Maternal Immune Activation Induces Neuroinflammation and Cortical Synaptic Deficits in the Adolescent Rat Offspring

 ,
,  ,
,  , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
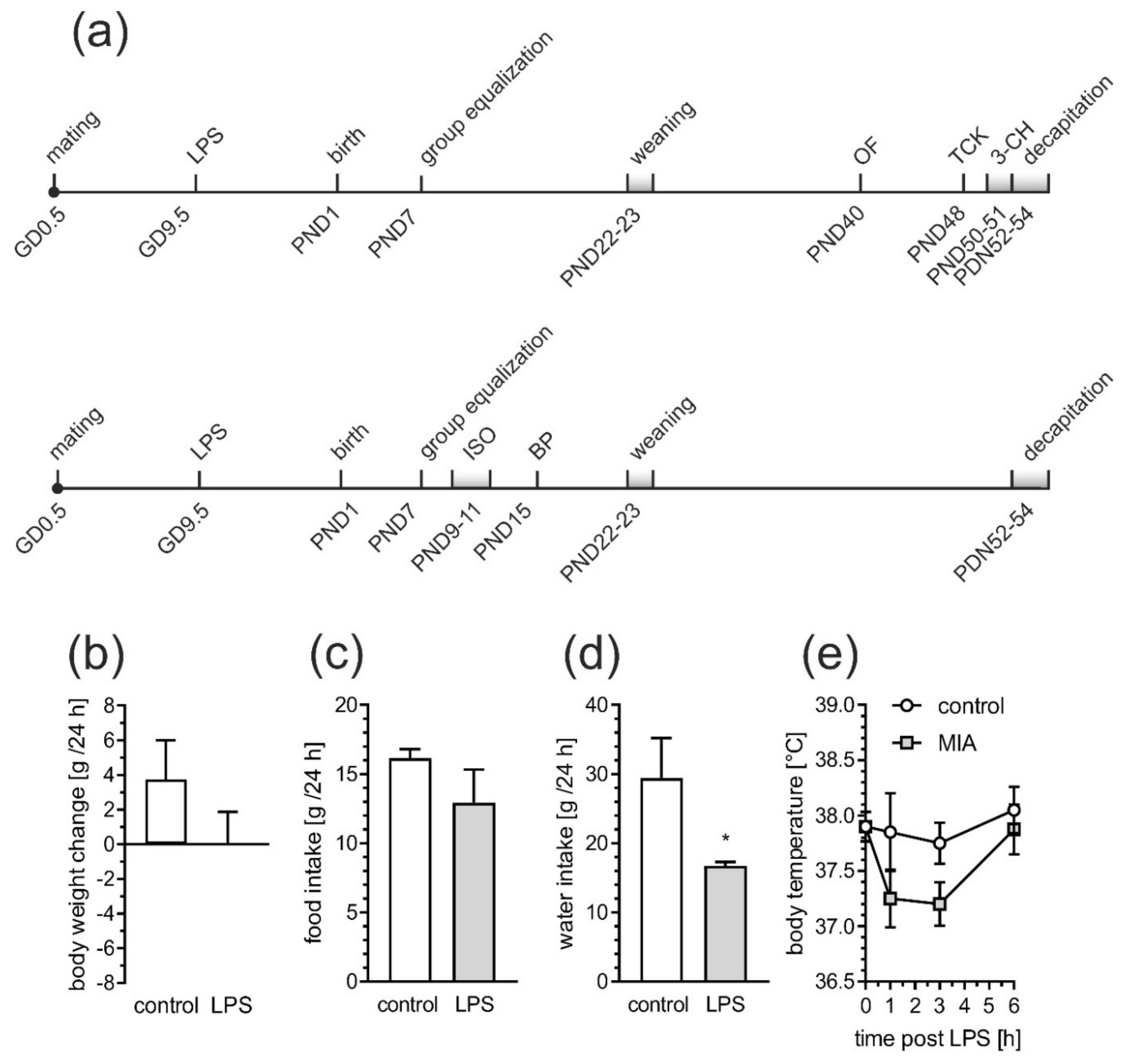
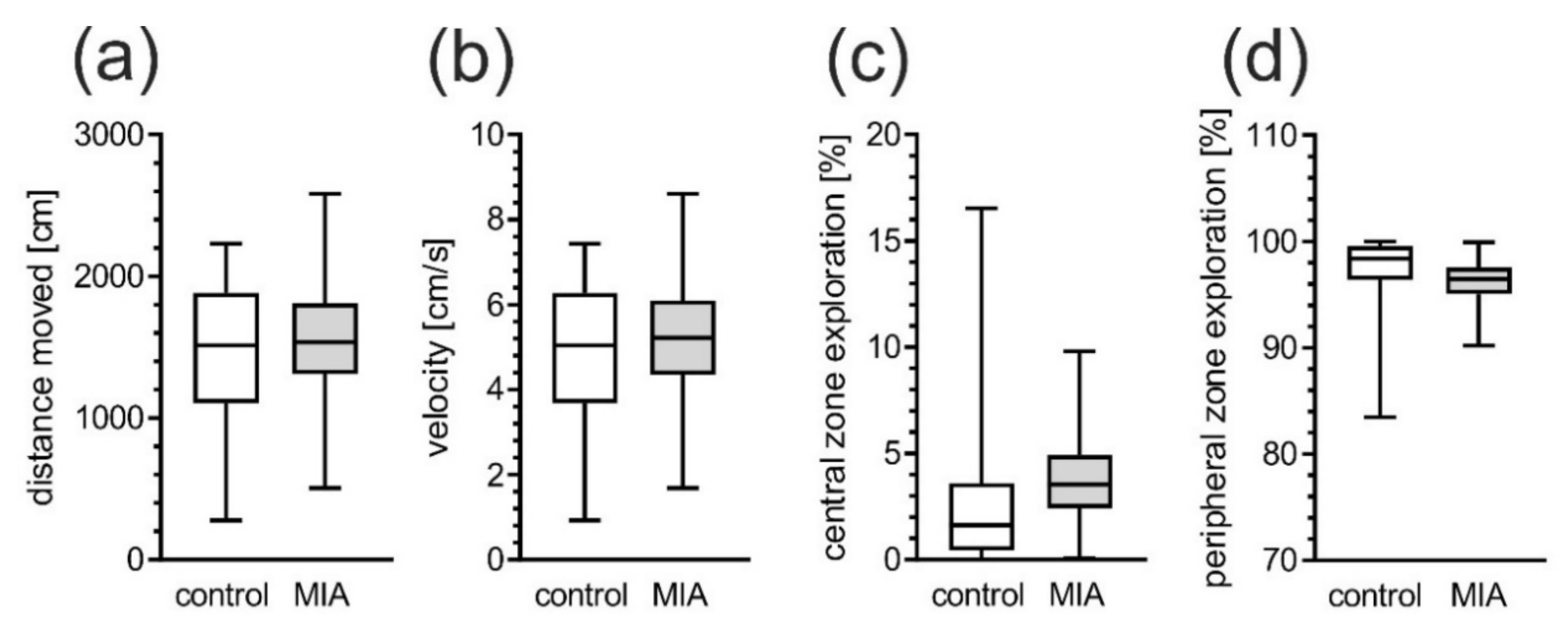
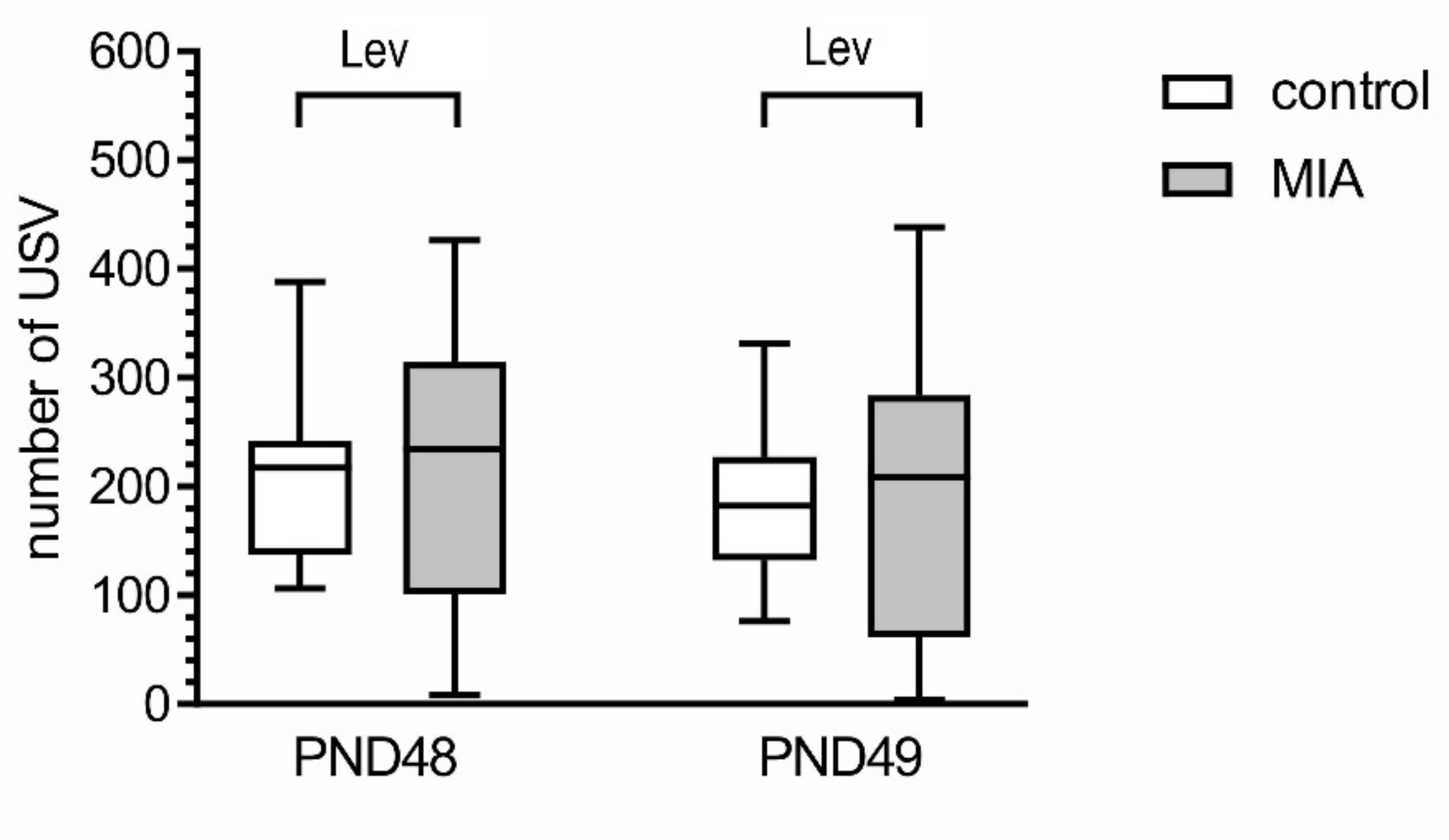
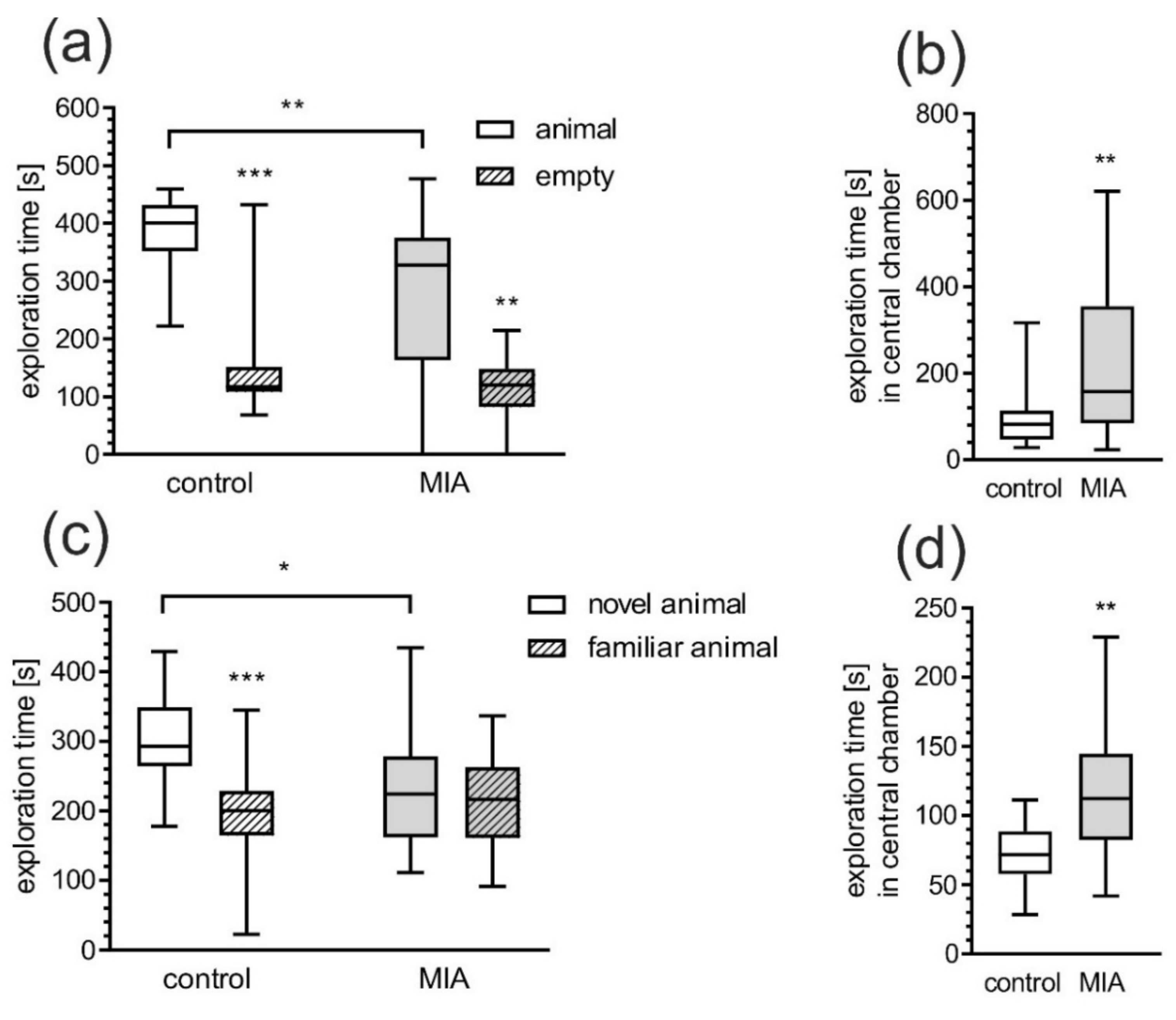
2.1. Maternal Immune Activation Alters Behavioral Phenotypes in Rat Offspring
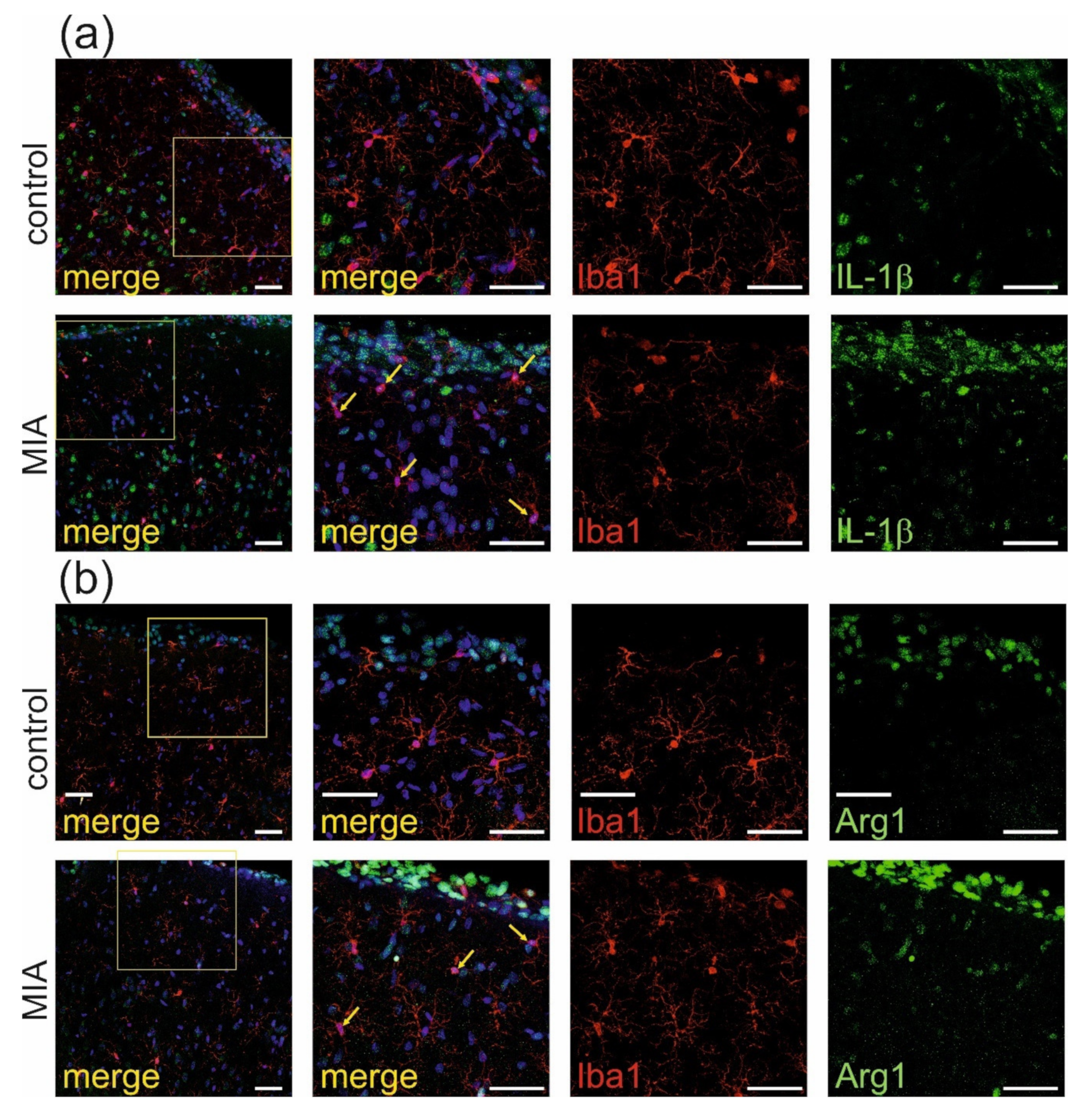
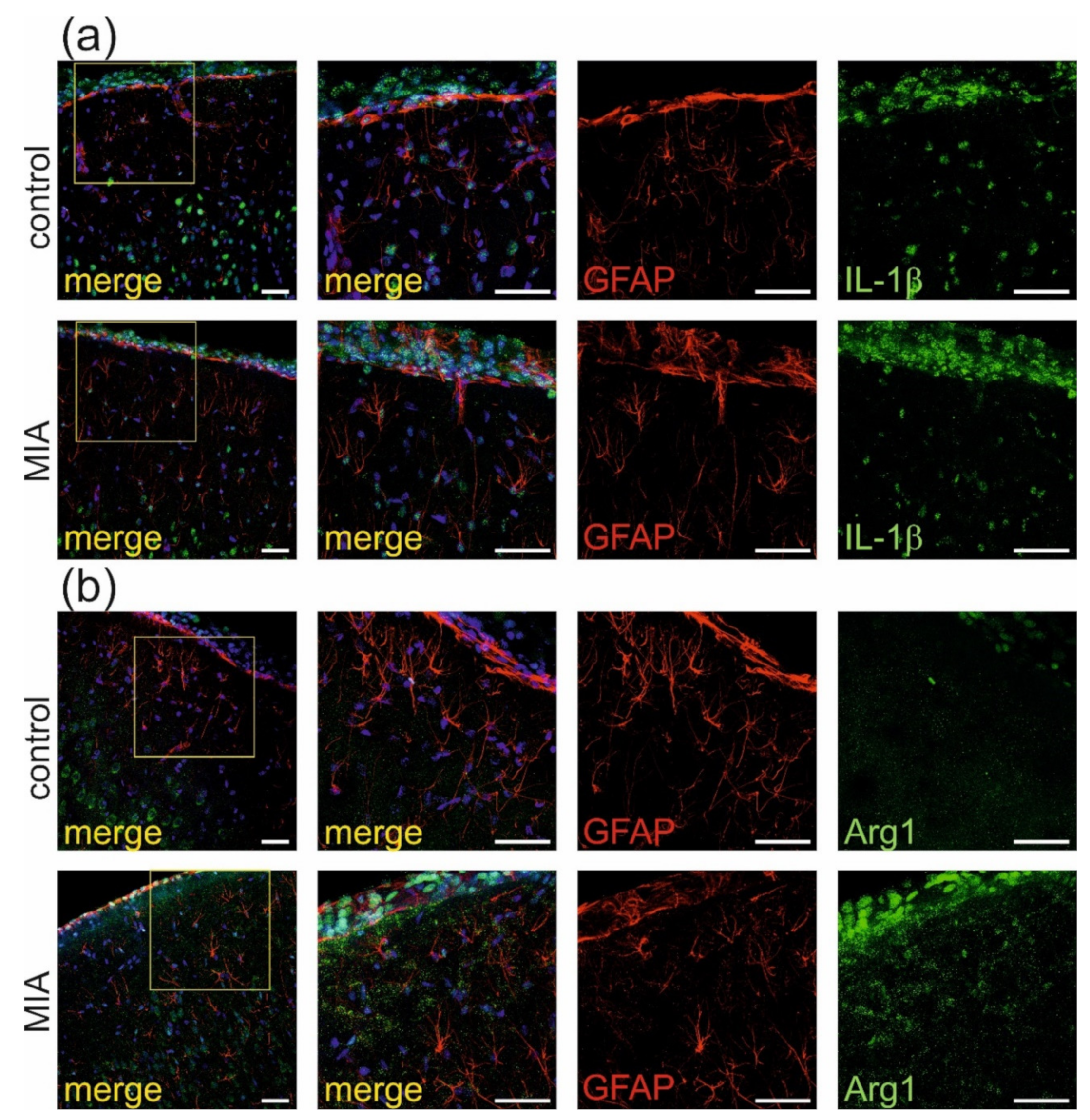
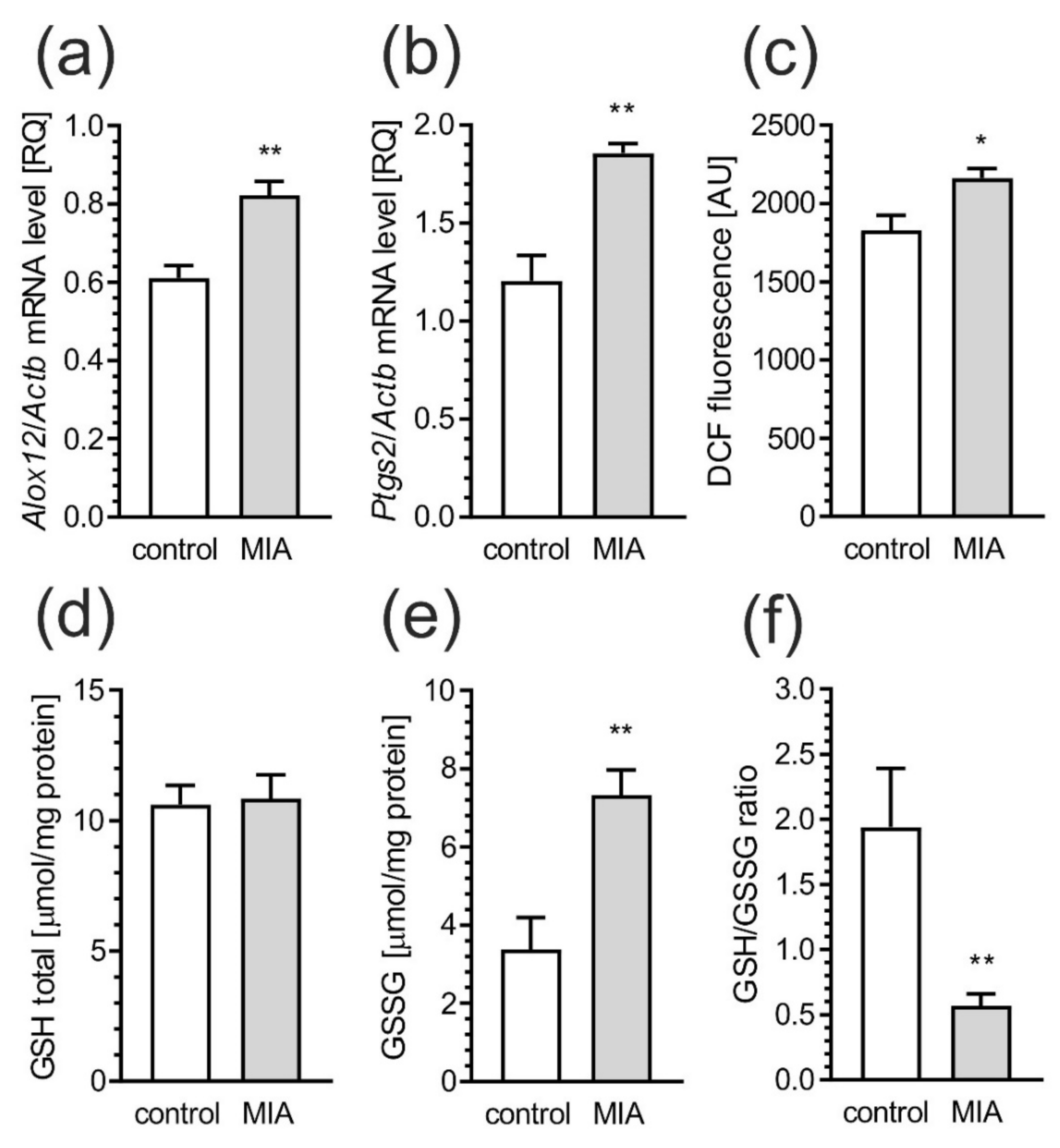
2.2. Maternal Immune Activation Increases Neuroinflammation and Oxidative Stress in Cerebral Cortex of Rat Offspring
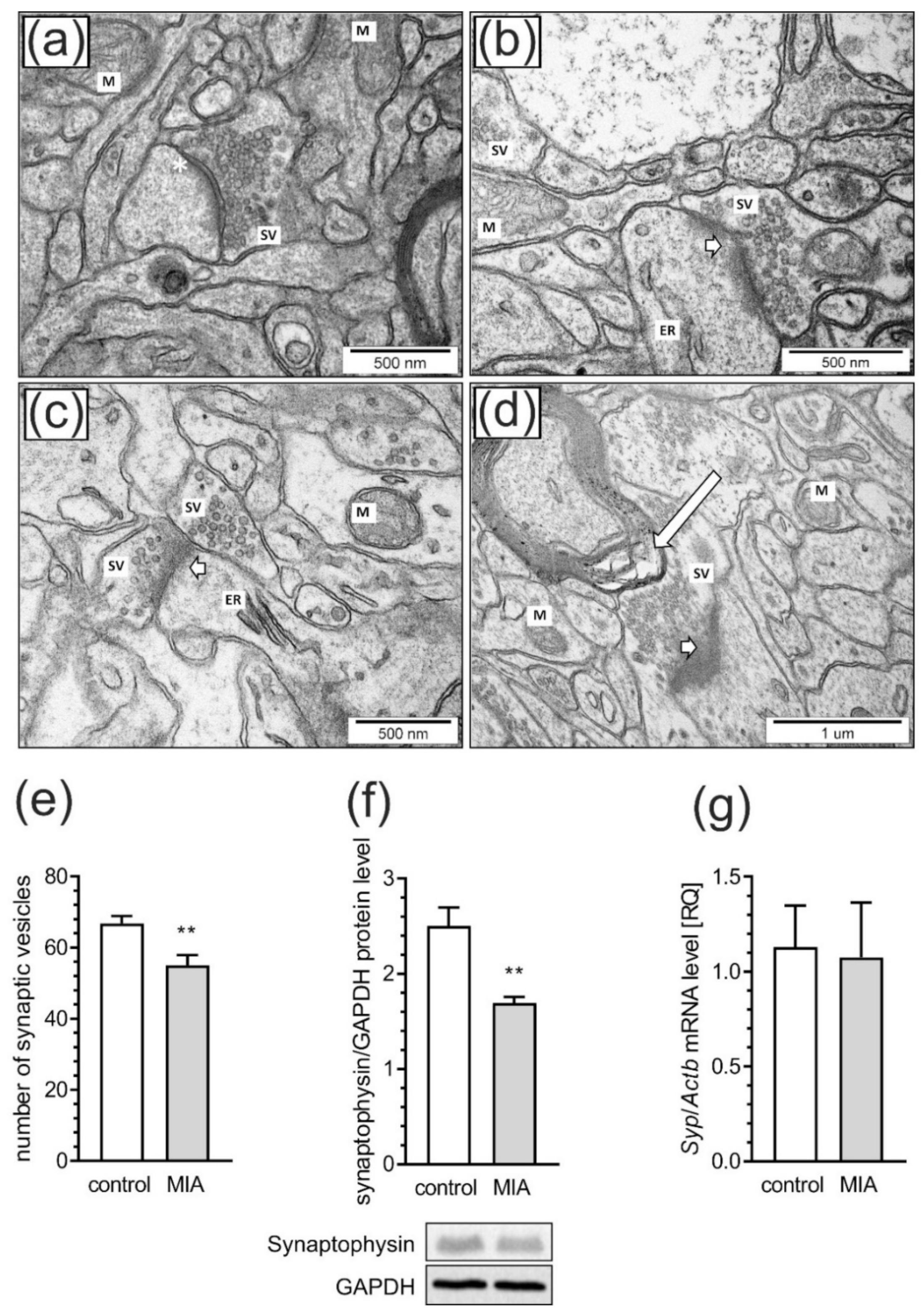
2.3. Maternal Immune Activation Induces Ultrastructural Changes in the Cerebral Cortex of Rat Offspring
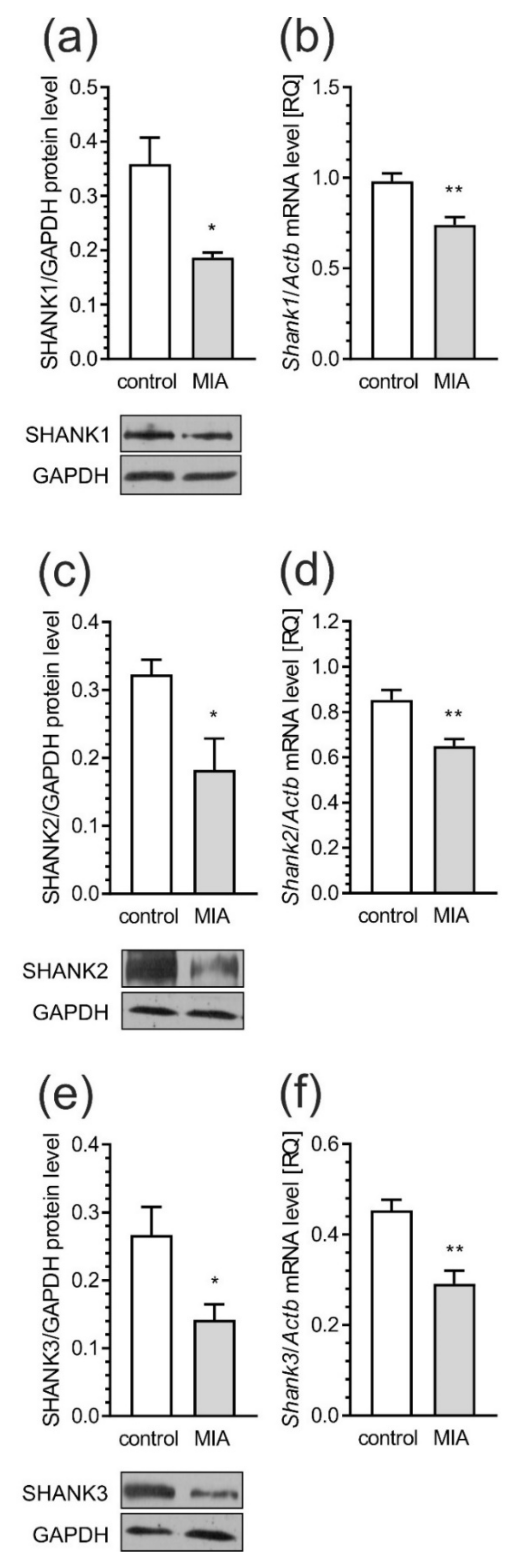
2.4. Maternal Immune Activation Alters Pre- and Postsynaptic Protein Levels in the Cerebral Cortex of Rat Offspring
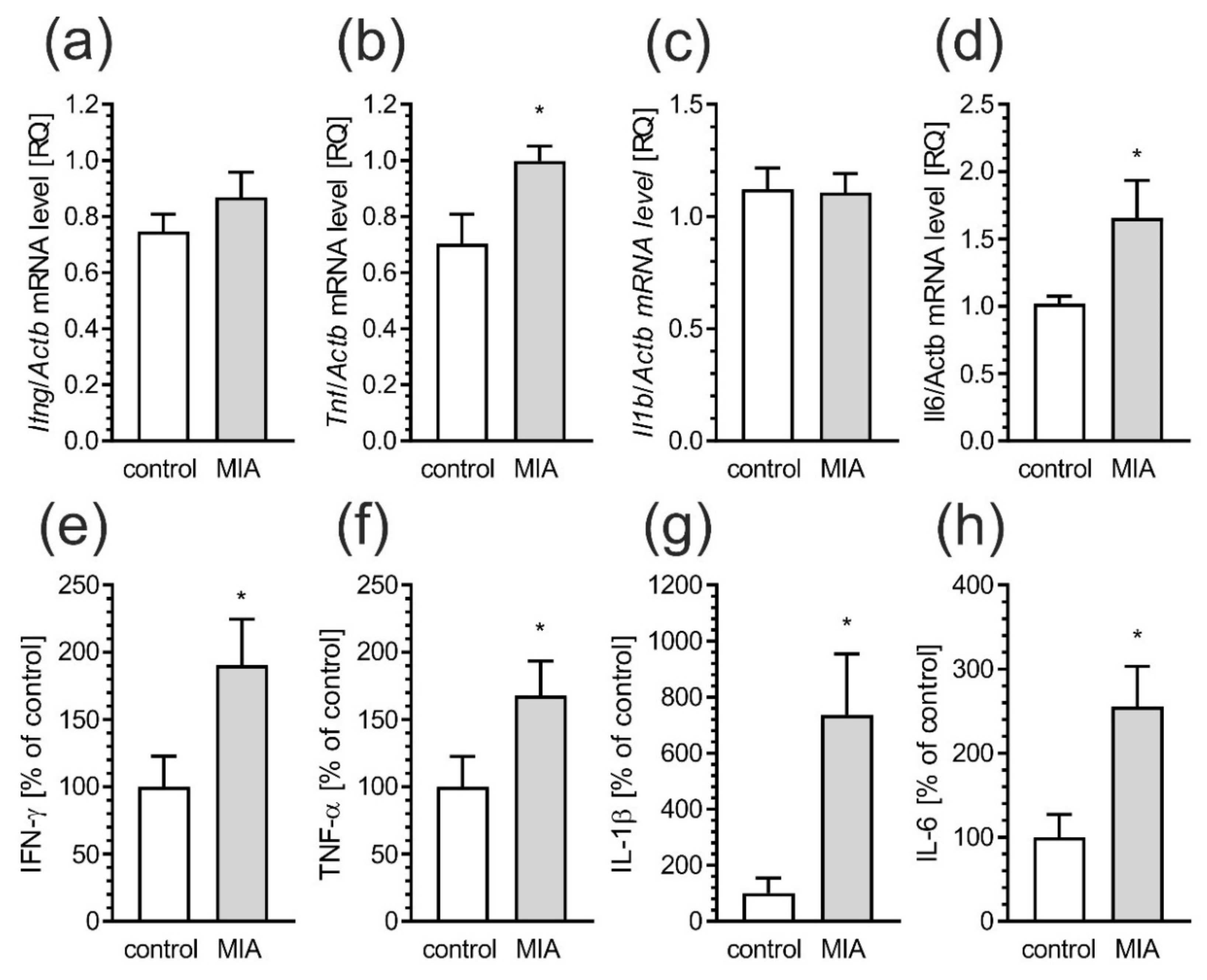
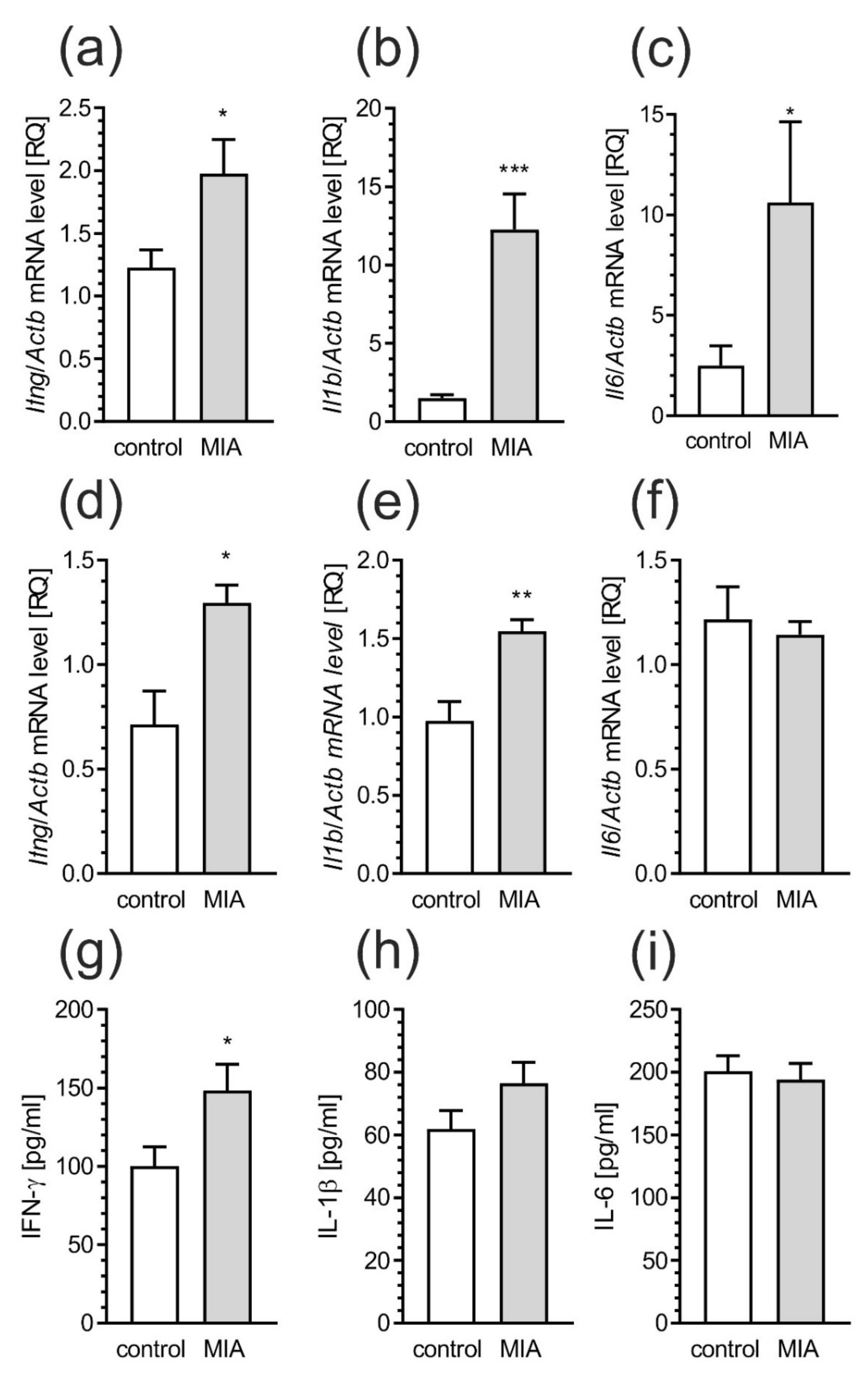
2.5. Maternal Immune Activation Induces Neuroinflammatory Responses in Fetuses and in the Offspring Pups
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Animals and the MIA Model
4.3. Behavioral Analysis
4.3.1. Open Field Test
4.3.2. Play Behavior (Tickling, TCK)
4.3.3. 3-Chamber Social Interaction Test (Crawley’s Sociability and Preference for Social Novelty Test)
4.4. Transmission Electron Microscopy (TEM) Analysis
4.5. Determination of Gene Expression (Real-Time PCR)
4.6. Measurement of Cytokine Levels in Brain Tissue Extract
4.7. Confocal Laser Scanning Analysis (Immunohistochemistry)
4.8. Measurement of the Reactive Oxygen Species (ROS) Level
4.9. Determination of Glutathione Levels
4.10. Determination of Protein Level (BCA Method)
4.11. Immunochemical Determination of Protein Levels (Western Blot)
4.12. Statistical Analysis of Biochemical and Behavioral Results
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 12-LOX. | 12-lipoxygenase |
| ASD | autism spectrum disorders |
| COX-2 | cyclooxygenase-2 |
| DCF | 2′,7′-dichlorofluorescein |
| DCFH | 2′,7′-dichlorodihydrofluorescein |
| DCFH-DA | 2′,7′-dichlorodihydrofluorescein diacetate |
| EM | electron microscopy |
| GSH | glutathione |
| GSSG | oxidized glutathione |
| IL-6 | interleukin 6 |
| ISO | juvenile isolation |
| LPS | lipopolysaccharide |
| MIA | maternal immune activation |
| NF-κB | nuclear factor κB |
| PND | postnatal day |
| poly(I:C) | polyinosinic-polycytidylic acid |
| PSD | post-synaptic density |
| PSD-95 | post-synaptic density protein 95 |
| ROS | reactive oxygen species |
| SHANK | SH3 and multiple ankyrin repeat domains protein |
| SNAP-25 | synaptosomal-associated protein 25 |
| SNARE | soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| Stx-1 | syntaxin-1 |
| SV | synaptic vesicle(s) |
| Syp | synaptophysin |
| TCK | tickling test |
| TEM | transmission electron microscopy |
| TNF-α | tumor necrosis factor alpha |
| USV | ultrasonic vocalizations |
| VAMP1/2 | vesicle-associated membrane protein 1/2 |
References
