Beyond Macrophages and T Cells: B Cells and Immunoglobulins Determine the Fate of the Atherosclerotic Plaque


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. B Cells—The Underestimated Players
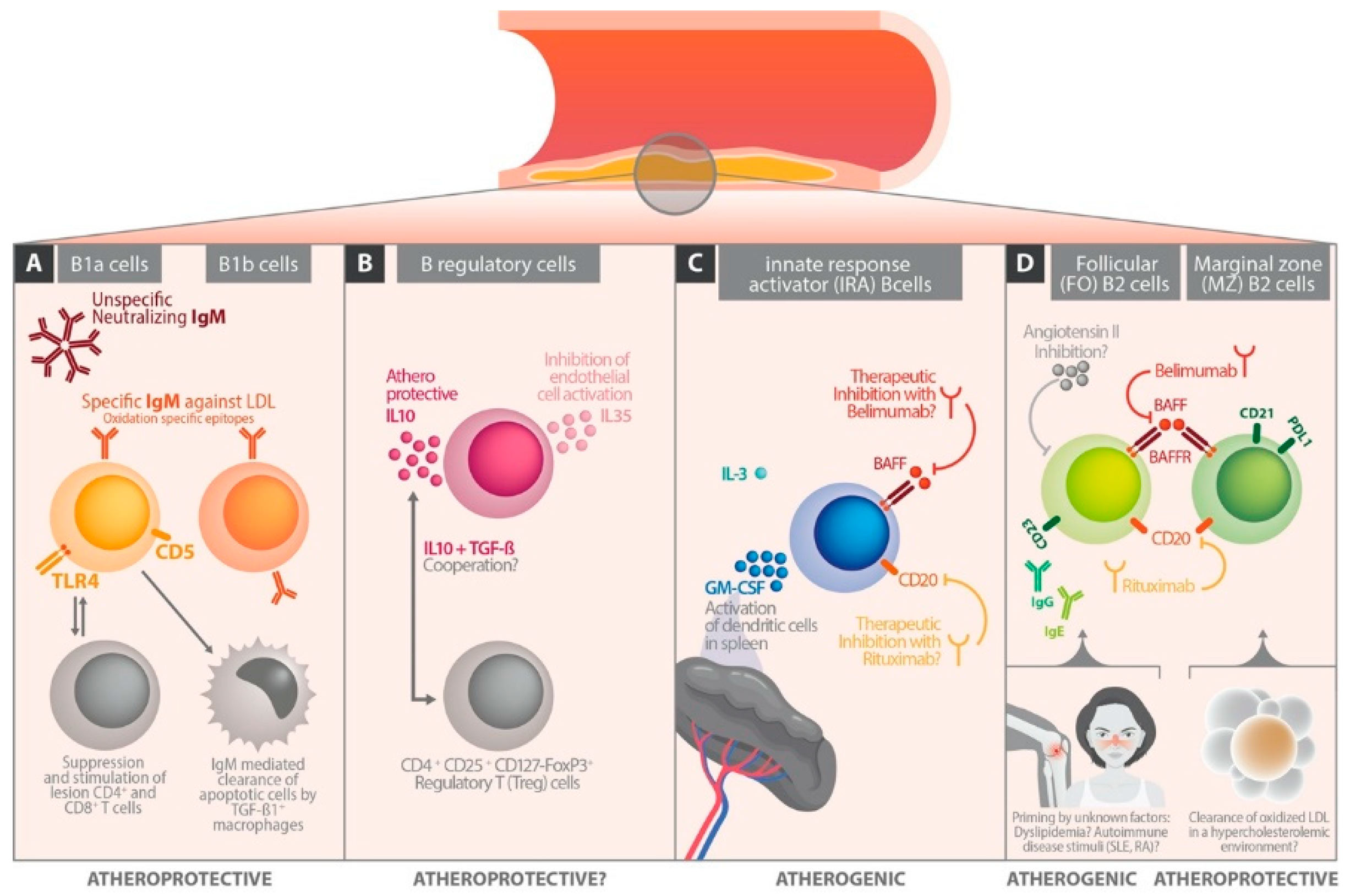
3. B Cell Subsets
4. B Cells in Experimental Atherosclerosis Models
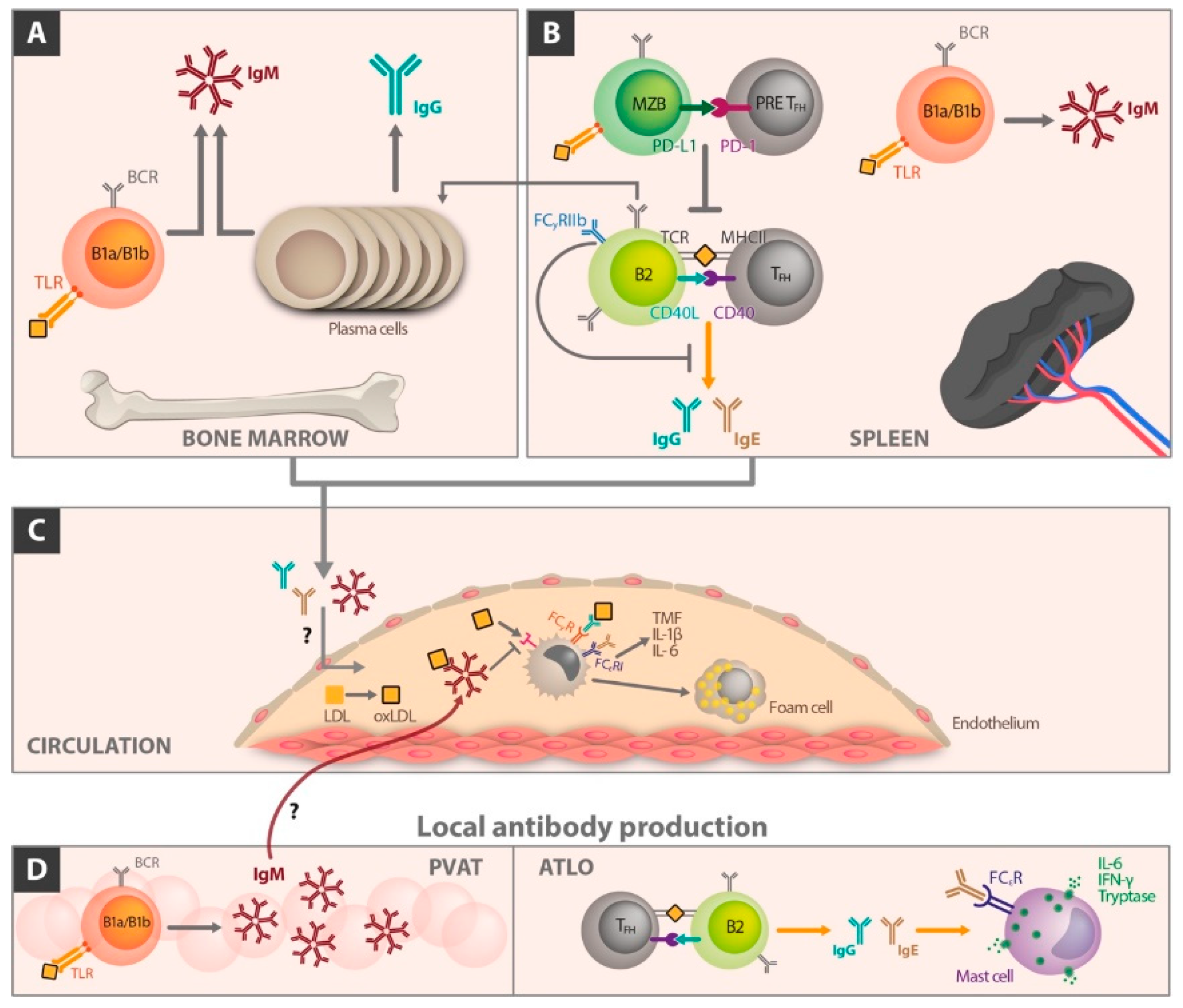
5. The Different Role of Immunoglobulin Classes in Atherosclerosis
6. The Influence of B Cell Differentiation on Atherosclerotic Perpetuation
6.1. Follicular B Cells
6.2. Marginal Zone B Cells
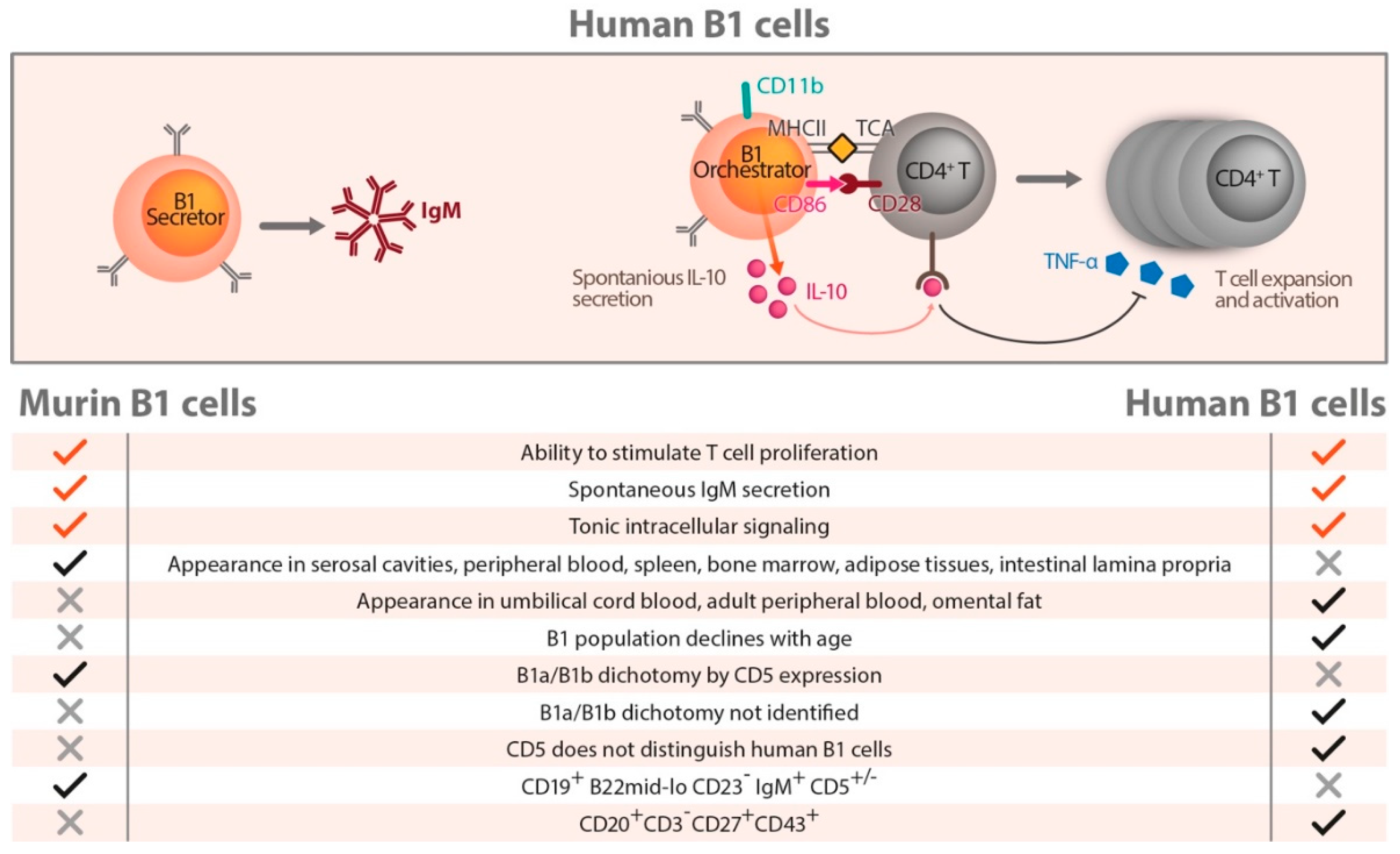
7. Human B Cells
8. Conclusions
Funding
Conflicts of Interest
References
- Chistiakov, D.A.; Kashirskikh, D.A.; Khotina, V.A.; Grechko, A.V.; Orekhov, A.N. Immune-Inflammatory Responses in Atherosclerosis: The Role of Myeloid Cells. J. Clin. Med. 2019, 8, 1798. [Google Scholar] [CrossRef] [PubMed]
- Nus, M.; Tsiantoulas, D.; Mallat, Z. Plan B (-cell) in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Mangge, H.; Almer, G.; Stelzer, I.; Reininghaus, E.; Prassl, R. Laboratory medicine for molecular imaging of atherosclerosis. Clin. Chim. Acta 2014, 437, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Mangge, H.; Hubmann, H.; Pilz, S.; Schauenstein, K.; Renner, W.; Marz, W. Beyond cholesterol--inflammatory cytokines, the key mediators in atherosclerosis. Clin. Chem. Lab. Med. 2004, 42, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Mangge, H.; Becker, K.; Fuchs, D.; Gostner, J.M. Antioxidants, inflammation and cardiovascular disease. World J. Cardiol. 2014, 6, 462–477. [Google Scholar] [CrossRef]
- Mangge, H.; Almer, G.; Truschnig-Wilders, M.; Schmidt, A.; Gasser, R.; Fuchs, D. Inflammation, adiponectin, obesity and cardiovascular risk. Curr. Med. Chem. 2010, 17, 4511–4520. [Google Scholar] [CrossRef]
- Gambardella, J.; Sardu, C.; Sacra, C.; Del Giudice, C.; Santulli, G. Quit smoking to outsmart atherogenesis: Molecular mechanisms underlying clinical evidence. Atherosclerosis 2017, 257, 242–245. [Google Scholar] [CrossRef]
