]
- Matteï, P.-J.; Faudry, E.; Job, V.; Izore, T.; Attree, I.; Dessen, A. Membrane targeting and pore formation by the type III secretion system translocon. FEBS J. 2010, 278, 414–426. [Google Scholar] [CrossRef]
- Ono, S. A plague of actin disassembly. J. Boil. Chem. 2017, 292, 8101–8102. [Google Scholar] [CrossRef] [PubMed]
- Ratner, D.; Orning, P.; Proulx, M.K.; Wang, D.; Gavrilin, M.A.; Wewers, M.D.; Alnemri, E.S.; Johnson, P.; Lee, B.; Mecsas, J.; et al. The Yersinia pestis Effector YopM Inhibits Pyrin Inflammasome Activation. PLoS Pathog. 2016, 12, e1006035. [Google Scholar] [CrossRef] [PubMed]
- Croxen, M.; Finlay, B.B. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Genet. 2009, 8, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Demeure, C.; Dussurget, O.; Fiol, G.M.; le Guern, A.-S.; Savin, C.; Pizarro-Cerdá, J. Yersinia pestis and plague: An updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics. Microbes Infect. 2019, 21, 202–212. [Google Scholar] [CrossRef]
- Bulgin, R.; Arbeloa, A.; Goulding, D.; Dougan, G.; Crepin, V.F.; Raymond, B.; Frankel, G. The T3SS Effector EspT Defines a New Category of Invasive Enteropathogenic E. coli (EPEC) Which Form Intracellular Actin Pedestals. PLoS Pathog. 2009, 5, e1000683. [Google Scholar] [CrossRef]
- Pacheco, V.C.; Yamamoto, D.; Abe, C.M.; Hernandes, R.T.; Mora, A.; Blanco, J.; Gomes, T.A. Invasion of differentiated intestinal Caco-2 cells is a sporadic property among atypical enteropathogenic Escherichia colis trains carrying common intimin subtypes. Pathog. Dis. 2013, 70, 167–175. [Google Scholar] [CrossRef]
- Ye, J.; Pan, Q.; Shang, Y.; Wei, X.; Peng, Z.; Chen, W.; Chen, L.; Wang, R. Core 2 mucin-type O-glycan inhibits EPEC or EHEC O157:H7 invasion into HT-29 epithelial cells. Gut Pathog. 2015, 7, 31. [Google Scholar] [CrossRef]
- Zychlinsky, A.; Prévost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Agbor, T.A.; McCormick, B.A. Salmonella effectors: Important players modulating host cell function during infection. Cell. Microbiol. 2011, 13, 1858–1869. [Google Scholar] [CrossRef]
- Wagner, C.; Hensel, M. Adhesive Mechanisms of Salmonella enterica. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2011; pp. 17–34. [Google Scholar]
- Stevens, M.P.; Stevens, J.M.; Jeng, R.L.; Taylor, L.A.; Wood, M.W.; Hawes, P.; Monaghan, P.; Welch, M.D.; Galyov, E.E. Identification of a bacterial factor required for actin-based motility of Burkholderia pseudomallei. Mol. Microbiol. 2005, 56, 40–53. [Google Scholar] [CrossRef]
- Benanti, E.L.; Nguyen, C.M.; Welch, M.D. Virulent Burkholderia species mimic host actin polymerases to drive actin-based motility. Cell 2015, 161, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Haglund, C.M.; Choe, J.E.; Skau, C.T.; Kovar, D.R.; Welch, M.D. Rickettsia Sca2 is a bacterial formin-like mediator of actin-based motility. Nature 2010, 12, 1057–1063. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
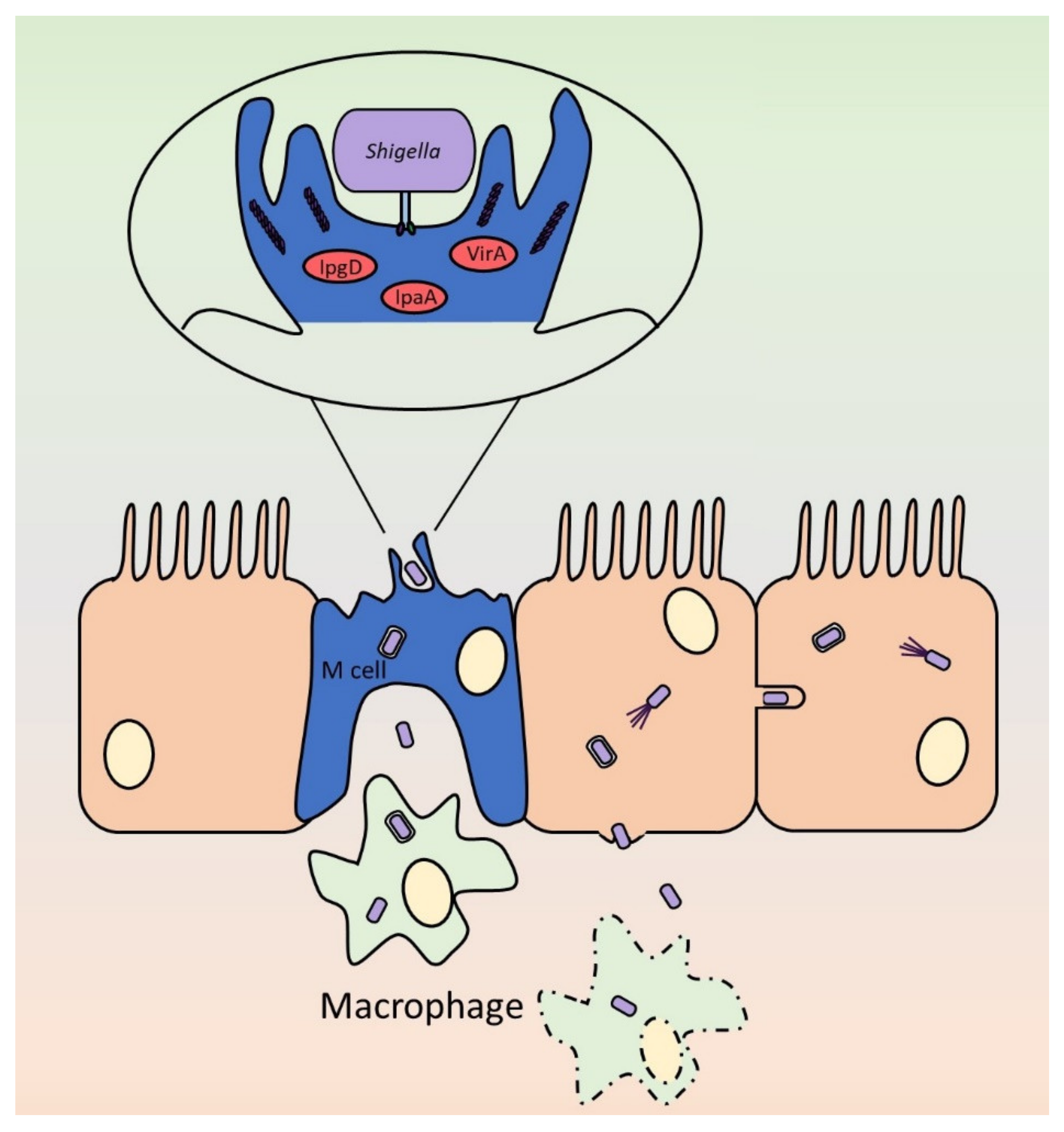{kind=link}
