Embryonic Program Activated during Blast Crisis of Chronic Myelogenous Leukemia (CML) Implicates a TCF7L2 and MYC Cooperative Chromatin Binding

 , , and
, , and 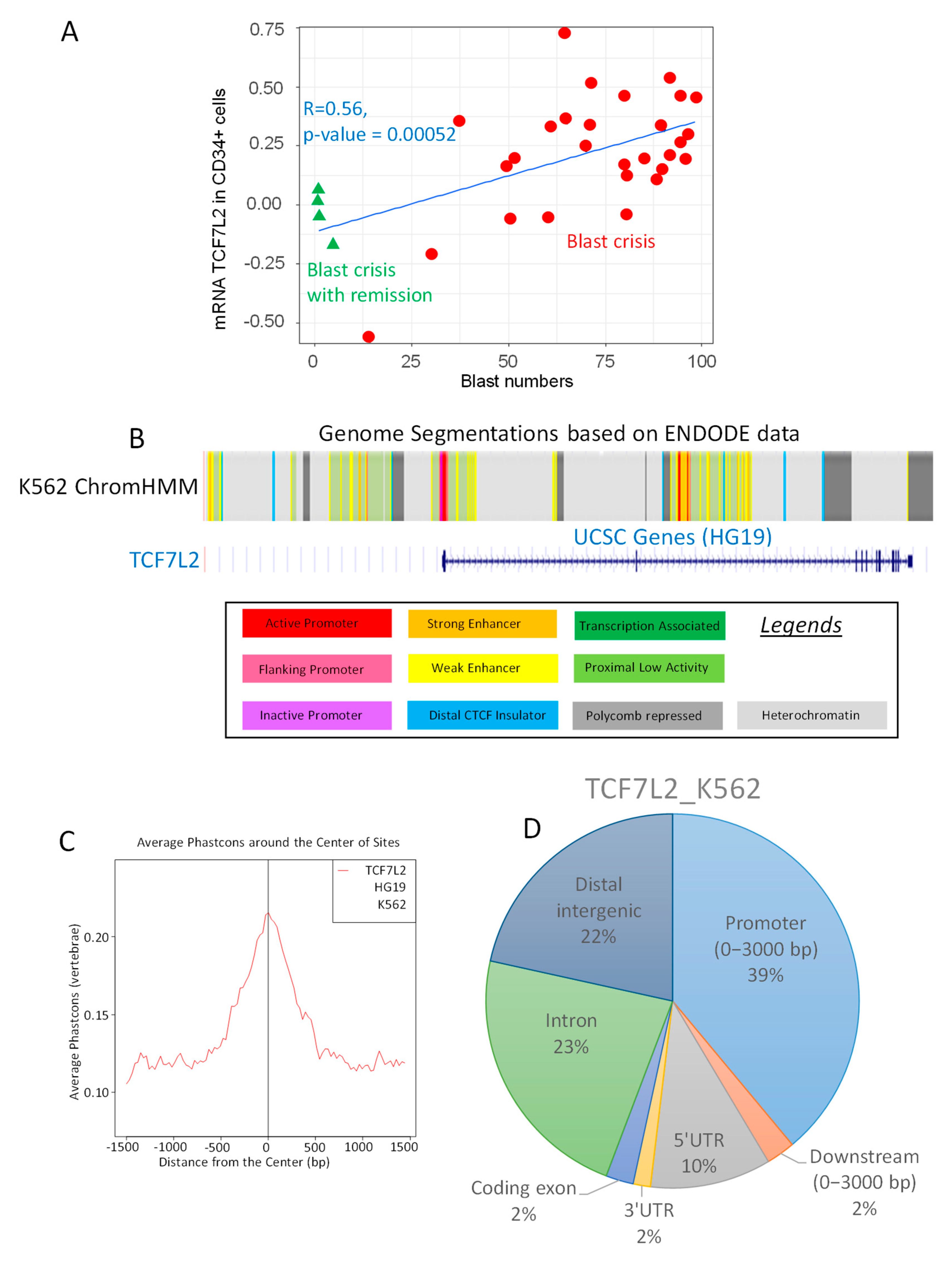{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. TCF7L2 Expression is Positively Correlated to the Number of Blast Cells in CML-BC
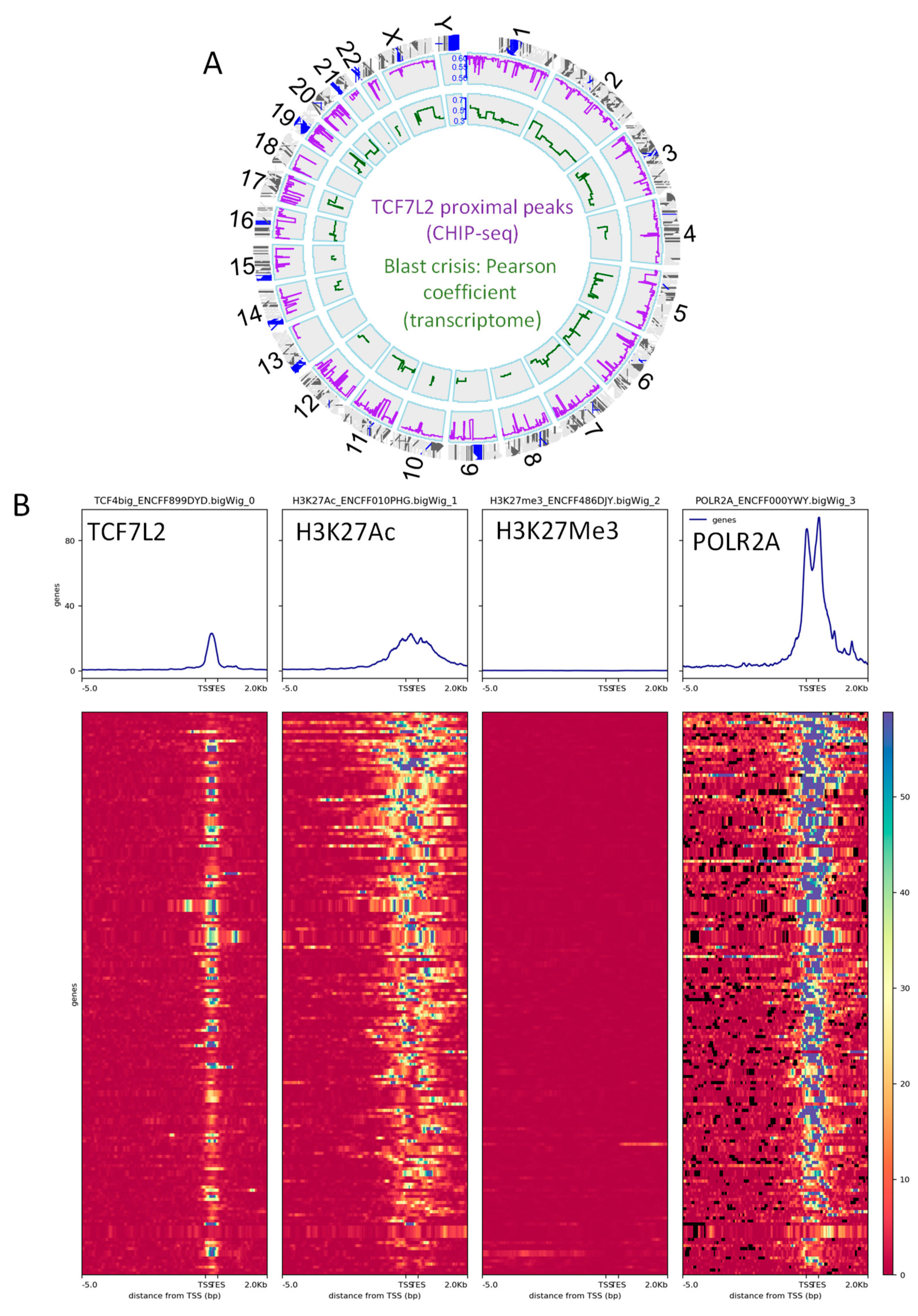
2.2. TCF7L2 Transcriptional Program is Active during CML Blast Crisis
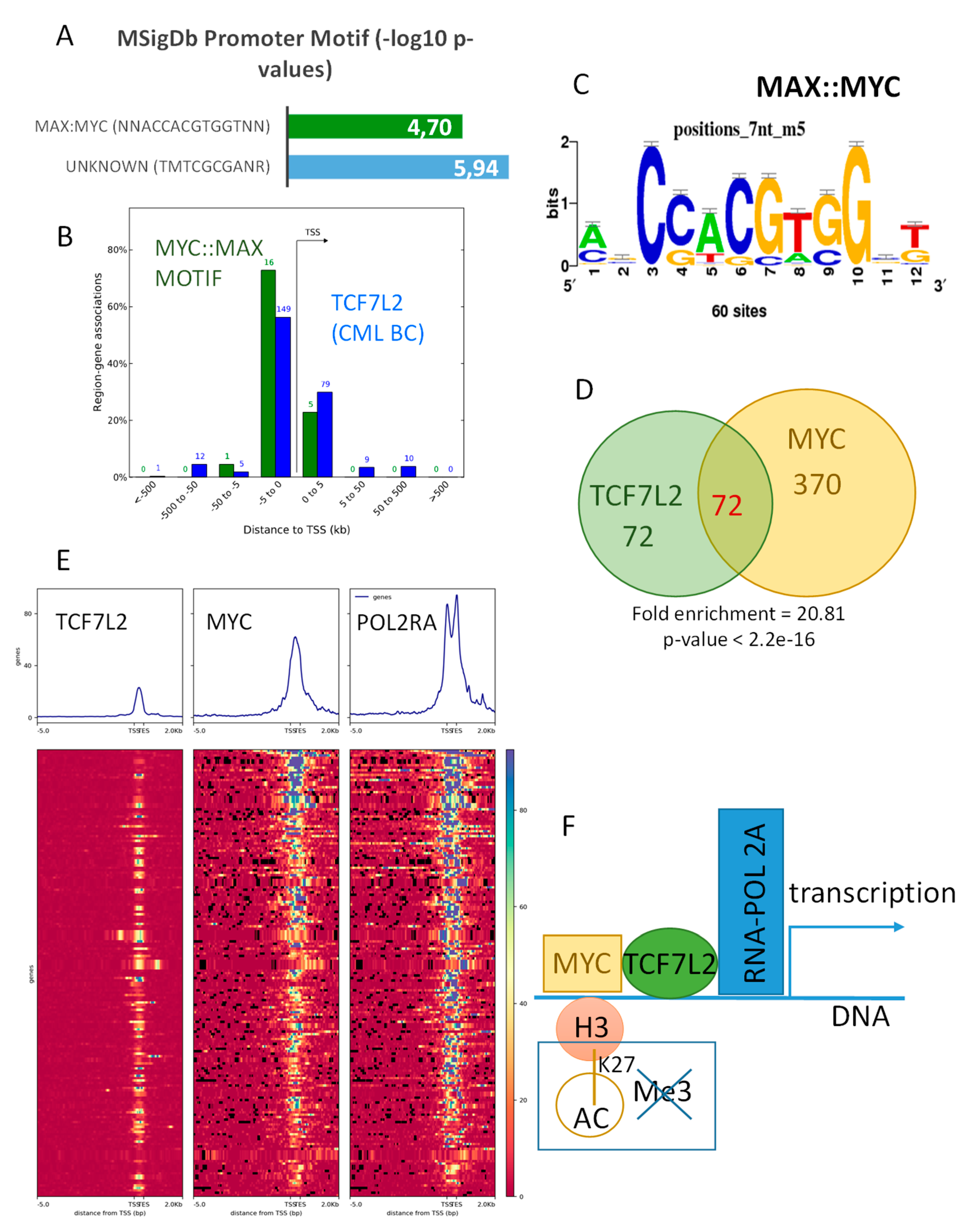
2.3. TCF7L2 Program Activated in CML-BC Shared with MYC Binding on Chromatin
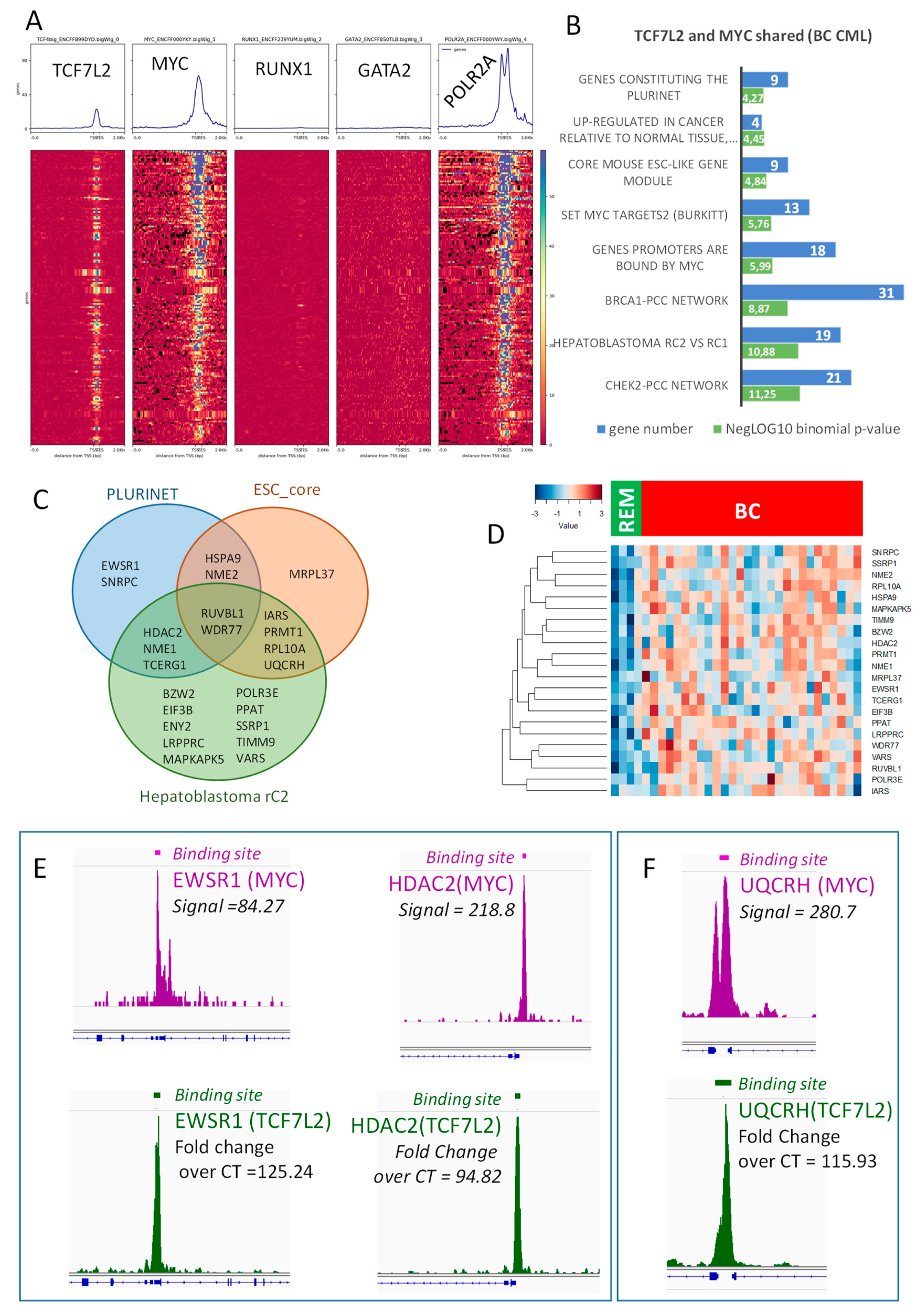
2.4. Transcriptional Program Shared between TCF7L2 and MYC during Blast Crisis is Independent of the Hematopoietic Profile
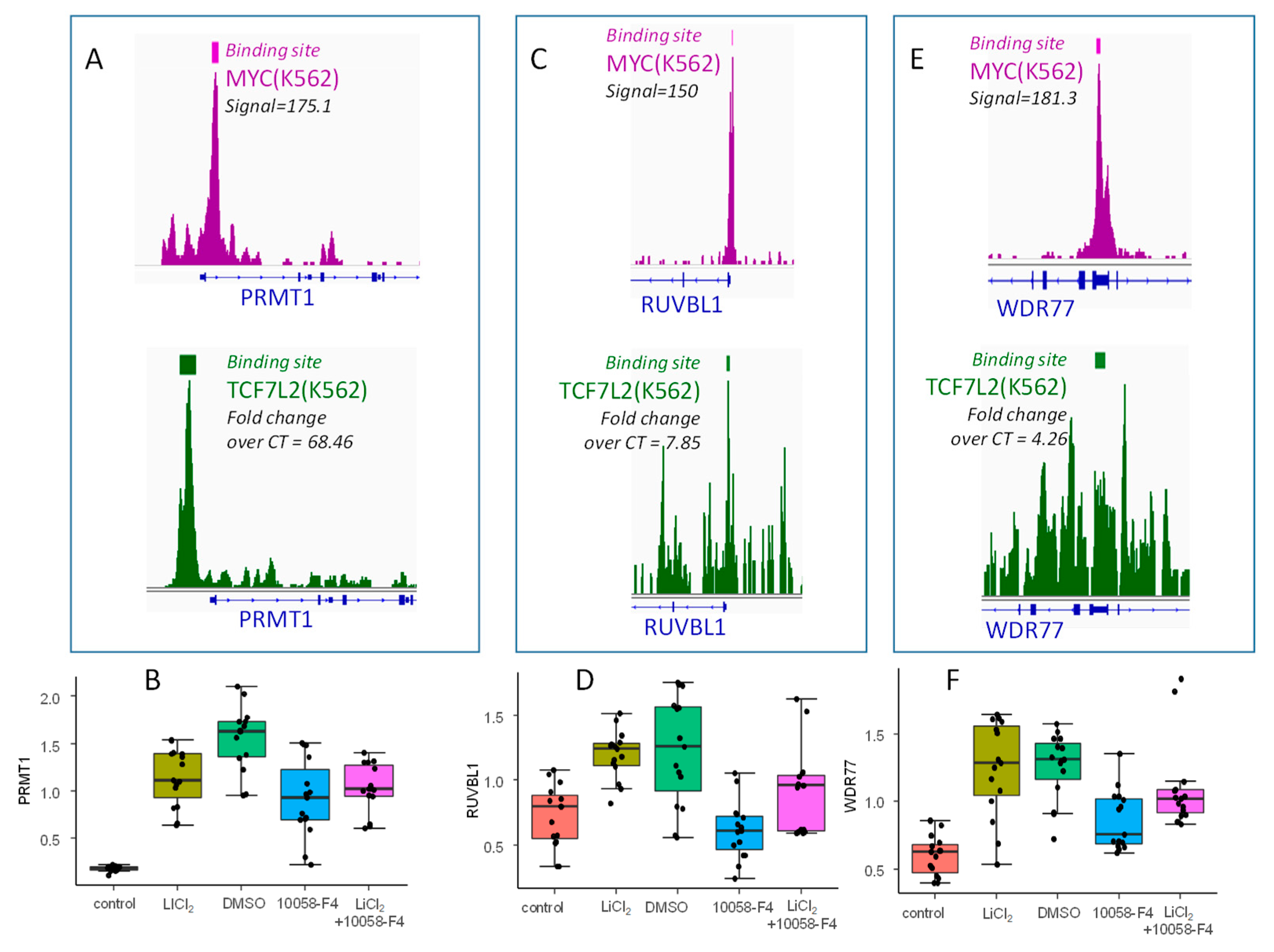
2.5. TCF7L2/MYC Cooperation is a Druggable Target
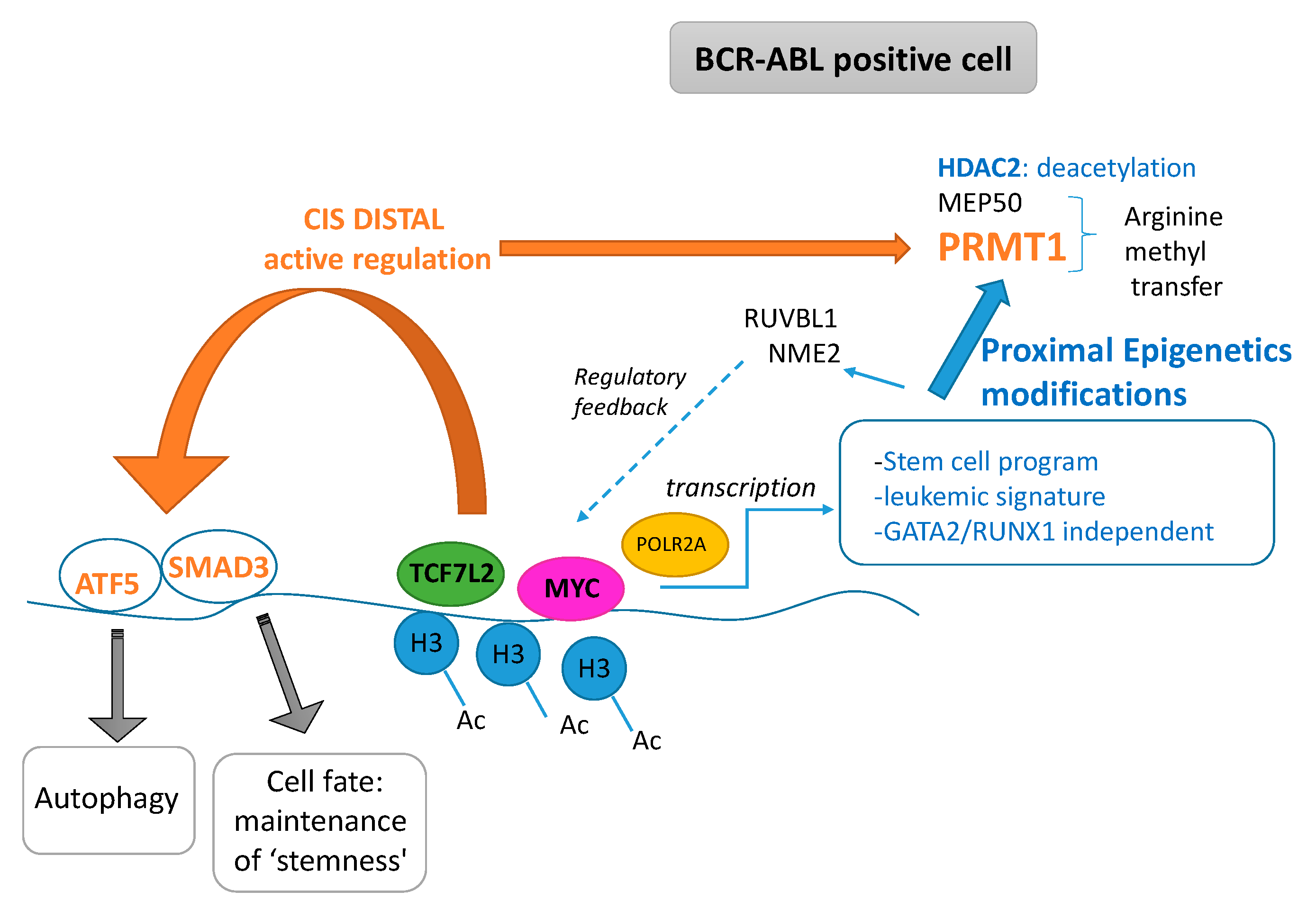
3. Discussion
4. Materials and Methods
4.1. Datasets Bioinformatics Integration
4.1.1. Public Chip-sequencing Data
4.1.2. Public Transcriptome Datasets
4.2. Epigenetic Integrative Analyses
4.3. Hematopoietic Cell Culture
4.4. RNA Extraction and Quantitative Reverse Transcription Polymerase Chain Reaction (QRT-PCR)
