Ionising Radiation Induces Promoter DNA Hypomethylation and Perturbs Transcriptional Activity of Genes Involved in Morphogenesis during Gastrulation in Zebrafish



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
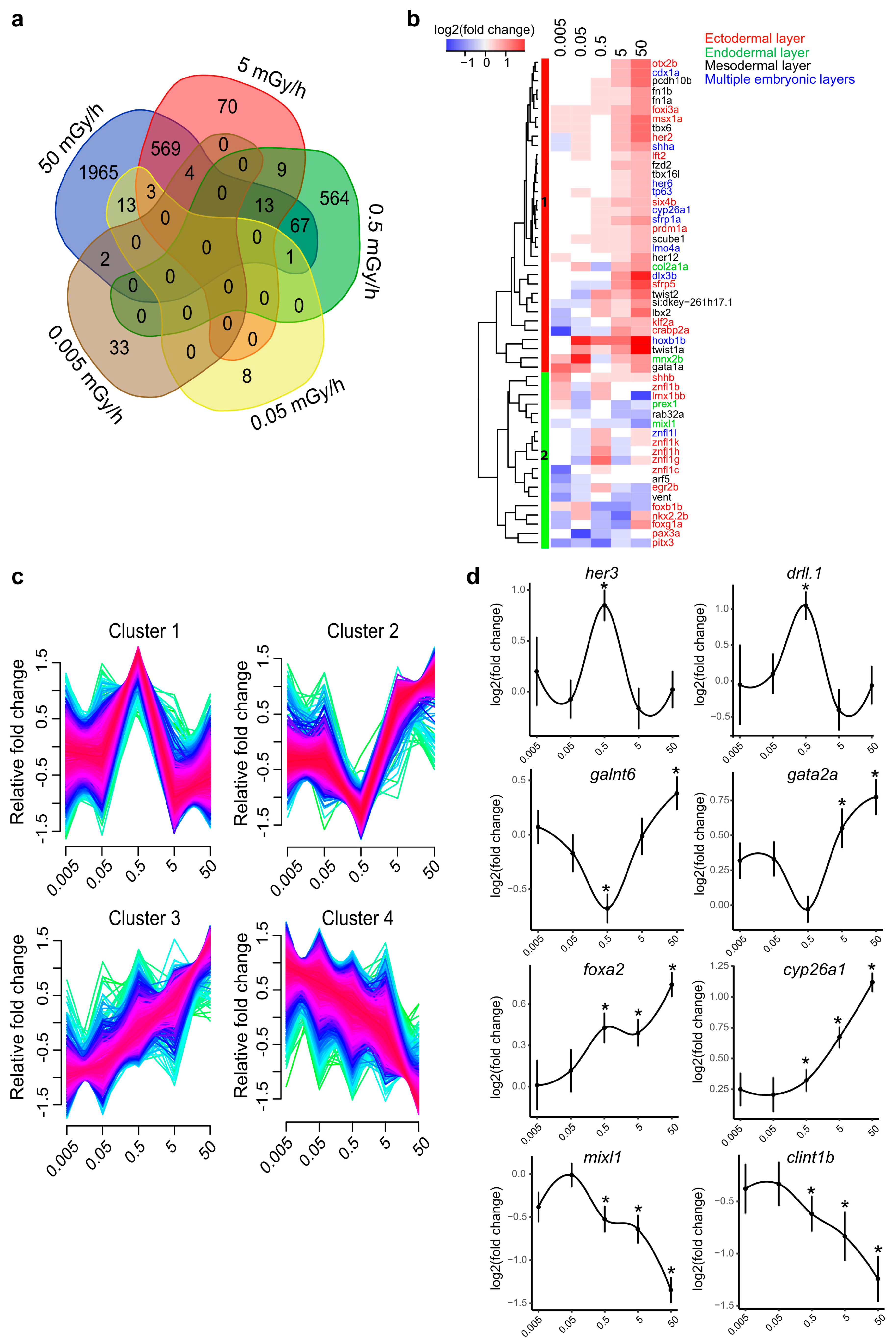
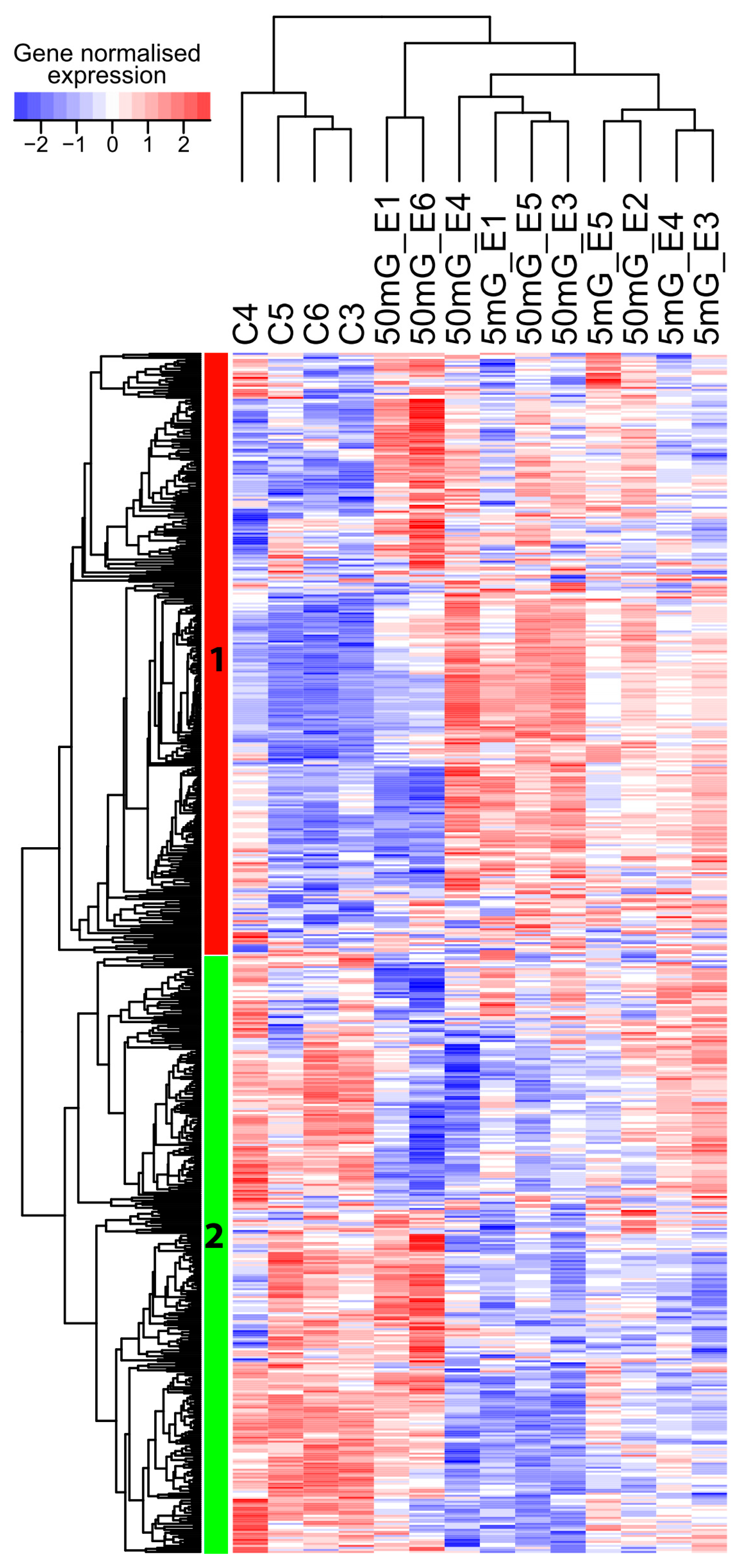
2.1. A Transcriptome-Wide Analysis Reveals Changes in the Expression of Genes Involved in Morphogenesis after Exposure to IR
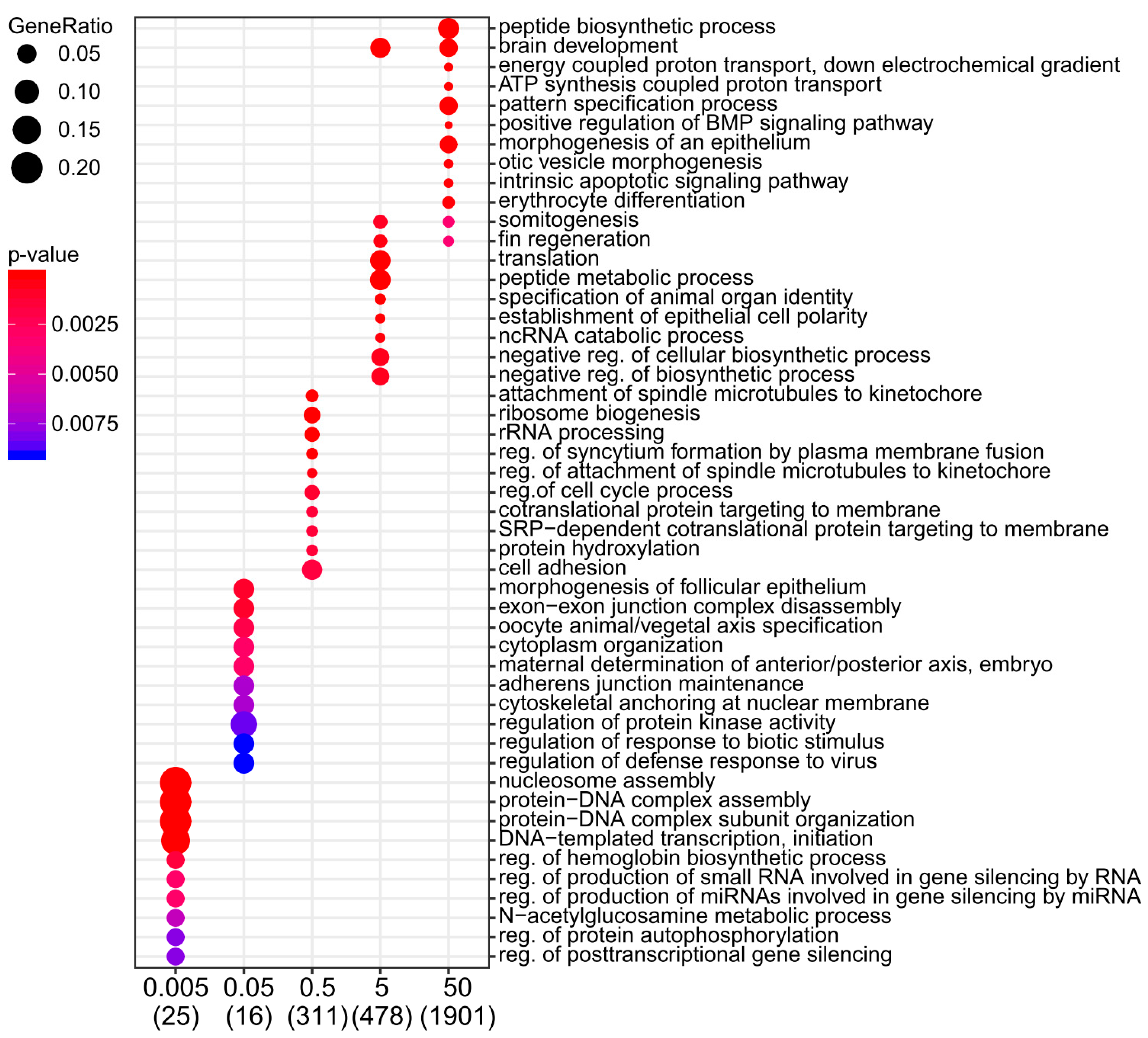
2.2. Moderate and High Dose Rates of IR Impact Biological Pathways Involved in Ectoderm and Mesoderm Development
2.3. Promoter Analysis Reveals Enrichment of Master Regulators Expressed in the Three Germ Layers and Involved in Mitochondria Energetic Metabolism
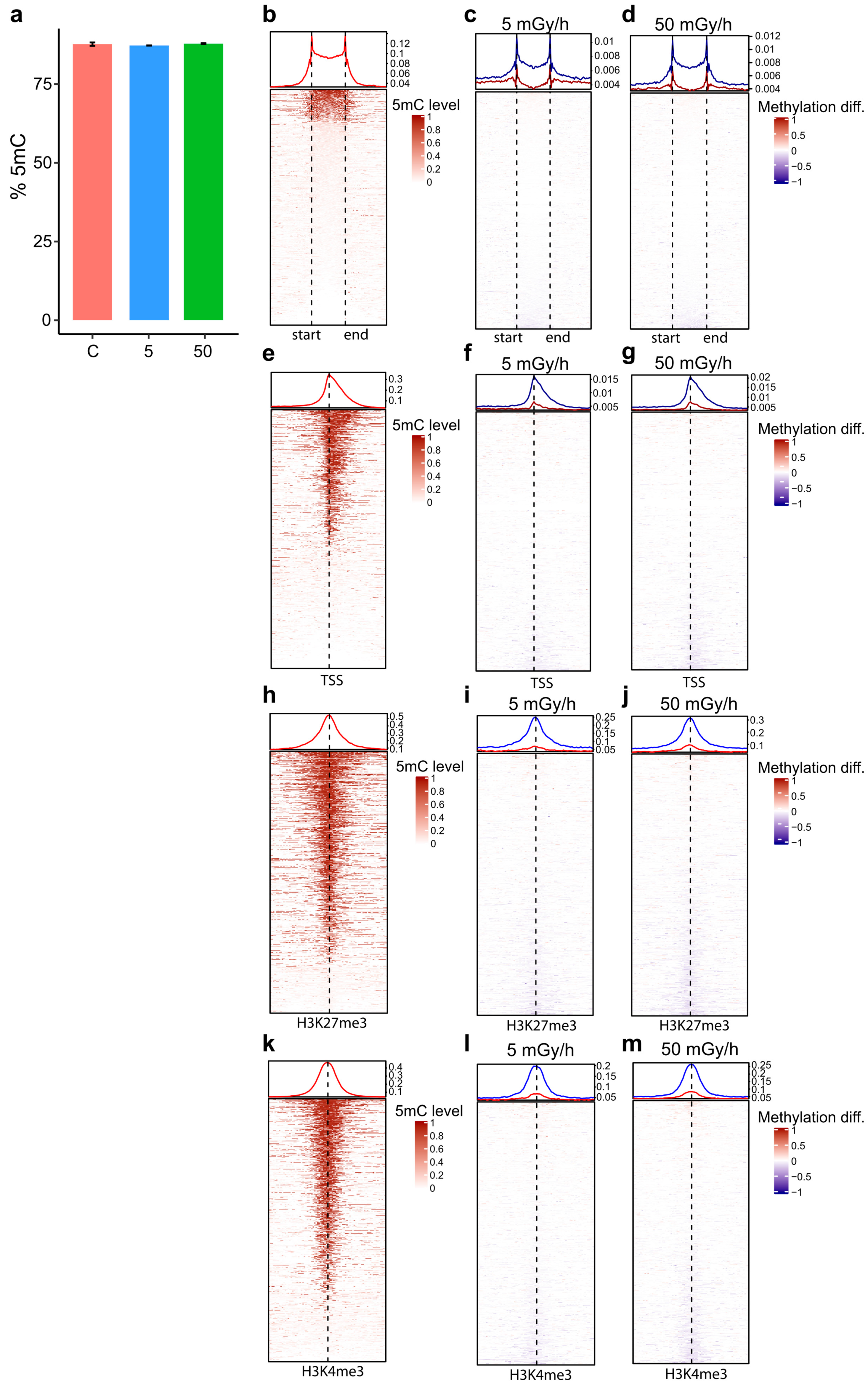
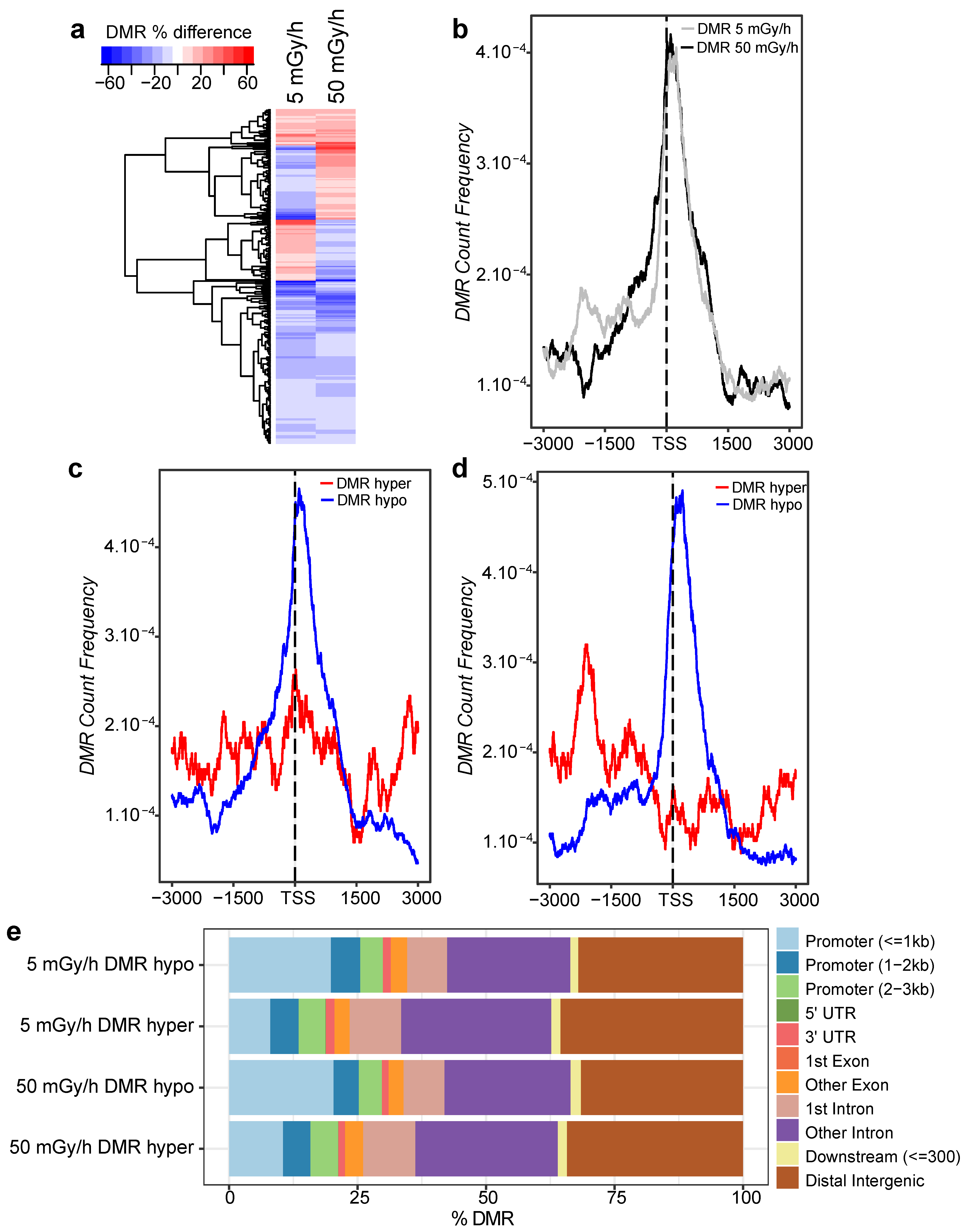
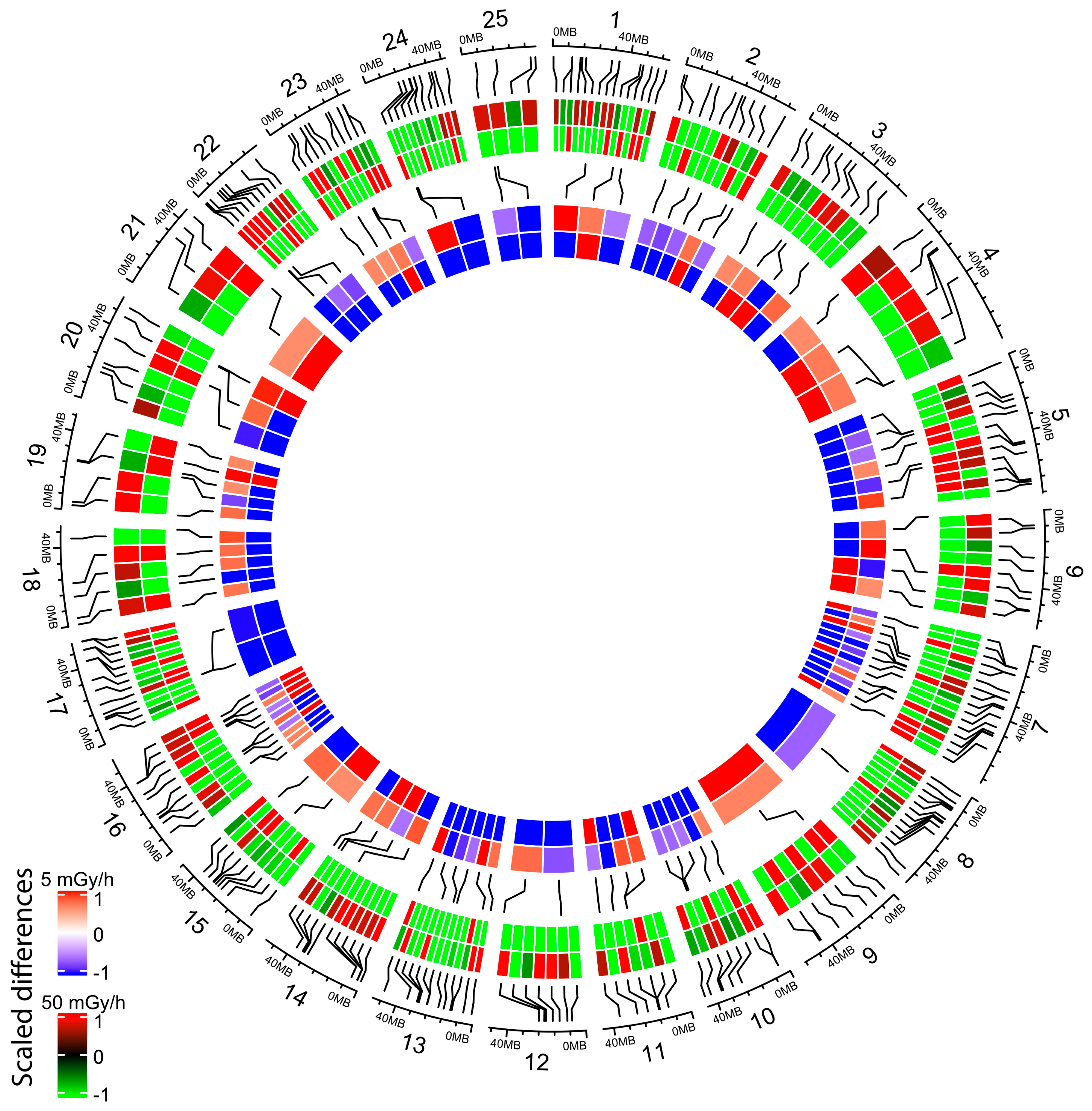
2.4. Embryonic Exposure to Moderate and High Dose Rates of IR Induces Promoter Hypomethylation
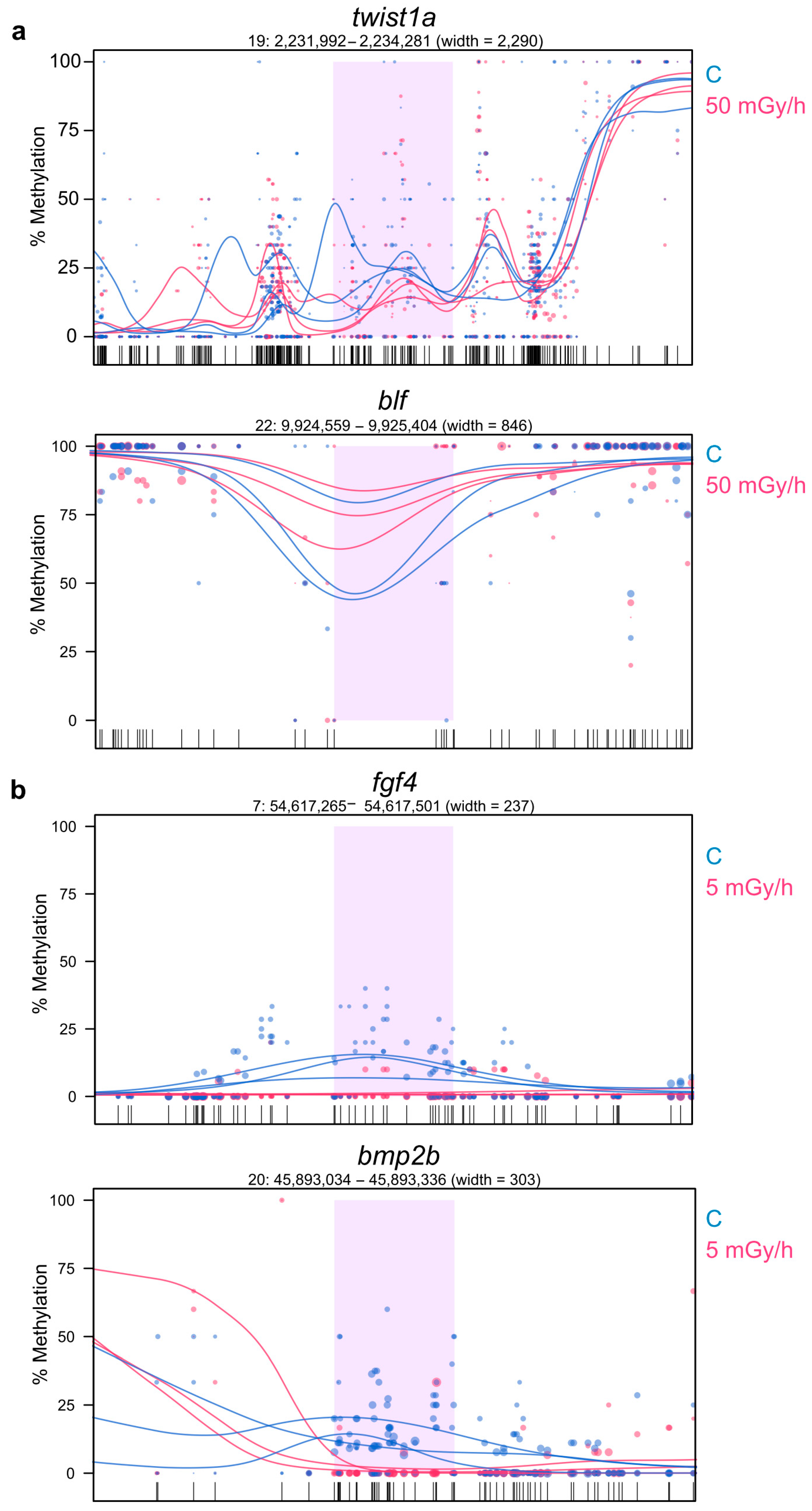
2.5. Promoter Hypomethylation after Exposure to Moderate and High Dose Rates of IR Modulates Transcriptional Activity of Important Developmental Genes
3. Discussion
4. Methods
4.1. Animal Experimentation and Ethics
4.2. Irradiation
4.3. Sequencing Libraries Preparation and Data Generation for Transcriptomic Analysis
4.4. Gene Ontology Analysis
4.5. TF-Binding Sites Enrichment
4.6. Purification of Genomic DNA
4.7. Bisulphite Conversion and Production of WGBS Data
4.8. Bioinformatic Analysis of WGBS Data
4.9. Association with Genomic Features
4.10. Analysis of H3K3me3 and H3K27me3 ChIP Data
5. Declarations
5.1. Ethics Approval and Consent to Participate
5.2. Availability of Data and Materials
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Husseneder, C.; Donaldson, J.R.; Foil, L.D. Impact of the 2010 Deepwater Horizon oil spill on population size and genetic structure of horse flies in Louisiana marshes. Sci. Rep. 2016, 6, 18968. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, A.; Dubansky, B.; Bodinier, C.; Garcia, T.I.; Miles, S.; Pilley, C.; Raghunathan, V.; Roach, J.L.; Walker, N.; Walter, R.B.; et al. Genomic and physiological footprint of the Deepwater Horizon oil spill on resident marsh fishes. Proc. Natl. Acad. Sci. USA 2012, 109, 20298–20302. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.H.; Rice, S.D.; Short, J.W.; Esler, D.; Bodkin, J.L.; Ballachey, B.E.; Irons, D.B. Long-Term Ecosystem Response to the Exxon Valdez Oil Spill. Science 2003, 302, 2082–2086. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Astudillo, V.; Henning, J.; Valenza, L.; Knott, L.; McKinnon, A.; Larkin, R.; Allavena, R. A Necropsy Study of Disease and Comorbidity Trends in Morbidity and Mortality in the Koala (Phascolarctos cinereus) in South-East Queensland, Australia. Sci. Rep. 2019, 9, 17494. [Google Scholar] [CrossRef]
- Lair, S.; Measures, L.N.; Martineau, D. Pathologic Findings and Trends in Mortality in the Beluga (Delphinapterus leucas) Population of the St Lawrence Estuary, Quebec, Canada, From 1983 to 2012. Vet. Pathol. 2016, 53, 22–36. [Google Scholar] [CrossRef]
- Pesavento, P.A.; Agnew, D.; Keel, M.K.; Woolard, K.D. Cancer in wildlife: Patterns of emergence. Nat. Rev. Cancer 2018, 18, 646–661. [Google Scholar] [CrossRef]
- McAloose, D.; Newton, A.L. Wildlife cancer: A conservation perspective. Nat. Rev. Cancer 2009, 9, 517–526. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 2011, 31, 363–373. [Google Scholar] [CrossRef]
- Rice, D.; Barone, S., Jr. Critical Periods of Vulnerability for the Developing Nervous System: Evidence from Humans and Animal Models. Environ. Health Perspect. 2000, 108, 511–533. [Google Scholar] [PubMed]
- UNSCEAR. Sources and Effecs of Ionizing Radiation; United Nations Office: Vienna, Austria, 2000. [Google Scholar]
- Schier, A.F. The maternal-zygotic transition: Death and birth of RNAs. Science 2007, 316, 406–407. [Google Scholar] [CrossRef] [PubMed]
- Haberle, V.; Li, N.; Hadzhiev, Y.; Plessy, C.; Previti, C.; Nepal, C.; Gehrig, J.; Dong, X.; Akalin, A.; Suzuki, A.M.; et al. Two independent transcription initiation codes overlap on vertebrate core promoters. Nature 2014, 507, 381–385. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef]
- Potok, M.E.; Nix, D.A.; Parnell, T.J.; Cairns, B.R. Reprogramming the Maternal Zebrafish Genome after Fertilization to Match the Paternal Methylation Pattern. Cell 2013, 153, 759–772. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, J.; Wang, J.-J.; Wang, L.; Zhang, L.; Li, G.; Yang, X.; Ma, X.; Sun, X.; Cai, J.; et al. Sperm, but Not Oocyte, DNA Methylome Is Inherited by Zebrafish Early Embryos. Cell 2013, 153, 773–784. [Google Scholar] [CrossRef]