{kind=link}
| Sct Name a | Shigella and EIEC | Yersiniab | EPEC and EHEC | Proposed Function | Reference |
|---|---|---|---|---|---|
| − | − | − | EspA | Filamentous bridge from needle to host | [22,59] |
| − | − | − | EspF | Phagocytosis inhibition, microvillus effacement, induction of apoptosis | [53,60] |
| − | − | − | EspFU | Actin pedestal formation (EHEC only) | [61] |
| EspH | Phagocytosis inhibition via indirect Rho GTPase subversion | [53] | |||
| − | − | − | EspJ | Phagocytosis inhibition via ADP ribosylation of host kinases | [62] |
| − | − | − | EspZ | Regulation of effector translocation | [63] |
| − | IcsB | − | − | Autophagosome escape | [64] |
| − | IpaA | − | − | Binds/activates vinculin | [65] |
| SctE | IpaB | YopB | EspD | Translocon (pore forming) | [42,66,67] |
| SctB | IpaC | YopD | EspB | Translocon (pore forming; secreted effector) | [42,68,69] |
| SctA | IpaD | LcrV | − | Translocon (regulation of secretion) | [70,71] |
| − | IpaH | YopM | − | Immune subversion via E3 ubiquitin ligase activity | [72,73] |
| − | IpaJ | − | − | Golgi body fragmentation | [74] |
| − | IpgB1/2 | − | EspM/EspT/Map | Host cytoskeleton remodelling via Rho GTPase activity | [75,76] |
| − | IpgD | − | − | Host cytoskeleton remodelling via ARF6 activation | [77] |
| − | − | − | LifA/Efa1 (lymphostatin) | Adhesin/toxin | [78] |
| − | OspB | − | − | Reduced cytokine production | [79] |
| − | OspC1 | − | − | Increased neutrophil migration (bacterial access to submucosa) | [80] |
| − | OspC3 | − | − | Pyroptosis inhibition via caspase-4 interference | [81] |
| − | OspD1 | − | − | Regulation of secretion | [82] |
| − | OspD2 | − | EspL c | Pyroptosis inhibition via regulation of VirA translocation | [83,84] |
| − | OspD3 (ShET2) | − | EspL2 d | Enterotoxin | [85,86] |
| − | OspE1/2 | − | EspO1/2 | Adhesin; actin stabilisation | [87,88] |
| − | OspF | − | − | Reduced cytokine production and increased neutrophil migration | [79,80] |
| − | OspG | YspK | NleH1/2 | NF-κB attenuation via ubiquitin binding | [89] |
| − | OspI | CHYP | Cif | Inhibits TRAF6 activation via UBC13 deamidation | [90,91] |
| − | OspZ | NleE | Increased neutrophil migration (bacterial access to submucosa) | [92] | |
| − | − | − | Tir | Actin pedestal formation | [93] |
| − | VirA | − | EspG | Autophagosome escape | [64] |
| − | − | YopE | − | Regulation; Rho GTPase activation | [94] |
| − | − | YopH | − | Immune subversion via phosphotyrosine phosphatase activity | [95] |
| − | − | YopP (YopJ) | − | Immune subversion via NF-κB attenuation | [96] |
| − | − | YopO (YpkA) | − | Prevents phagocytosis via phosphorylation of actin regulators | [97] |
| − | − | YopT | − | Immune subversion via Rho GTPase cleavage | [98] |
| Organisms | Autotransporters | T5SS Subclass | Proposed Function | Reference |
|---|---|---|---|---|
| EHEC, EPEC | ||||
| Cah | 5a | Autoaggregation and biofilm formation | [107] | |
| EhaA | 5a | Autoaggregation and biofilm formation | [108] | |