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Daley, G.Q.; Van Etten, R.A.; Baltimore, D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990, 247, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Chomel, J.-C.; Bonnet, M.-L.; Sorel, N.; Bertrand, A.; Meunier, M.-C.; Fichelson, S.; Melkus, M.; Bennaceur-Griscelli, A.; Guilhot, F.; Turhan, A.G. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood 2011, 118, 3657–3660. [Google Scholar] [CrossRef]
- Chomel, J.C.; Bonnet, M.-L.; Sorel, N.; Sloma, I.; Bennaceur-Griscelli, A.; Rea, D.; Legros, L.; Marfaing-Koka, A.; Bourhis, J.-H.; Ame, S.; et al. Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget 2016, 7, 35293–35301. [Google Scholar] [CrossRef]
- Staal, F.J.T.; Chhatta, A.; Mikkers, H. Caught in a Wnt storm: Complexities of Wnt signaling in hematopoiesis. Exp. Hematol. 2016, 44, 451–457. [Google Scholar] [CrossRef]
- Staal, F.J.T.; Famili, F.; Garcia Perez, L.; Pike-Overzet, K. Aberrant Wnt Signaling in Leukemia. Cancers 2016, 8. [Google Scholar] [CrossRef]
- Abraham, S.A.; Hopcroft, L.E.M.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.K.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef]
- Spicuglia, S.; Vanhille, L. Chromatin signatures of active enhancers. Nucl. Austin Tex. 2012, 3, 126–131. [Google Scholar] [CrossRef]
- Sawyers, C.L.; Callahan, W.; Witte, O.N. Dominant negative MYC blocks transformation by ABL oncogenes. Cell 1992, 70, 901–910. [Google Scholar] [CrossRef]