- Kiecker, C.; Bates, T.; Bell, E. Molecular specification of germ layers in vertebrate embryos. Cell. Mol. Life Sci. 2016, 73, 923–947. [Google Scholar] [CrossRef]
- Farrell, J.A.; Wang, Y.; Riesenfeld, S.J.; Shekhar, K.; Regev, A.; Schier, A.F. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science (80-) 2018, 360, 3131. [Google Scholar] [CrossRef]
- Loree, J.; Koturbash, I.; Kutanzi, K.; Baker, M.; Pogribny, I.; Kovalchuk, O. Radiation-induced molecular changes in rat mammary tissue: Possible implications for radiation-induced carcinogenesis. Int. J. Radiat. Biol. 2006, 82, 805–815. [Google Scholar] [CrossRef]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J.; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Landscape of somatic retrotransposition in human cancers. Science (80-) 2012, 337, 967–971. [Google Scholar] [CrossRef]
- Gama-sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Bernal, A.J.; Dolinoy, D.C.; Huang, D.; Skaar, D.A.; Weinhouse, C.; Jirtle, R.L. Adaptive radiation-induced epigenetic alterations mitigated by antioxidants. FASEB J. 2013, 27, 665–671. [Google Scholar] [CrossRef]
- Lindeman, L.C.; Kamstra, J.H.; Ballangby, J.; Hurem, S.; Martín, L.M.; Brede, D.A.; Teien, H.C.; Oughton, D.H.; Salbu, B.; Lyche, J.L.; et al. Gamma radiation induces locus specific changes to histone modification enrichment in zebrafish and Atlantic salmon. PLoS ONE 2019, 14, e0212123. [Google Scholar] [CrossRef]
- Murat El Houdigui, S.; Adam-Guillermin, C.; Loro, G.; Arcanjo, C.; Frelon, S.; Floriani, M.; Dubourg, N.; Baudelet, E.; Audebert, S.; Camoin, L.; et al. A systems biology approach reveals neuronal and muscle developmental defects after chronic exposure to ionising radiation in zebrafish. Sci. Rep. 2019, 9, 20241. [Google Scholar] [CrossRef]
- Garnier-Laplace, J.; Della-Vedova, C.; Andersson, P.; Copplestone, D.; Cailes, C.; Beresford, N.A.; Howard, B.J.; Howe, P.; Whitehouse, P. A multi-criteria weight of evidence approach for deriving ecological benchmarks for radioactive substances. J. Radiol. Prot. 2012, 30, 215–233. [Google Scholar] [CrossRef]
- ICRP. Annex d. Radiation effects in reference animals and plants. Ann. ICRP 2008, 38, 179–229. [Google Scholar] [CrossRef]