- Reus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.; Pearlman, D.M.; Alper, K.; Najjar, A.; Devinsky, O. Neuroinflammation and psychiatric illness. J. Neuroinflamm. 2013, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.; Norton, S.; Fox, N.; Kusnecov, A.W. Maternal immune activation with staphylococcal enterotoxin A produces unique behavioral changes in C57BL/6 mouse offspring. Brain Behav. Immun. 2019, 75, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Stolp, H.B. Neuropoietic cytokines in normal brain development and neurodevelopmental disorders. Mol. Cell. Neurosci. 2013, 53, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.M.; Cowan, M.; Moonah, S.N.; Petri, W.A., Jr. The Impact of Systemic Inflammation on Neurodevelopment. Trends Mol. Med. 2018, 24, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Abbott, P.W.; Gumusoglu, S.B.; Bittle, J.; Beversdorf, D.Q.; Stevens, H.E. Prenatal stress and genetic risk: How prenatal stress interacts with genetics to alter risk for psychiatric illness. Psychoneuroendocrinology 2018, 90, 9–21. [Google Scholar] [CrossRef]
- Angelidou, A.; Asadi, S.; Alysandratos, K.D.; Karagkouni, A.; Kourembanas, S.; Theoharides, T.C. Perinatal stress, brain inflammation and risk of autism-review and proposal. BMC Pediatrics 2012, 12, 89. [Google Scholar] [CrossRef]
- Meyer, U.; Feldon, J.; Dammann, O. Schizophrenia and autism: Both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr. Res. 2011, 69, 26r–33r. [Google Scholar] [CrossRef]
- Parker-Athill, E.C.; Tan, J. Maternal immune activation and autism spectrum disorder: Interleukin-6 signaling as a key mechanistic pathway. Neuro-Signals 2010, 18, 113–128. [Google Scholar] [CrossRef]
- Kirsten, T.B.; Bernardi, M.M. Prenatal lipopolysaccharide induces hypothalamic dopaminergic hypoactivity and autistic-like behaviors: Repetitive self-grooming and stereotypies. Behav. Brain Res. 2017, 331, 25–29. [Google Scholar] [CrossRef]
- Kirsten, T.B.; Chaves-Kirsten, G.P.; Bernardes, S.; Scavone, C.; Sarkis, J.E.; Bernardi, M.M.; Felicio, L.F. Lipopolysaccharide Exposure Induces Maternal Hypozincemia, and Prenatal Zinc Treatment Prevents Autistic-Like Behaviors and Disturbances in the Striatal Dopaminergic and mTOR Systems of Offspring. PLoS ONE 2015, 10, e0134565. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, T.B.; Chaves-Kirsten, G.P.; Chaible, L.M.; Silva, A.C.; Martins, D.O.; Britto, L.R.; Dagli, M.L.; Torrao, A.S.; Palermo-Neto, J.; Bernardi, M.M. Hypoactivity of the central dopaminergic system and autistic-like behavior induced by a single early prenatal exposure to lipopolysaccharide. J. Neurosci. Res. 2012, 90, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Missault, S.; Van den Eynde, K.; Vanden Berghe, W.; Fransen, E.; Weeren, A.; Timmermans, J.P.; Kumar-Singh, S.; Dedeurwaerdere, S. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav. Immun. 2014, 42, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, S.; Khan, D.; Kong, E.; Berger, A.; Pollak, A.; Pollak, D.D. The poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol. Ther. 2015, 149, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Wischhof, L.; Irrsack, E.; Osorio, C.; Koch, M. Prenatal LPS-exposure—A neurodevelopmental rat model of schizophreni—Differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 57, 17–30. [Google Scholar] [CrossRef]
- Tetreault, N.A.; Hakeem, A.Y.; Jiang, S.; Williams, B.A.; Allman, E.; Wold, B.J.; Allman, J.M. Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 2012, 42, 2569–2584. [Google Scholar] [CrossRef]
- Radewicz, K.; Garey, L.J.; Gentleman, S.M.; Reynolds, R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J. Neuropathol. Exp. Neurol. 2000, 59, 137–150. [Google Scholar] [CrossRef]