- Srutova, K.; Curik, N.; Burda, P.; Savvulidi, F.; Silvestri, G.; Trotta, R.; Klamova, H.; Pecherkova, P.; Sovova, Z.; Koblihova, J.; et al. BCR-ABL1 mediated miR-150 downregulation through MYC contributed to myeloid differentiation block and drug resistance in chronic myeloid leukemia. Haematologica 2018, 103, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.; Ye, B.; Li, X.; Margulies, K.B.; Xu, H.; Wang, X.; Li, F. Transcription Factor 7-like 2 Mediates Canonical Wnt/β-Catenin Signaling and c-Myc Upregulation in Heart Failure. Circ. Heart Fail. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Habener, J.F. Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J. Biol. Chem. 2008, 283, 8723–8735. [Google Scholar] [CrossRef] [PubMed]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Hatzis, P.; van der Flier, L.G.; van Driel, M.A.; Guryev, V.; Nielsen, F.; Denissov, S.; Nijman, I.J.; Koster, J.; Santo, E.E.; Welboren, W.; et al. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol. Cell. Biol. 2008, 28, 2732–2744. [Google Scholar] [CrossRef]
- Sheng, Z.; Ma, L.; Sun, J.E.; Zhu, L.J.; Green, M.R. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood 2011, 118, 2840–2848. [Google Scholar] [CrossRef]
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef]
- Wilson, N.K.; Foster, S.D.; Wang, X.; Knezevic, K.; Schütte, J.; Kaimakis, P.; Chilarska, P.M.; Kinston, S.; Ouwehand, W.H.; Dzierzak, E.; et al. Combinatorial transcriptional control in blood stem/progenitor cells: Genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 2010, 7, 532–544. [Google Scholar] [CrossRef]