- Zabetakis, I.; Lordan, R.; Norton, C.; Tsoupras, A. COVID-19: The Inflammation Link and the Role of Nutrition in Potential Mitigation. Nutrients 2020, 12, 1466. [Google Scholar] [CrossRef]
- Gambardella, J.; Santulli, G. Integrating diet and inflammation to calculate cardiovascular risk. Atherosclerosis 2016, 253, 258–261. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Nikiforov, N.N.; Ivanova, E.A.; Sobenin, I.A. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. J. Clin. Med. 2020, 9, 978. [Google Scholar] [CrossRef]
- Alfaidi, M.; Acosta, C.H.; Wang, D.; Traylor, J.G.; Orr, A.W. Selective role of Nck1 in atherogenic inflammation and plaque formation. J. Clin. Invest. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tsiantoulas, D.; Diehl, C.J.; Witztum, J.L.; Binder, C.J. B cells and humoral immunity in atherosclerosis. Circ. Res. 2014, 114, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Upadhye, A.; Sturek, J.M.; McNamara, C.A. 2019 Russell Ross Memorial Lecture in Vascular Biology: B Lymphocyte-Mediated Protective Immunity in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, H.; Li, Y.; Kanellakis, P.; Tay, C.; Cao, A.; Liu, E.; Peter, K.; Tipping, P.; Toh, B.H.; Bobik, A.; et al. Toll-Like Receptor (TLR)4 and MyD88 are Essential for Atheroprotection by Peritoneal B1a B Cells. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, S.M.; Perry, H.M.; Gonen, A.; Prohaska, T.A.; Srikakulapu, P.; Grewal, S.; Das, D.; McSkimming, C.; Taylor, A.M.; Tsimikas, S.; et al. B-1b Cells Secrete Atheroprotective IgM and Attenuate Atherosclerosis. Circ. Res. 2015, 117, 28–39. [Google Scholar] [CrossRef]
- Upadhye, A.; Srikakulapu, P.; Gonen, A.; Hendrikx, S.; Perry, H.M.; Nguyen, A.; McSkimming, C.; Marshall, M.A.; Garmey, J.C.; Taylor, A.M.; et al. Diversification and CXCR4-Dependent Establishment of the Bone Marrow B-1a Cell Pool Governs Atheroprotective IgM Production Linked to Human Coronary Atherosclerosis. Circ. Res. 2019, 125, 55–70. [Google Scholar] [CrossRef]
- Doring, Y.; Jansen, Y.; Cimen, I.; Aslani, M.; Gencer, S.; Peters, L.J.F.; Duchene, J.; Weber, C.; van der Vorst, E.P.C. B-Cell-Specific CXCR4 Protects Against Atherosclerosis Development and Increases Plasma IgM Levels. Circ. Res. 2020, 126, 787–788. [Google Scholar] [CrossRef]