- Beversdorf, D.Q. Phenotyping, Etiological Factors, and Biomarkers: Toward Precision Medicine in Autism Spectrum Disorders. J. Dev. Behav. Pediatr. JDBP 2016, 37, 659–673. [Google Scholar] [CrossRef]
- Blundell, J.; Blaiss, C.A.; Etherton, M.R.; Espinosa, F.; Tabuchi, K.; Walz, C.; Bolliger, M.F.; Sudhof, T.C.; Powell, C.M. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 2115–2129. [Google Scholar] [CrossRef]
- Giovedi, S.; Corradi, A.; Fassio, A.; Benfenati, F. Involvement of synaptic genes in the pathogenesis of autism spectrum disorders: The case of synapsins. Front. Pediatr. 2014, 2, 94. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y. Postnatal neurodevelopmental disorders: Meeting at the synapse? Science (New York, N.Y.) 2003, 302, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C. Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Res. 2011, 1380, 42–77. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef]
- Shishido, E. Autism spectrum disorder and genes for synaptic proteins. Brain Nerve = Shinkei Kenkyu No Shinpo 2012, 64, 65–70. [Google Scholar]
- Veenstra-VanderWeele, J.; Blakely, R.D. Networking in autism: Leveraging genetic, biomarker and model system findings in the search for new treatments. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2012, 37, 196–212. [Google Scholar] [CrossRef]
- Betancur, C.; Sakurai, T.; Buxbaum, J.D. The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 2009, 32, 402–412. [Google Scholar] [CrossRef]
- Craig, A.M.; Kang, Y. Neurexin-neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 2007, 17, 43–52. [Google Scholar] [CrossRef]
- Iasevoli, F.; Tomasetti, C.; de Bartolomeis, A. Scaffolding proteins of the post-synaptic density contribute to synaptic plasticity by regulating receptor localization and distribution: Relevance for neuropsychiatric diseases. Neurochem. Res. 2013, 38, 1–22. [Google Scholar] [CrossRef]
- Sheng, M.; Kim, E. The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Verpelli, C.; Schmeisser, M.J.; Sala, C.; Boeckers, T.M. Scaffold proteins at the postsynaptic density. Adv. Exp. Med. Biol. 2012, 970, 29–61. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, M.V.; Moon, H.M.; Su, J.; Palmer, T.D.; Courchesne, E.; Pramparo, T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry 2018, 23, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Haida, O.; Al Sagheer, T.; Balbous, A.; Francheteau, M.; Matas, E.; Soria, F.; Fernagut, P.O.; Jaber, M. Sex-dependent behavioral deficits and neuropathology in a maternal immune activation model of autism. Transl. Psychiatry 2019, 9, 124. [Google Scholar] [CrossRef]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef]
- Atladottir, H.O.; Henriksen, T.B.; Schendel, D.E.; Parner, E.T. Autism after infection, febrile episodes, and antibiotic use during pregnancy: An exploratory study. Pediatrics 2012, 130, e1447–e1454. [Google Scholar] [CrossRef]
- Brown, A.S.; Sourander, A.; Hinkka-Yli-Salomaki, S.; McKeague, I.W.; Sundvall, J.; Surcel, H.M. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol. Psychiatry 2014, 19, 259–264. [Google Scholar] [CrossRef]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef]
- Canetta, S.; Sourander, A.; Surcel, H.M.; Hinkka-Yli-Salomaki, S.; Leiviska, J.; Kellendonk, C.; McKeague, I.W.; Brown, A.S. Elevated maternal C-reactive protein and increased risk of schizophrenia in a national birth cohort. Am. J. Psychiatry 2014, 171, 960–968. [Google Scholar] [CrossRef]