| EhaB | 5a | Binding to extracellular matrix (ECM) molecules, biofilm formation | [109] | |
| EhaD a | 5a | Biofilm formation | [108] | |
| EhaG | 5c | Binding to ECM molecules, biofilm formation | [110] | |
| EhaJ | 5a | Binding to ECM molecules, biofilm formation | [111] | |
| EspC b | 5a | Regulation of T3SS secretion, cytotoxin | [112] | |
| EspP a | 5a | Regulation of T3SS, cleavage of coagulation and complement factors, formation of rope-like adhesive aggregates | [112] | |
| FdeC | 5e | Binding to host cells and ECM molecules, autoaggregation and biofilm formation | [113] | |
| IatB | 5e | Biofilm formation | [114] | |
| IatD | 5e | Biofilm formation | [114] | |
| Intimin | 5e | Binding to Tir; actin pedestal formation | [115] | |
| YeeJ | 5e | Biofilm formation | [116] | |
| EIEC, Shigella sp. | ||||
| IcsA | 5a | Adhesion to host cells; actin-based intracellular motility | [117] | |
| Pic | 5a | Mucin degradation, serum resistance | [112] | |
| Sat c | 5a | Cytotoxin | [112] | |
| SepA c | 5a | Disruption of apical pole of polarized epithelial cells, facilitation of invasion | [112] | |
| SigA c | 5a | Cytotoxin | [112] | |
| Yersinia enterocolitica, Y. pseudotuberculosis | ||||
| Invasin (InvA) | 5e | Binding to β1 integrins; invasion of M cells | [118] | |
| Ifp (InvB) d | 5e | Binding to and invasion of epithelial cells | [119] | |
| InvC | 5e | Binding to epithelial cells | [120] | |
| InvD d | 5e | Binding to Fab fragment of immunoglobulins, binding to B cells | [121] | |
| YadA | 5c | Binding to ECM molecules and serum components; host cell adhesion and immune evasion | [122] | |
| YapC d | 5a | Autoaggregation and biofilm formation, binding to host cells | [123] | |
| YapE | 5a | Autoaggregation, binding to host cells | [124] | |
| YapV d | 5a | Binding to N-WASP and ECM molecules | [125] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whelan, R.; McVicker, G.; Leo, J.C. Staying out or Going in? The Interplay between Type 3 and Type 5 Secretion Systems in Adhesion and Invasion of Enterobacterial Pathogens. Int. J. Mol. Sci. 2020, 21, 4102. https://doi.org/10.3390/ijms21114102
Whelan R, McVicker G, Leo JC. Staying out or Going in? The Interplay between Type 3 and Type 5 Secretion Systems in Adhesion and Invasion of Enterobacterial Pathogens. International Journal of Molecular Sciences. 2020; 21(11):4102. https://doi.org/10.3390/ijms21114102
Chicago/Turabian StyleWhelan, Rachel, Gareth McVicker, and Jack C. Leo. 2020. "Staying out or Going in? The Interplay between Type 3 and Type 5 Secretion Systems in Adhesion and Invasion of Enterobacterial Pathogens" International Journal of Molecular Sciences 21, no. 11: 4102. https://doi.org/10.3390/ijms21114102
APA StyleWhelan, R., McVicker, G., & Leo, J. C. (2020). Staying out or Going in? The Interplay between Type 3 and Type 5 Secretion Systems in Adhesion and Invasion of Enterobacterial Pathogens. International Journal of Molecular Sciences, 21(11), 4102. https://doi.org/10.3390/ijms21114102