- Fillatreau, S. Regulatory roles of B cells in infectious diseases. Clin. Exp. Rheumatol. 2016, 34, 1–5. [Google Scholar]
- Li, X.; Shao, Y.; Sha, X.; Fang, P.; Kuo, Y.M.; Andrews, A.J.; Li, Y.; Yang, W.Y.; Maddaloni, M.; Pascual, D.W.; et al. IL-35 (Interleukin-35) Suppresses Endothelial Cell Activation by Inhibiting Mitochondrial Reactive Oxygen Species-Mediated Site-Specific Acetylation of H3K14 (Histone 3 Lysine 14). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Zhang, B.; Wang, Z.; Joehanes, R.; Zhu, J.; Johnson, A.D.; Ying, S.; Munson, P.J.; Raghavachari, N.; Wang, R.; et al. A systems biology framework identifies molecular underpinnings of coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Mantani, P.T.; Ljungcrantz, I.; Andersson, L.; Alm, R.; Hedblad, B.; Bjorkbacka, H.; Nilsson, J.; Fredrikson, G.N. Circulating CD40+ and CD86+ B cell subsets demonstrate opposing associations with risk of stroke. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 211–218. [Google Scholar] [CrossRef]
- Caligiuri, G.; Nicoletti, A.; Poirier, B.; Hansson, G.K. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J. Clin. Invest. 2002, 109, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Major, A.S.; Fazio, S.; Linton, M.F. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Chousterman, B.G.; Swirski, F.K. Innate response activator B cells: Origins and functions. Int. Immunol. 2015, 27, 537–541. [Google Scholar] [CrossRef]
- Tsiantoulas, D.; Bot, I.; Ozsvar-Kozma, M.; Goderle, L.; Perkmann, T.; Hartvigsen, K.; Conrad, D.H.; Kuiper, J.; Mallat, Z.; Binder, C.J. Increased Plasma IgE Accelerate Atherosclerosis in Secreted IgM Deficiency. Circ. Res. 2017, 120, 78–84. [Google Scholar] [CrossRef]
- Baumgarth, N. The double life of a B-1 cell: Self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 2011, 11, 34–46. [Google Scholar] [CrossRef]