- Canetta, S.E.; Bao, Y.; Co, M.D.; Ennis, F.A.; Cruz, J.; Terajima, M.; Shen, L.; Kellendonk, C.; Schaefer, C.A.; Brown, A.S. Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am. J. Psychiatry 2014, 171, 557–563. [Google Scholar] [CrossRef]
- Parboosing, R.; Bao, Y.; Shen, L.; Schaefer, C.A.; Brown, A.S. Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry 2013, 70, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Fernandez de Cossio, L.; Guzman, A.; van der Veldt, S.; Luheshi, G.N. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav. Immun. 2017, 63, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Giovanoli, S.; Weber-Stadlbauer, U.; Schedlowski, M.; Meyer, U.; Engler, H. Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain Behav. Immun. 2016, 55, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Nyffeler, M.; Yee, B.K.; Knuesel, I.; Feldon, J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav. Immun. 2008, 22, 469–486. [Google Scholar] [CrossRef]
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 551–563. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; O’Neill, L.A. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 2004, 113, 153–162. [Google Scholar] [CrossRef]
- Enayati, M.; Solati, J.; Hosseini, M.H.; Shahi, H.R.; Saki, G.; Salari, A.A. Maternal infection during late pregnancy increases anxiety- and depression-like behaviors with increasing age in male offspring. Brain Res. Bull. 2012, 87, 295–302. [Google Scholar] [CrossRef]
- Meyer, U.; Nyffeler, M.; Engler, A.; Urwyler, A.; Schedlowski, M.; Knuesel, I.; Yee, B.K.; Feldon, J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 4752–4762. [Google Scholar] [CrossRef]
- Morgan, K.N.; Thayer, J.E.; Frye, C.A. Prenatal stress suppresses rat pup ultrasonic vocalization and myoclonic twitching in response to separation. Dev. Psychobiol. 1999, 34, 205–215. [Google Scholar] [CrossRef]
- Pendyala, G.; Chou, S.; Jung, Y.; Coiro, P.; Spartz, E.; Padmashri, R.; Li, M.; Dunaevsky, A. Maternal Immune Activation Causes Behavioral Impairments and Altered Cerebellar Cytokine and Synaptic Protein Expression. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2017, 42, 1435–1446. [Google Scholar] [CrossRef]
- Tordjman, S.; Drapier, D.; Bonnot, O.; Graignic, R.; Fortes, S.; Cohen, D.; Millet, B.; Laurent, C.; Roubertoux, P.L. Animal models relevant to schizophrenia and autism: Validity and limitations. Behav. Genet. 2007, 37, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Schwendener, S.; Meyer, U.; Feldon, J. Deficient maternal care resulting from immunological stress during pregnancy is associated with a sex-dependent enhancement of conditioned fear in the offspring. J. Neurodev. Disord. 2009, 1, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Schaafsma, W.; Basterra, L.B.; Jacobs, S.; Brouwer, N.; Meerlo, P.; Schaafsma, A.; Boddeke, E.; Eggen, B.J.L. Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol. Dis. 2017, 106, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Baharnoori, M.; Bhardwaj, S.K.; Srivastava, L.K. Neonatal behavioral changes in rats with gestational exposure to lipopolysaccharide: A prenatal infection model for developmental neuropsychiatric disorders. Schizophr. Bull. 2012, 38, 444–456. [Google Scholar] [CrossRef]
- Hava, G.; Vered, L.; Yael, M.; Mordechai, H.; Mahoud, H. Alterations in behavior in adult offspring mice following maternal inflammation during pregnancy. Dev. Psychobiol. 2006, 48, 162–168. [Google Scholar] [CrossRef]