- Schier, A.F.; Talbot, W.S. Molecular Genetics of Axis Formation in Zebrafish. Annu. Rev. Genet. 2005, 39, 561–613. [Google Scholar] [CrossRef]
- Sukonina, V.; Ma, H.; Zhang, W.; Bartesaghi, S.; Subhash, S.; Heglind, M.; Foyn, H.; Betz, M.J.; Nilsson, D.; Lidell, M.E.; et al. FOXK1 and FOXK2 regulate aerobic glycolysis. Nature 2019, 566, 279–283. [Google Scholar] [CrossRef]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef]
- Lindeman, L.C.; Andersen, I.S.; Reiner, A.H.; Li, N.; Aanes, H.; Østrup, O.; Winata, C.; Mathavan, S.; Müller, F.; Aleström, P.; et al. Prepatterning of Developmental Gene Expression by Modified Histones before Zygotic Genome Activation. Dev. Cell 2011, 21, 993–1004. [Google Scholar] [CrossRef]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.-N. Comparative Analysis of Transposable Elements Highlights Mobilome Diversity and Evolution in Vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Olek, A.; Walter, J. The pre-implantation ontogeny of the H19 methylation imprint. Nat. Genet. 1997, 17, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.W.; Skvarca, L.B.; Thisse, C.; Thisse, B.; Hukriede, N.A.; Davidson, A.J. BMP and retinoic acid regulate anterior-posterior patterning of the non-axial mesoderm across the dorsal-ventral axis. Nat. Commun. 2016, 7, 12197. [Google Scholar] [CrossRef] [PubMed]
- Sumanas, S.; Zhang, B.; Dai, R.; Lin, S. 15-Zinc finger protein Bloody Fingers is required for zebrafish morphogenetic movements during neurulation. Dev. Biol. 2005, 283, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.H.; Schmid, B.; Trout, J.; Connors, S.A.; Ekker, M.; Mullins, M.C. Ventral and lateral regions of the zebrafish gastrula, including the Neural crest progenitors, are established by a bmp2b/swirl pathway of genes. Dev. Biol. 1998, 199, 93–110. [Google Scholar] [CrossRef]
- Bladen, C.L.; Lam, W.K.; Dynan, W.S.; Kozlowski, D.J. DNA damage response and Ku80 function in the vertebrate embryo. Nucleic Acids Res. 2005, 33, 3002–3010. [Google Scholar] [CrossRef]
- Zaichkina, S.I.; Rozanova, O.M.; Aptikaeva, G.F.; Achmadieva, A.C.; Klokov, D.Y. Low Doses of Gamma-Radiation Induce Nonlinear Dose Responses in Mammalian and Plant Cells. Nonlinearity Biol. Toxicol. Med. 2004, 2, 154014204905198. [Google Scholar] [CrossRef]
