Chrysosplenol d, a Flavonol from Artemisia annua, Induces ERK1/2-Mediated Apoptosis in Triple Negative Human Breast Cancer Cells


 , , , and
, , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Ingredients of the Momundo Artemisia Annua Dietary Supplement
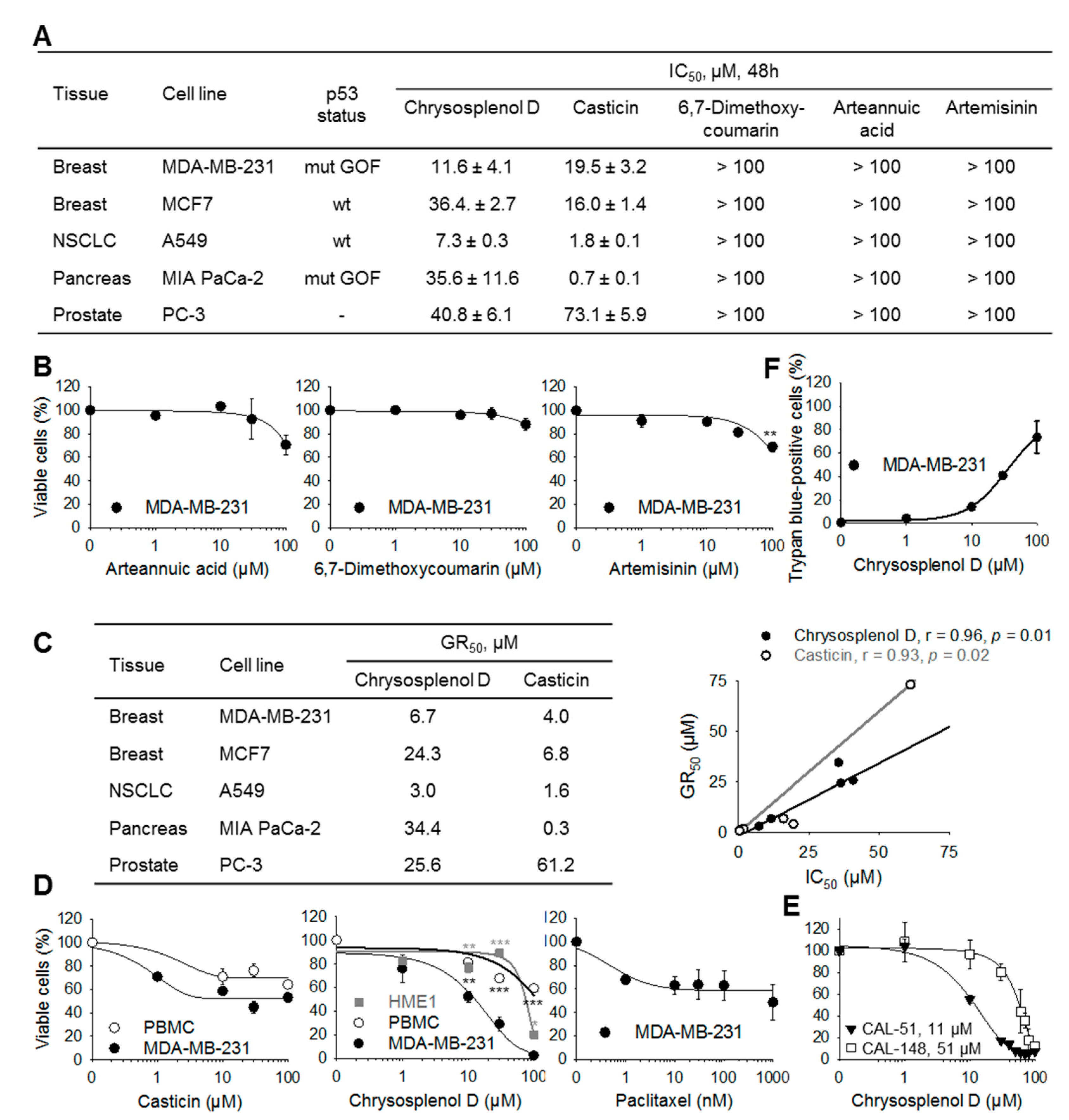
2.2. Chrysosplenol d and Casticin Selectively Inhibit the Viability of Several Cancer Cell Lines
2.3. Chrysosplenol d and Casticin Inhibit the Cell Cycle Progression of MDA-MB-231 Cells in Vitro
2.4. Chrysosplenol d and Casticin Induce Apoptosis in Breast Cancer Cells
2.5. Chrysosplenol d and Casticin Inhibit Tumor Growth of Breast Cancer Xenografts in Vivo
2.6. Chrysosplenol d and Casticin Induce Mitochondrial Membrane Permeabilization in Breast Cancer Cells
2.7. Chrysosplenol d Activates ERK1/2
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Analytical Fingerprint of Momundo Artemisia annua Dietary Supplement by HPLC-DAD-MS Analysis
4.3. Quantification of Artemisinin by HPLC-MS/MS
4.4. Cell Culture
4.5. Analysis of Cell Viability
4.6. Cell-Cycle Analysis
4.7. Breast Cancer Xenografts
4.8. Analysis of Apoptosis
4.9. Analysis of ROS, Mitochondria, and Lysosomes
4.10. Human Phospho-Kinase Array and Western Immunoblotting
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| AKT | Protein kinase B |
| AZKIM | Academic Center for Complementary and Integrative Medicine |
| BCI | (E)-2-benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one |
| BCL2 | B-cell lymphoma 2 |
| CAM | Chorioallantoic membrane |
| DMSO | Dimethyl sulfoxide |
| ERK | Extracellular signal-regulated kinase |
| ESI | Electrospray ionisation |
| FBS | Fetal bovine serum |
| ΔΨm | Mitochondrial membrane potential |
| NMR | Nuclear magnetic resonance |
| GFP | Green fluorescent protein |
| GOF | Gain of function |
| GR50 | Half-maximal growth rate inhibition |
| HPLC-DAD-MS | High-performance liquid chromatography–diode array & mass spectrometric detection |
| HPLC-MS/MS | High-performance liquid chromatography-tandem mass spectrometry |
| IC50 | Half-maximal inhibitory concentration |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| LC3II | Lipid-modified form of microtubule-associated proteins 1A/1B light chain 3B |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| MEK | Mitogen-activated protein kinase kinase |
| MRM | Multiple-reaction monitoring |
| NSCLC | Non-small-cell lung carcinaoma |
| PBMC | Peripheral blood mononuclear cells |
| PI3K | Phosphoinositide 3-kinase |
| Ras | Rat sarcoma oncogen |
| ROS | Reactive oxygen species |
| TNBC | Triple negative breast cancer |