- Goldman, S.; O’Brien, L.M.; Filipek, P.A.; Rapin, I.; Herbert, M.R. Motor stereotypies and volumetric brain alterations in children with Autistic Disorder. Res. Autism Spectr. Disord. 2013, 7, 82–92. [Google Scholar] [CrossRef]
- Mody, M.; Belliveau, J.W. Speech and Language Impairments in Autism: Insights from Behavior and Neuroimaging. N. Am. J. Med. Sci. 2013, 5, 157–161. [Google Scholar] [CrossRef]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; Van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef]
- Molloy, C.A.; Morrow, A.L.; Meinzen-Derr, J.; Schleifer, K.; Dienger, K.; Manning-Courtney, P.; Altaye, M.; Wills-Karp, M. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol. 2006, 172, 198–205. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Ling, Z.; Gayle, D.A.; Ma, S.Y.; Lipton, J.W.; Tong, C.W.; Hong, J.S.; Carvey, P.M. In utero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neurons in the postnatal rat midbrain. Mov. Disord. Off. J. Mov. Disord. Soc. 2002, 17, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Ashdown, H.; Dumont, Y.; Ng, M.; Poole, S.; Boksa, P.; Luheshi, G.N. The role of cytokines in mediating effects of prenatal infection on the fetus: Implications for schizophrenia. Mol. Psychiatry 2006, 11, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Liverman, C.S.; Kaftan, H.A.; Cui, L.; Hersperger, S.G.; Taboada, E.; Klein, R.M.; Berman, N.E. Altered expression of pro-inflammatory and developmental genes in the fetal brain in a mouse model of maternal infection. Neurosci. Lett. 2006, 399, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Oskvig, D.B.; Elkahloun, A.G.; Johnson, K.R.; Phillips, T.M.; Herkenham, M. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav. Immun. 2012, 26, 623–634. [Google Scholar] [CrossRef]
- Urakubo, A.; Jarskog, L.F.; Lieberman, J.A.; Gilmore, J.H. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr. Res. 2001, 47, 27–36. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; Patterson, P.H. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun. 2011, 25, 604–615. [Google Scholar] [CrossRef]
- Lundberg, A.M.; Drexler, S.K.; Monaco, C.; Williams, L.M.; Sacre, S.M.; Feldmann, M.; Foxwell, B.M. Key differences in TLR3/poly I:C signaling and cytokine induction by human primary cells: A phenomenon absent from murine cell systems. Blood 2007, 110, 3245–3252. [Google Scholar] [CrossRef]
- Kimura, M.; Toth, L.A.; Agostini, H.; Cady, A.B.; Majde, J.A.; Krueger, J.M. Comparison of acute phase responses induced in rabbits by lipopolysaccharide and double-stranded RNA. Am. J. Physiol. 1994, 267, R1596–R1605. [Google Scholar] [CrossRef]
- Chez, M.G.; Dowling, T.; Patel, P.B.; Khanna, P.; Kominsky, M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatric Neurol. 2007, 36, 361–365. [Google Scholar] [CrossRef]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef]
- Wei, H.; Alberts, I.; Li, X. Brain IL-6 and autism. Neuroscience 2013, 252, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry 2014, 75, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K. Plasma increase of interleukin-12 and interferon-gamma. Pathological significance in autism. J. Neuroimmunol. 1996, 66, 143–145. [Google Scholar] [CrossRef]
- Croonenberghs, J.; Bosmans, E.; Deboutte, D.; Kenis, G.; Maes, M. Activation of the inflammatory response system in autism. Neuropsychobiology 2002, 45, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tostes, M.H.F.S.; Teixeira, H.C.; Gattaz, W.F.; Brandão, M.A.F.; Raposo, N.R.B. Altered neurotrophin, neuropeptide, cytokines and nitric oxide levels in autism. Pharmacopsychiatry 2012, 45, 241–243. [Google Scholar] [CrossRef]