- Miller, R.K.; McCrea, P.D. Wnt to build a tube: Contributions of Wnt signaling to epithelial tubulogenesis. Dev. Dyn. 2010, 239, 77–93. [Google Scholar] [CrossRef]
- Bier, E.; De Robertis, E.M. BMP gradients: A paradigm for morphogen-mediated developmental patterning. Science 2015, 348, aaa5838. [Google Scholar] [CrossRef]
- Siebel, C.; Lendahl, U. Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef]
- Gagnaire, B.; Cavalié, I.; Pereira, S.; Floriani, M.; Dubourg, N.; Camilleri, V.; Adam-Guillermin, C. External gamma irradiation-induced effects in early-life stages of zebrafish, Danio rerio. Aquat. Toxicol. 2015, 169, 69–78. [Google Scholar] [CrossRef]
- El Ghouzzi, V.; Bianchi, F.T.; Molineris, I.; Mounce, B.C.; Berto, G.E.; Rak, M.; Lebon, S.; Aubry, L.; Tocco, C.; Gai, M.; et al. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly and p53. Cell Death Dis. 2016, 7, e2440. [Google Scholar] [CrossRef]
- Miller, R.W.; Blot, W.J. Small head size after in-utero exposure to atomic radiation. Lancet 1972, 2, 784–787. [Google Scholar] [CrossRef]
- Otake, M.; Schull, W.J. In utero exposure to A-bomb radiation and mental retardation; a reassessment. Br. J. Radiol. 1984, 57, 409–414. [Google Scholar] [CrossRef]
- Otake, M.; Schull, W.J.; Lee, S. Threshold for radiation-related severe mental retardation in prenatally exposed A-bomb survivors: A re-analysis. Int. J. Radiat. Biol. 1996, 70, 755–763. [Google Scholar] [CrossRef]
- Hayama, S.-I.; Tsuchiya, M.; Ochiai, K.; Nakiri, S.; Nakanishi, S.; Ishii, N.; Kato, T.; Tanaka, A.; Konno, F.; Kawamoto, Y.; et al. Small head size and delayed body weight growth in wild Japanese monkey fetuses after the Fukushima Daiichi nuclear disaster. Sci. Rep. 2017, 7, 3528. [Google Scholar] [CrossRef]
- Møller, A.P.; Bonisoli-Alquati, A.; Rudolfsen, G.; Mousseau, T.A. Chernobyl birds have smaller brains. PLoS ONE 2011, 6, e16862. [Google Scholar] [CrossRef]
- Hurem, S.; Martín, L.M.; Brede, D.A.; Skjerve, E.; Nourizadeh-Lillabadi, R.; Lind, O.C.; Christensen, T.; Berg, V.; Teien, H.-C.; Salbu, B.; et al. Dose-dependent effects of gamma radiation on the early zebrafish development and gene expression. PLoS ONE 2017, 12, e0179259. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Raiche, J.; Rodriguez-Juarez, R.; Pogribny, I.; Kovalchuk, O. Sex- and tissue-specific expression of maintenance and de novo DNA methyltransferases upon low dose X-irradiation in mice. Biochem. Biophys. Res. Commun. 2004, 325, 39–47. [Google Scholar] [CrossRef]