| XTT | 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2h-tetrazolium-5-carboxanilide |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. Ca-Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Jitariu, A.; Cîmpean, a.; Ribatti, D.; Raica, M. Triple negative breast cancer: The kiss of death. Oncotarget 2017, 8, 46652–46662. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Modulation of apoptosis by natural products for cancer therapy. Planta. Med. 2010, 76, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef]
- Efferth, T.; Herrmann, F.; Tahrani, A.; Wink, M. Cytotoxic activity of secondary metabolites derived from Artemisia annua L. towards cancer cells in comparison to its designated active constituent artemisinin. Phytomedicine 2011, 18, 959–969. [Google Scholar] [CrossRef]
- Ferreira, J.F.; Luthria, D.L.; Sasaki, T.; Heyerick, A. Flavonoids from Artemisia annua L. as antioxidants and their potential synergism with artemisinin against malaria and cancer. Molecules 2010, 15, 3135–3170. [Google Scholar] [CrossRef]
- Lang, S.J.; Schmiech, M.; Hafner, S.; Paetz, C.; Steinborn, C.; Huber, R.; El Gaafary, M.; Werner, K.; Schmidt, C.Q.; Syrovets, T.; et al. Antitumor activity of an Artemisia annua herbal preparation and identification of active ingredients. Phytomedicine 2019, 62, 152962. [Google Scholar] [CrossRef]
- Haidara, K.; Zamir, L.; Shi, Q.W.; Batist, G. The flavonoid Casticin has multiple mechanisms of tumor cytotoxicity action. Cancer Lett. 2006, 242, 180–190. [Google Scholar] [CrossRef]
- Liu, L.P.; Cao, X.C.; Liu, F.; Quan, M.F.; Sheng, X.F.; Ren, K.Q. Casticin induces breast cancer cell apoptosis by inhibiting the expression of forkhead box protein M1. Oncol. Lett. 2014, 7, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Amina, M.; Alam, P.; Parvez, M.K.; Al-Musayeib, N.M.; Al-Hwaity, S.A.; Al-Rashidi, N.S.; Al-Dosari, M.S. Isolation and validated HPTLC analysis of four cytotoxic compounds, including a new sesquiterpene from aerial parts of Plectranthus cylindraceus. Nat. Prod. Res. 2018, 32, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Kawaii, S.; Tomono, Y.; Katase, E.; Ogawa, K.; Yano, M. Antiproliferative activity of flavonoids on several cancer cell lines. Biosci. Biotechnol. Biochem. 1999, 63, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Syrovets, T.; Gschwend, J.E.; Büchele, B.; Laumonnier, Y.; Zugmaier, W.; Genze, F.; Simmet, T. Inhibition of IκB kinase activity by acetyl-boswellic acids promotes apoptosis in androgen-independent PC-3 prostate cancer cells in vitro and in vivo. J. Biol. Chem. 2005, 280, 6170–6180. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Molina, G.; Vogt, A.; Bakan, A.; Dai, W.; Queiroz de Oliveira, P.; Znosko, W.; Smithgall, T.E.; Bahar, I.; Lazo, J.S.; Day, B.W.; et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 2009, 5, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.E.; Gelmann, E.P.; Lippman, M.E.; Dickson, R.B. Epidermal growth factor receptor gene expression in estrogen receptor-positive and negative human breast cancer cell lines. Mol. Endocrinol. 1987, 1, 216–223. [Google Scholar] [CrossRef]
- Lee, W.J.; Hsiao, M.; Chang, J.L.; Yang, S.F.; Tseng, T.H.; Cheng, C.W.; Chow, J.M.; Lin, K.H.; Lin, Y.W.; Liu, C.C.; et al. Quercetin induces mitochondrial-derived apoptosis via reactive oxygen species-mediated ERK activation in HL-60 leukemia cells and xenograft. Arch. Toxicol. 2015, 89, 1103–1117. [Google Scholar] [CrossRef]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P.; Keyomarsi, K.; Yarden, Y.; Seger, R. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 2001, 20, 147–155. [Google Scholar] [CrossRef]
- El Gaafary, M.; Büchele, B.; Syrovets, T.; Agnolet, S.; Schneider, B.; Schmidt, C.Q.; Simmet, T. An α-acetoxy-tirucallic acid isomer inhibits Akt/mTOR signaling and induces oxidative stress in prostate cancer cells acid isomer inhibits Akt/mTOR signaling and induces oxidative stress in prostate cancer cells. J. Pharmacol. Exp. Ther. 2015, 352, 33–42. [Google Scholar] [CrossRef]
- Estrada, A.C.; Syrovets, T.; Pitterle, K.; Lunov, O.; Büchele, B.; Schimana-Pfeifer, J.; Schmidt, T.; Morad, S.A.; Simmet, T. Tirucallic acids are novel pleckstrin homology domain-dependent Akt inhibitors inducing apoptosis in prostate cancer cells. Mol. Pharmacol. 2010, 77, 378–387. [Google Scholar] [CrossRef]
- Deschenes-Simard, X.; Kottakis, F.; Meloche, S.; Ferbeyre, G. ERKs in cancer: Friends or foes? Cancer Res. 2014, 74, 412–419. [Google Scholar] [CrossRef]
- De Luca, A.; Maiello, M.R.; D′Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Tar. 2012, 16 (Suppl. 2), S17–S27. [Google Scholar] [CrossRef]
- Cook, S.J.; Stuart, K.; Gilley, R.; Sale, M.J. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017, 284, 4177–4195. [Google Scholar] [CrossRef]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Berglind, H.; Pawitan, Y.; Kato, S.; Ishioka, C.; Soussi, T. Analysis of p53 mutation status in human cancer cell lines: A paradigm for cell line cross-contamination. Cancer Biol. Ther. 2008, 7, 699–708. [Google Scholar] [CrossRef]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef]
- Yan, W.; Chen, X. Identification of GRO1 as a critical determinant for mutant p53 gain of function. J. Biol. Chem. 2009, 284, 12178–12187. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef] [PubMed]