- Kyaw, T.; Tay, C.; Khan, A.; Dumouchel, V.; Cao, A.; To, K.; Kehry, M.; Dunn, R.; Agrotis, A.; Tipping, P.; et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J. Immunol. 2010, 185, 4410–4419. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Herbin, O.; Bouaziz, J.D.; Binder, C.J.; Uyttenhove, C.; Laurans, L.; Taleb, S.; Van Vre, E.; Esposito, B.; Vilar, J.; et al. B cell depletion reduces the development of atherosclerosis in mice. J. Exp. Med. 2010, 207, 1579–1587. [Google Scholar] [CrossRef]
- Kyaw, T.; Cui, P.; Tay, C.; Kanellakis, P.; Hosseini, H.; Liu, E.; Rolink, A.G.; Tipping, P.; Bobik, A.; Toh, B.H. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE(−/−) mice. PLoS ONE 2013, 8, e60430. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Tsiantoulas, D.; Baker, L.; Harrison, J.; Masters, L.; Murphy, D.; Loinard, C.; Binder, C.J.; Mallat, Z. BAFF receptor deficiency reduces the development of atherosclerosis in mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, T.; Tay, C.; Hosseini, H.; Kanellakis, P.; Gadowski, T.; MacKay, F.; Tipping, P.; Bobik, A.; Toh, B.H. Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS ONE 2012, 7, e29371. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.; Liu, Y.H.; Hosseini, H.; Kanellakis, P.; Cao, A.; Peter, K.; Tipping, P.; Bobik, A.; Toh, B.H.; Kyaw, T. B-cell-specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc. Res. 2016, 111, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, T.; Tipping, P.; Bobik, A.; Toh, B.H. Protective role of natural IgM-producing B1a cells in atherosclerosis. Trends Cardiovasc. Med. 2012, 22, 48–53. [Google Scholar] [CrossRef]
- Ponnuswamy, P.; Joffre, J.; Herbin, O.; Esposito, B.; Laurans, L.; Binder, C.J.; Tedder, T.F.; Zeboudj, L.; Loyer, X.; Giraud, A.; et al. Angiotensin II synergizes with BAFF to promote atheroprotective regulatory B cells. Sci. Rep. 2017, 7, 4111. [Google Scholar] [CrossRef]
- Rincon-Arevalo, H.; Quintero, J.C.; Fortich, F.; Rojas, M.; Vasquez, G.; Castano, D.; Yassin, L.M. Low frequency of IL-10(+) B cells in patients with atherosclerosis is related with inflammatory condition. Heliyon 2020, 6, e03441. [Google Scholar] [CrossRef]
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.; et al. Angiotensin-converting enzyme-2 (ACE2), SARS-CoV-2 and pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020. [Google Scholar] [CrossRef]