- Baubec, T.; Schübeler, D. Genomic patterns and context specific interpretation of DNA methylation. Curr. Opin. Genet. Dev. 2014, 25, 85–92. [Google Scholar] [CrossRef]
- Vastenhouw, N.L.; Zhang, Y.; Woods, I.G.; Imam, F.; Regev, A.; Liu, X.S.; Rinn, J.; Schier, A.F. Chromatin signature of embryonic pluripotency is established during genome activation. Nature 2010, 464, 922–926. [Google Scholar] [CrossRef]
- Kalinich, J.F.; Catravas, G.N.; Snyder, S.L. The Effect of gamma Radiation on DNA Methylation. Radiat. Res. 1989, 197, 185–197. [Google Scholar] [CrossRef]
- Pogribny, I.; Koturbash, I.; Tryndyak, V.; Hudson, D.; Stevenson, S.M.L.; Sedelnikova, O.; Bonner, W.; Kovalchuk, O. Fractionated low-dose radiation exposure leads to accumulation of DNA damage and profound alterations in DNA and histone methylation in the murine thymus. Mol. Cancer Res. 2005, 3, 553–561. [Google Scholar] [CrossRef]
- Miousse, I.R.; Kutanzi, K.R.; Koturbash, I. Effects of ionizing radiation on DNA methylation: From experimental biology to clinical applications. Int. J. Radiat. Biol. 2017, 93, 457–469. [Google Scholar] [CrossRef]
- Kovalchuk, O.; Burke, P.; Besplug, J.; Slovack, M.; Filkowski, J.; Pogribny, I. Methylation changes in muscle and liver tissues of male and female mice exposed to acute and chronic low-dose X-ray-irradiation. Mutat. Res. Mol. Mech. Mutagen. 2004, 548, 75–84. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. DNA methylation and genetic instability in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 1997, 94, 2545–2550. [Google Scholar] [CrossRef]
- Chen, R.Z.; Pettersson, U.; Beard, C.; Jackson-Grusby, L.; Jaenisch, R. DNA hypomethylation leads to elevated mutation rates. Nature 1998, 395, 89–93. [Google Scholar] [CrossRef]