- Zhang, S.-J.; Ma, L.-Y.; Huang, Q.-H.; Li, G.; Gu, B.-W.; Gao, X.-D.; Shi, J.-Y.; Wang, Y.-Y.; Gao, L.; Cai, X.; et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 2076–2081. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tsuzuki, S.; Minami, Y.; Yamamoto, Y.; Abe, A.; Ohshima, K.; Seto, M.; Naoe, T. Functionally deregulated AML1/RUNX1 cooperates with BCR-ABL to induce a blastic phase-like phenotype of chronic myelogenous leukemia in mice. PLoS ONE 2013, 8, e74864. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.-J.; Laurent, L.C.; Kostka, D.; Ulitsky, I.; Williams, R.; Lu, C.; Park, I.-H.; Rao, M.S.; Shamir, R.; Schwartz, P.H.; et al. Regulatory networks define phenotypic classes of human stem cell lines. Nature 2008, 455, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Cairo, S.; Armengol, C.; De Reyniès, A.; Wei, Y.; Thomas, E.; Renard, C.-A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Leng, Y.; Yang, X.; Gong, Y.; Sun, H.; Wang, K.; Xu, W.; Zheng, Y.; Naren, D.; Shi, R. High-expressing cystic fibrosis transmembrane conductance regulator interacts with histone deacetylase 2 to promote the development of Ph+ leukemia through the HDAC2-mediated PTEN pathway. Leuk. Res. 2017, 57, 9–19. [Google Scholar] [CrossRef]