- Nurmohamed, M.T.; Heslinga, M.; Kitas, G.D. Cardiovascular comorbidity in rheumatic diseases. Nat. Rev. Rheumatol. 2015, 11, 693–704. [Google Scholar] [CrossRef]
- Nurmohamed, M.T. Editorial: Treat to target in rheumatoid arthritis: Good for the joints as well as the heart? Arthritis Rheumatol. 2015, 67, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, M.; Bao, Y.; Signorovitch, J.; Trahey, A.; Mulani, P.; Furst, D.E. Longer durations of antitumour necrosis factor treatment are associated with reduced risk of cardiovascular events in patients with rheumatoid arthritis. RMD Open 2015, 1, e000080. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Cariappa, A.; Moran, S.T. Marginal zone B cells. Annu. Rev. Immunol. 2005, 23, 161–196. [Google Scholar] [CrossRef]
- Cinamon, G.; Zachariah, M.A.; Lam, O.M.; Foss, F.W., Jr.; Cyster, J.G. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 2008, 9, 54–62. [Google Scholar] [CrossRef]
- Cerutti, A.; Cols, M.; Puga, I. Marginal zone B cells: Virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 2013, 13, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Grasset, E.K.; Duhlin, A.; Agardh, H.E.; Ovchinnikova, O.; Hagglof, T.; Forsell, M.N.; Paulsson-Berne, G.; Hansson, G.K.; Ketelhuth, D.F.; Karlsson, M.C. Sterile inflammation in the spleen during atherosclerosis provides oxidation-specific epitopes that induce a protective B-cell response. Proc. Natl. Acad. Sci. USA 2015, 112, E2030–E2038. [Google Scholar] [CrossRef]
- Zhang, P.; Li, W.; Wang, Y.; Hou, L.; Xing, Y.; Qin, H.; Wang, J.; Liang, Y.; Han, H. Identification of CD36 as a new surface marker of marginal zone B cells by transcriptomic analysis. Mol. Immunol. 2007, 44, 332–337. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid. Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef]
- Fletcher, C.A.; Sutherland, A.P.; Groom, J.R.; Batten, M.L.; Ng, L.G.; Gommerman, J.; Mackay, F. Development of nephritis but not sialadenitis in autoimmune-prone BAFF transgenic mice lacking marginal zone B cells. Eur. J. Immunol. 2006, 36, 2504–2514. [Google Scholar] [CrossRef]