- Monteiro, S.; Roque, S.; Marques, F.; Correia-Neves, M.; Cerqueira, J.J. Brain interference: Revisiting the role of IFNγ in the central nervous system. Prog. Neurobiol. 2017, 156, 149–163. [Google Scholar] [CrossRef]
- Czapski, G.A.; Czubowicz, K.; Strosznajder, J.B.; Strosznajder, R.P. The Lipoxygenases: Their Regulation and Implication in Alzheimer’s Disease. Neurochem. Res. 2016, 41, 243–257. [Google Scholar] [CrossRef]
- Roy, A.; Jana, A.; Yatish, K.; Freidt, M.B.; Fung, Y.K.; Martinson, J.A.; Pahan, K. Reactive oxygen species up-regulate CD11b in microglia via nitric oxide: Implications for neurodegenerative diseases. Free Radic. Biol. Med. 2008, 45, 686–699. [Google Scholar] [CrossRef]
- Wilkinson, B.L.; Landreth, G.E. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J. Neuroinflamm. 2006, 3, 30. [Google Scholar] [CrossRef][Green Version]
- Chauhan, A.; Audhya, T.; Chauhan, V. Brain region-specific glutathione redox imbalance in autism. Neurochem. Res. 2012, 37, 1681–1689. [Google Scholar] [CrossRef]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: Systematic review and meta-analyses. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, S.; Fu, Y.; Li, X. Synaptic proteins and receptors defects in autism spectrum disorders. Front. Cell. Neurosci. 2014, 8, 276. [Google Scholar] [CrossRef] [PubMed]
- Frankle, W.G.; Lerma, J.; Laruelle, M. The synaptic hypothesis of schizophrenia. Neuron 2003, 39, 205–216. [Google Scholar] [CrossRef]
- Martinez-Cerdeno, V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol. 2017, 77, 393–404. [Google Scholar] [CrossRef]
- Cheung, C.; Yu, K.; Fung, G.; Leung, M.; Wong, C.; Li, Q.; Sham, P.; Chua, S.; McAlonan, G. Autistic disorders and schizophrenia: Related or remote? An anatomical likelihood estimation. PLoS ONE 2010, 5, e12233. [Google Scholar] [CrossRef]
- Wang, Q.; Deng, W.; Huang, C.; Li, M.; Ma, X.; Wang, Y.; Jiang, L.; Lui, S.; Huang, X.; Chua, S.E.; et al. Abnormalities in connectivity of white-matter tracts in patients with familial and non-familial schizophrenia. Psychol. Med. 2011, 41, 1691–1700. [Google Scholar] [CrossRef]
- Sasson, N.J.; Pinkham, A.E.; Carpenter, K.L.; Belger, A. The benefit of directly comparing autism and schizophrenia for revealing mechanisms of social cognitive impairment. J. Neurodev. Disord. 2011, 3, 87–100. [Google Scholar] [CrossRef]
- Budd, J.M.; Kisvarday, Z.F. Communication and wiring in the cortical connectome. Front. Neuroanat. 2012, 6, 42. [Google Scholar] [CrossRef]
- Dickerson, D.D.; Bilkey, D.K. Aberrant neural synchrony in the maternal immune activation model: Using translatable measures to explore targeted interventions. Front. Behav. Neurosci. 2013, 7, 217. [Google Scholar] [CrossRef]
- Ecker, C. The neuroanatomy of autism spectrum disorder: An overview of structural neuroimaging findings and their translatability to the clinical setting. Autism: Int. J. Res. Pract. 2017, 21, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Mensen, V.T.; Wierenga, L.M.; van Dijk, S.; Rijks, Y.; Oranje, B.; Mandl, R.C.W.; Durston, S. Development of cortical thickness and surface area in autism spectrum disorder. Neuroimage Clin. 2016, 13, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Corradi, A.; Fadda, M.; Piton, A.; Patry, L.; Marte, A.; Rossi, P.; Cadieux-Dion, M.; Gauthier, J.; Lapointe, L.; Mottron, L.; et al. SYN2 is an autism predisposing gene: Loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum. Mol. Genet. 