- Siriboonpiputtana, T.; Zeisig, B.B.; Zarowiecki, M.; Fung, T.K.; Mallardo, M.; Tsai, C.-T.; Lau, P.N.I.; Hoang, Q.C.; Veiga, P.; Barnes, J.; et al. Transcriptional memory of cells of origin overrides β-catenin requirement of MLL cancer stem cells. EMBO J. 2017, 36, 3139–3155. [Google Scholar] [CrossRef]
- Cheung, N.; Fung, T.K.; Zeisig, B.B.; Holmes, K.; Rane, J.K.; Mowen, K.A.; Finn, M.G.; Lenhard, B.; Chan, L.C.; So, C.W.E. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 2016, 29, 32–48. [Google Scholar] [CrossRef]
- Ho, M.-C.; Wilczek, C.; Bonanno, J.B.; Xing, L.; Seznec, J.; Matsui, T.; Carter, L.G.; Onikubo, T.; Kumar, P.R.; Chan, M.K.; et al. Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS ONE 2013, 8, e57008. [Google Scholar] [CrossRef]
- Antonysamy, S.; Bonday, Z.; Campbell, R.M.; Doyle, B.; Druzina, Z.; Gheyi, T.; Han, B.; Jungheim, L.N.; Qian, Y.; Rauch, C.; et al. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 17960–17965. [Google Scholar] [CrossRef]
- Gao, S.; Wu, H.; Wang, F.; Wang, Z. Altered differentiation and proliferation of prostate epithelium in mice lacking the androgen receptor cofactor p44/WDR77. Endocrinology 2010, 151, 3941–3953. [Google Scholar] [CrossRef]
- Zhou, L.; Wu, H.; Lee, P.; Wang, Z. Roles of the androgen receptor cofactor p44 in the growth of prostate epithelial cells. J. Mol. Endocrinol. 2006, 37, 283–300. [Google Scholar] [CrossRef]