- Jackson, S.W.; Scharping, N.E.; Jacobs, H.M.; Wang, S.; Chait, A.; Rawlings, D.J. Cutting Edge: BAFF Overexpression Reduces Atherosclerosis via TACI-Dependent B Cell Activation. J. Immunol. 2016, 197, 4529–4534. [Google Scholar] [CrossRef]
- Nus, M.; Sage, A.P.; Lu, Y.; Masters, L.; Lam, B.Y.H.; Newland, S.; Weller, S.; Tsiantoulas, D.; Raffort, J.; Marcus, D.; et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 2017, 23, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Guedj, K.; Andreata, F.; Morvan, M.; Bey, L.; Khallou-Laschet, J.; Gaston, A.T.; Delbosc, S.; Alsac, J.M.; Bruneval, P.; et al. Control of the T follicular helper-germinal center B-cell axis by CD8(+) regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation 2015, 131, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.M.; Wilson, P.C.; James, J.A.; Capra, J.D. Human B cell subsets. Adv. Immunol. 2008, 98, 151–224. [Google Scholar] [CrossRef]
- Liu, Y.J.; Arpin, C.; de Bouteiller, O.; Guret, C.; Banchereau, J.; Martinez-Valdez, H.; Lebecque, S. Sequential triggering of apoptosis, somatic mutation and isotype switch during germinal center development. Semin Immunol. 1996, 8, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.O.; Holodick, N.E.; Rothstein, T.L. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J. Exp. Med. 2011, 208, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, T.L.; Griffin, D.O.; Holodick, N.E.; Quach, T.D.; Kaku, H. Human B-1 cells take the stage. Ann. N. Y. Acad. Sci. 2013, 1285, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, T.L.; Quach, T.D. The human counterpart of mouse B-1 cells. Ann. N. Y. Acad. Sci. 2015, 1362, 143–152. [Google Scholar] [CrossRef]
- Covens, K.; Verbinnen, B.; Geukens, N.; Meyts, I.; Schuit, F.; Van Lommel, L.; Jacquemin, M.; Bossuyt, X. Characterization of proposed human B-1 cells reveals pre-plasmablast phenotype. Blood 2013, 121, 5176–5183. [Google Scholar] [CrossRef] [PubMed]
- Descatoire, M.; Weill, J.C.; Reynaud, C.A.; Weller, S. A human equivalent of mouse B-1 cells? J. Exp. Med. 2011, 208, 2563–2564. [Google Scholar] [CrossRef]