2014, 23, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Cupertino, R.B.; Kappel, D.B.; Bandeira, C.E.; Schuch, J.B.; da Silva, B.S.; Muller, D.; Bau, C.H.; Mota, N.R. SNARE complex in developmental psychiatry: Neurotransmitter exocytosis and beyond. J. Neural Transm. (Vienna, Austria: 1996) 2016, 123, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Tejero, R.; Arancillo, M.; Vardar, G.; Korotkova, T.; Kintscher, M.; Schmitz, D.; Ponomarenko, A.; Tabares, L.; Rosenmund, C. Syntaxin 1B is important for mouse postnatal survival and proper synaptic function at the mouse neuromuscular junctions. J. Neurophysiol. 2015, 114, 2404–2417. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.; Keers, R.; Lester, K.J.; Coleman, J.R.; Breen, G.; Arendt, K.; Blatter-Meunier, J.; Cooper, P.; Creswell, C.; Fjermestad, K.; et al. HPA axis related genes and response to psychological therapies: Genetics and epigenetics. Depress. Anxiety 2015, 32, 861–870. [Google Scholar] [CrossRef]
- Li, X.W.; Cao, L.; Wang, F.; Yang, Q.G.; Tong, J.J.; Li, X.Y.; Chen, G.H. Maternal inflammation linearly exacerbates offspring age-related changes of spatial learning and memory, and neurobiology until senectitude. Behav. Brain Res. 2016, 306, 178–196. [Google Scholar] [CrossRef]
- Chang, S.; Zhang, W.; Gao, L.; Wang, J. Prioritization of candidate genes for attention deficit hyperactivity disorder by computational analysis of multiple data sources. Protein Cell 2012, 3, 526–534. [Google Scholar] [CrossRef][Green Version]
- Nakamura, K.; Iwata, Y.; Anitha, A.; Miyachi, T.; Toyota, T.; Yamada, S.; Tsujii, M.; Tsuchiya, K.J.; Iwayama, Y.; Yamada, K.; et al. Replication study of Japanese cohorts supports the role of STX1A in autism susceptibility. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 454–458. [Google Scholar] [CrossRef]
- Coley, A.A.; Gao, W.J. PSD95: A synaptic protein implicated in schizophrenia or autism? Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 82, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Catts, V.S.; Derminio, D.S.; Hahn, C.G.; Weickert, C.S. Postsynaptic density levels of the NMDA receptor NR1 subunit and PSD-95 protein in prefrontal cortex from people with schizophrenia. NPJ Schizophr. 2015, 1, 15037. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- El-Husseini, A.E.; Schnell, E.; Chetkovich, D.M.; Nicoll, R.A.; Bredt, D.S. PSD-95 involvement in maturation of excitatory synapses. Science (New York, N.Y.) 2000, 290, 1364–1368. [Google Scholar]
- Yoo, J.; Bakes, J.; Bradley, C.; Collingridge, G.L.; Kaang, B.K. Shank mutant mice as an animal model of autism. Philos. Trans. R. Soc. London Ser. B Biol. Sci. 2014, 369, 20130143. [Google Scholar] [CrossRef]
- Monteiro, P.; Feng, G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef]
- Zhou, Y.; Kaiser, T.; Monteiro, P.; Zhang, X.; Van der Goes, M.S.; Wang, D.; Barak, B.; Zeng, M.; Li, C.; Lu, C.; et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 2016, 89, 147–162. [Google Scholar] [CrossRef]
- Mei, Y.; Monteiro, P.; Zhou, Y.; Kim, J.A.; Gao, X.; Fu, Z.; Feng, G. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 2016, 530, 481–484. [Google Scholar] [CrossRef]