- Jin, Y.; Zhou, J.; Xu, F.; Jin, B.; Cui, L.; Wang, Y.; Du, X.; Li, J.; Li, P.; Ren, R.; et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Invest. 2016, 126, 3961–3980. [Google Scholar] [CrossRef] [PubMed]
- Furuno, K.; Masatsugu, T.; Sonoda, M.; Sasazuki, T.; Yamamoto, K. Association of Polycomb group SUZ12 with WD-repeat protein MEP50 that binds to histone H2A selectively in vitro. Biochem. Biophys. Res. Commun. 2006, 345, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Migliori, V.; Müller, J.; Phalke, S.; Low, D.; Bezzi, M.; Mok, W.C.; Sahu, S.K.; Gunaratne, J.; Capasso, P.; Bassi, C.; et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 2012, 19, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, P.; Vaites, L.P.; Kim, J.K.; Mellert, H.; Gurung, B.; Nakagawa, H.; Herlyn, M.; Hua, X.; Rustgi, A.K.; McMahon, S.B.; et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 2010, 18, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Gallant, P. Control of transcription by Pontin and Reptin. Trends Cell Biol. 2007, 17, 187–192. [Google Scholar] [CrossRef]
- Huber, O.; Ménard, L.; Haurie, V.; Nicou, A.; Taras, D.; Rosenbaum, J. Pontin and reptin, two related ATPases with multiple roles in cancer. Cancer Res. 2008, 68, 6873–6876. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, J.; Yan, L.; Tang, Y.; Zhang, W.; Li, D.; Zang, Y.; Kong, F.; Xu, Z. Cytoplasmic expression of pontin in renal cell carcinoma correlates with tumor invasion, metastasis and patients’ survival. PLoS ONE 2015, 10, e0118659. [Google Scholar] [CrossRef]