- Perez-Andres, M.; Grosserichter-Wagener, C.; Teodosio, C.; van Dongen, J.J.; Orfao, A.; van Zelm, M.C. The nature of circulating CD27+CD43+ B cells. J. Exp. Med. 2011, 208, 2565–2566. [Google Scholar] [CrossRef]
- Weller, S.; Braun, M.C.; Tan, B.K.; Rosenwald, A.; Cordier, C.; Conley, M.E.; Plebani, A.; Kumararatne, D.S.; Bonnet, D.; Tournilhac, O.; et al. Human blood IgM “memory“ B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 2004, 104, 3647–3654. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. From CANTOS to CIRT to COLCOT to Clinic: Will All Atherosclerosis Patients Soon Be Treated With Combination Lipid-Lowering and Inflammation-Inhibiting Agents? Circulation 2020, 141, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Fernando, S.; Schwarz, N.; Tan, J.T.; Bursill, C.A.; Psaltis, P.J. Inflammation as a Therapeutic Target in Atherosclerosis. J. Clin. Med. 2019, 8, 1109. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangge, H.; Prüller, F.; Schnedl, W.; Renner, W.; Almer, G. Beyond Macrophages and T Cells: B Cells and Immunoglobulins Determine the Fate of the Atherosclerotic Plaque. Int. J. Mol. Sci. 2020, 21, 4082. https://doi.org/10.3390/ijms21114082
Mangge H, Prüller F, Schnedl W, Renner W, Almer G. Beyond Macrophages and T Cells: B Cells and Immunoglobulins Determine the Fate of the Atherosclerotic Plaque. International Journal of Molecular Sciences. 2020; 21(11):4082. https://doi.org/10.3390/ijms21114082
Chicago/Turabian StyleMangge, Harald, Florian Prüller, Wolfgang Schnedl, Wilfried Renner, and Gunter Almer. 2020. "Beyond Macrophages and T Cells: B Cells and Immunoglobulins Determine the Fate of the Atherosclerotic Plaque" International Journal of Molecular Sciences 21, no. 11: 4082. https://doi.org/10.3390/ijms21114082
APA StyleMangge, H., Prüller, F., Schnedl, W., Renner, W., & Almer, G. (2020). Beyond Macrophages and T Cells: B Cells and Immunoglobulins Determine the Fate of the Atherosclerotic Plaque. International Journal of Molecular Sciences, 21(11), 4082. https://doi.org/10.3390/ijms21114082