- El Gaafary, M.; Ezzat, S.M.; El Sayed, A.M.; Sabry, O.M.; Hafner, S.; Lang, S.; Schmiech, M.; Syrovets, T.; Simmet, T. Acovenoside A induces mitotic catastrophe followed by apoptosis in non-small-cell lung cancer cells. J. Nat. Prod. 2017, 80, 3203–3210. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Laumonnier, Y.; Syrovets, T.; Simmet, T. Plasmin triggers cytokine induction in human monocyte-derived macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Schmiech, M.; Lang, S.J.; Ulrich, J.; Werner, K.; Rashan, L.J.; Syrovets, T.; Simmet, T. Comparative Investigation of Frankincense Nutraceuticals: Correlation of Boswellic and Lupeolic Acid Contents with Cytokine Release Inhibition and Toxicity against Triple-Negative Breast Cancer Cells. Nutrients 2019, 11, 2341. [Google Scholar] [CrossRef]
- El Gaafary, M.; Hafner, S.; Lang, S.J.; Jin, L.; Sabry, O.M.; Vogel, C.V.; Vanderwal, C.D.; Syrovets, T.; Simmet, T. A novel polyhalogenated monoterpene induces cell cycle arrest and apoptosis in breast cancer cells. Mar. Drugs 2019, 17, 437. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lang, S.J.; Schmiech, M.; Hafner, S.; Paetz, C.; Werner, K.; El Gaafary, M.; Schmidt, C.Q.; Syrovets, T.; Simmet, T. Chrysosplenol d, a Flavonol from Artemisia annua, Induces ERK1/2-Mediated Apoptosis in Triple Negative Human Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 4090. https://doi.org/10.3390/ijms21114090
Lang SJ, Schmiech M, Hafner S, Paetz C, Werner K, El Gaafary M, Schmidt CQ, Syrovets T, Simmet T. Chrysosplenol d, a Flavonol from Artemisia annua, Induces ERK1/2-Mediated Apoptosis in Triple Negative Human Breast Cancer Cells. International Journal of Molecular Sciences. 2020; 21(11):4090. https://doi.org/10.3390/ijms21114090
Chicago/Turabian StyleLang, Sophia J., Michael Schmiech, Susanne Hafner, Christian Paetz, Katharina Werner, Menna El Gaafary, Christoph Q. Schmidt, Tatiana Syrovets, and Thomas Simmet. 2020. "Chrysosplenol d, a Flavonol from Artemisia annua, Induces ERK1/2-Mediated Apoptosis in Triple Negative Human Breast Cancer Cells" International Journal of Molecular Sciences 21, no. 11: 4090. https://doi.org/10.3390/ijms21114090
APA StyleLang, S. J., Schmiech, M., Hafner, S., Paetz, C., Werner, K., El Gaafary, M., Schmidt, C. Q., Syrovets, T., & Simmet, T. (2020). Chrysosplenol d, a Flavonol from Artemisia annua, Induces ERK1/2-Mediated Apoptosis in Triple Negative Human Breast Cancer Cells. International Journal of Molecular Sciences, 21(11), 4090. https://doi.org/10.3390/ijms21114090