- Postel, E.H.; Berberich, S.J.; Flint, S.J.; Ferrone, C.A. Human c-myc transcription factor PuF identified as nm23-H2 nucleoside diphosphate kinase, a candidate suppressor of tumor metastasis. Science 1993, 261, 478–480. [Google Scholar] [CrossRef]
- Breig, O.; Bras, S.; Martinez Soria, N.; Osman, D.; Heidenreich, O.; Haenlin, M.; Waltzer, L. Pontin is a critical regulator for AML1-ETO-induced leukemia. Leukemia 2014, 28, 1271–1279. [Google Scholar] [CrossRef]
- Gertz, J.; Savic, D.; Varley, K.E.; Partridge, E.C.; Safi, A.; Jain, P.; Cooper, G.M.; Reddy, T.E.; Crawford, G.E.; Myers, R.M. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol. Cell 2013, 52, 25–36. [Google Scholar] [CrossRef]
- Radich, J.P.; Dai, H.; Mao, M.; Oehler, V.; Schelter, J.; Druker, B.; Sawyers, C.; Shah, N.; Stock, W.; Willman, C.L.; et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, P.; Noble, W.S. Analysis of strain and regional variation in gene expression in mouse brain. Genome Biol. 2001, 2, RESEARCH0042. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, H.; Ma, J.; Zang, C.; Wang, C.; Wang, J.; Tang, Q.; Meyer, C.A.; Zhang, Y.; Liu, X.S. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 2013, 8, 2502–2515. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desterke, C.; Hugues, P.; Hwang, J.W.; Bennaceur-Griscelli, A.; Turhan, A.G. Embryonic Program Activated during Blast Crisis of Chronic Myelogenous Leukemia (CML) Implicates a TCF7L2 and MYC Cooperative Chromatin Binding. Int. J. Mol. Sci. 2020, 21, 4057. https://doi.org/10.3390/ijms21114057
Desterke C, Hugues P, Hwang JW, Bennaceur-Griscelli A, Turhan AG. Embryonic Program Activated during Blast Crisis of Chronic Myelogenous Leukemia (CML) Implicates a TCF7L2 and MYC Cooperative Chromatin Binding. International Journal of Molecular Sciences. 2020; 21(11):4057. https://doi.org/10.3390/ijms21114057
Chicago/Turabian StyleDesterke, Christophe, Patricia Hugues, Jin Wook Hwang, Annelise Bennaceur-Griscelli, and Ali G. Turhan. 2020. "Embryonic Program Activated during Blast Crisis of Chronic Myelogenous Leukemia (CML) Implicates a TCF7L2 and MYC Cooperative Chromatin Binding" International Journal of Molecular Sciences 21, no. 11: 4057. https://doi.org/10.3390/ijms21114057
APA StyleDesterke, C., Hugues, P., Hwang, J. W., Bennaceur-Griscelli, A., & Turhan, A. G. (2020). Embryonic Program Activated during Blast Crisis of Chronic Myelogenous Leukemia (CML) Implicates a TCF7L2 and MYC Cooperative Chromatin Binding. International Journal of Molecular Sciences, 21(11), 4057. https://doi.org/10.3390/ijms21114057