- Tonkiss, J.; Harrison, R.H.; Galler, J.R. Differential effects of prenatal protein malnutrition and prenatal cocaine on a test of homing behavior in rat pups. Physiol. Behav. 1996, 60, 1013–1018. [Google Scholar] [CrossRef]
- Panksepp, J.; Burgdorf, J. “Laughing” rats and the evolutionary antecedents of human joy? Physiol. Behav. 2003, 79, 533–547. [Google Scholar] [CrossRef]
- Panksepp, J.; Burgdorf, J. 50-kHz chirping (laughter?) in response to conditioned and unconditioned tickle-induced reward in rats: Effects of social housing and genetic variables. Behav. Brain Res. 2000, 115, 25–38. [Google Scholar] [CrossRef]
- Wohr, M.; Kehl, M.; Borta, A.; Schanzer, A.; Schwarting, R.K.; Hoglinger, G.U. New insights into the relationship of neurogenesis and affect: Tickling induces hippocampal cell proliferation in rats emitting appetitive 50-kHz ultrasonic vocalizations. Neuroscience 2009, 163, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Schwarting, R.K.; Jegan, N.; Wohr, M. Situational factors, conditions and individual variables which can determine ultrasonic vocalizations in male adult Wistar rats. Behav. Brain Res. 2007, 182, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Ziemka-Nalecz, M.; Jaworska, J.; Sypecka, J.; Polowy, R.; Filipkowski, R.K.; Zalewska, T. Sodium Butyrate, a Histone Deacetylase Inhibitor, Exhibits Neuroprotective/Neurogenic Effects in a Rat Model of Neonatal Hypoxia-Ischemia. Mol. Neurobiol. 2017, 54, 5300–5318. [Google Scholar] [CrossRef] [PubMed]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Dominiak, A.; Wilkaniec, A.; Jesko, H.; Czapski, G.A.; Lenkiewicz, A.M.; Kurek, E.; Wroczynski, P.; Adamczyk, A. Selol, an organic selenium donor, prevents lipopolysaccharide-induced oxidative stress and inflammatory reaction in the rat brain. Neurochem. Int. 2017, 108, 66–77. [Google Scholar] [CrossRef]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cieślik, M.; Gąssowska-Dobrowolska, M.; Jęśko, H.; Czapski, G.A.; Wilkaniec, A.; Zawadzka, A.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Boguszewski, P.M.; et al. Maternal Immune Activation Induces Neuroinflammation and Cortical Synaptic Deficits in the Adolescent Rat Offspring. Int. J. Mol. Sci. 2020, 21, 4097. https://doi.org/10.3390/ijms21114097
Cieślik M, Gąssowska-Dobrowolska M, Jęśko H, Czapski GA, Wilkaniec A, Zawadzka A, Dominiak A, Polowy R, Filipkowski RK, Boguszewski PM, et al. Maternal Immune Activation Induces Neuroinflammation and Cortical Synaptic Deficits in the Adolescent Rat Offspring. International Journal of Molecular Sciences. 2020; 21(11):4097. https://doi.org/10.3390/ijms21114097
Chicago/Turabian StyleCieślik, Magdalena, Magdalena Gąssowska-Dobrowolska, Henryk Jęśko, Grzegorz A. Czapski, Anna Wilkaniec, Aleksandra Zawadzka, Agnieszka Dominiak, Rafał Polowy, Robert K. Filipkowski, Paweł M. Boguszewski, and et al. 2020. "Maternal Immune Activation Induces Neuroinflammation and Cortical Synaptic Deficits in the Adolescent Rat Offspring" International Journal of Molecular Sciences 21, no. 11: 4097. https://doi.org/10.3390/ijms21114097
APA StyleCieślik, M., Gąssowska-Dobrowolska, M., Jęśko, H., Czapski, G. A., Wilkaniec, A., Zawadzka, A., Dominiak, A., Polowy, R., Filipkowski, R. K., Boguszewski, P. M., Gewartowska, M., Frontczak-Baniewicz, M., Sun, G. Y., Beversdorf, D. Q., & Adamczyk, A. (2020). Maternal Immune Activation Induces Neuroinflammation and Cortical Synaptic Deficits in the Adolescent Rat Offspring. International Journal of Molecular Sciences, 21(11), 4097. https://doi.org/10.3390/ijms21114097