- Kamstra, J.H.; Hurem, S.; Martin, L.M.; Lindeman, L.C.; Legler, J.; Oughton, D.; Salbu, B.; Brede, D.A.; Lyche, J.L.; Aleström, P. Ionizing radiation induces transgenerational effects of DNA methylation in zebrafish. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Kam, W.W.Y.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Di Pietro, G.; Magno, L.A.V.; Rios-Santos, F. Glutathione S-transferases: An overview in cancer research. Expert Opin. Drug Metab. Toxicol. 2010, 6, 153–170. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Cyr, A.R.; Domann, F.E. The redox basis of epigenetic modifications: From mechanisms to functional consequences. Antioxid. Redox Signal. 2011, 15, 551–589. [Google Scholar] [CrossRef]
- Lee, D.H.; Jacobs, D.R.; Porta, M. Hypothesis: A unifying mechanism for nutrition and chemicals as lifelong modulators of DNA hypomethylation. Environ. Health Perspect. 2009, 117, 1799–1802. [Google Scholar] [CrossRef]
- Honjo, Y.; Ichinohe, T. Cellular responses to ionizing radiation change quickly over time during early development in zebrafish. Cell Biol. Int. 2019, 43, 516–527. [Google Scholar] [CrossRef]
- Kloypan, C.; Srisa-art, M.; Mutirangura, A.; Boonla, C. LINE-1 hypomethylation induced by reactive oxygen species is mediated via depletion of S-adenosylmethionine. Cell Biochem. Funct. 2015, 33, 375–384. [Google Scholar] [CrossRef]
- Niedzwiecki, M.M.; Hall, M.N.; Liu, X.; Oka, J.; Harper, K.N.; Slavkovich, V.; Ilievski, V.; Levy, D.; van Geen, A.; Mey, J.L.; et al. Blood glutathione redox status and global methylation of peripheral blood mononuclear cell DNA in Bangladeshi adults. Epigenetics 2013, 8, 730–738. [Google Scholar] [CrossRef]
- Lertratanangkoon, K.; Wu, C.J.; Savaraj, N.; Thomas, M.L. Alterations of DNA methylation by glutathione depletion. Cancer Lett. 1997, 120, 149–156. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Xu, K.; Mao, X.; Xue, L.; Liu, X.; Yu, H.; Chen, L.; Chu, X. Genome-wide screen of DNA methylation changes induced by low dose X-ray radiation in mice. PLoS ONE 2014, 9, e90804. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Futschik, M.E.; Carlisle, B. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 2005, 3, 965–988. [Google Scholar] [CrossRef] [PubMed]
- Adrian Alexa, J.R. TopGO: Enrichment Analysis for Gene Ontology, R package version 2.36.0; Bioconductor; 2019; Available online: http://www.bioconductor.org/ (accessed on 1 June 2020). [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2 Elegant Graphics for Data Analysis. In Ggplot2 1–7; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Kwon, A.T.; Arenillas, D.J.; Worsley Hunt, R.; Wasserman, W.W. oPOSSUM-3: Advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 (Bethesda) 2012, 2, 987–1002. [Google Scholar] [CrossRef]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Chèneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Feng, H.; Conneely, K.N.; Wu, H. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014, 42, e69. [Google Scholar] [CrossRef]
- Laufer, B.I.; Hwang, H.; Vogel Ciernia, A.; Mordaunt, C.E.; LaSalle, J.M. Whole genome bisulfite sequencing of Down syndrome brain reveals regional DNA hypermethylation and novel disorder insights. Epigenetics 2019, 14, 672–684. [Google Scholar] [CrossRef]
- Korthauer, K.; Chakraborty, S.; Benjamini, Y.; Irizarry, R.A. Detection and accurate false discovery rate control of differentially methylated regions from whole genome bisulfite sequencing. Biostatistics 2019, 20, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.D.; Langmead, B.; Irizarry, R.A. BSmooth: From whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012, 13, R83. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M.; Ishaque, N. EnrichedHeatmap: An R/Bioconductor package for comprehensive visualization of genomic signal associations. BMC Genom. 2018, 19, 234. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murat El Houdigui, S.; Adam-Guillermin, C.; Armant, O. Ionising Radiation Induces Promoter DNA Hypomethylation and Perturbs Transcriptional Activity of Genes Involved in Morphogenesis during Gastrulation in Zebrafish. Int. J. Mol. Sci. 2020, 21, 4014. https://doi.org/10.3390/ijms21114014
Murat El Houdigui S, Adam-Guillermin C, Armant O. Ionising Radiation Induces Promoter DNA Hypomethylation and Perturbs Transcriptional Activity of Genes Involved in Morphogenesis during Gastrulation in Zebrafish. International Journal of Molecular Sciences. 2020; 21(11):4014. https://doi.org/10.3390/ijms21114014
Chicago/Turabian StyleMurat El Houdigui, Sophia, Christelle Adam-Guillermin, and Olivier Armant. 2020. "Ionising Radiation Induces Promoter DNA Hypomethylation and Perturbs Transcriptional Activity of Genes Involved in Morphogenesis during Gastrulation in Zebrafish" International Journal of Molecular Sciences 21, no. 11: 4014. https://doi.org/10.3390/ijms21114014
APA StyleMurat El Houdigui, S., Adam-Guillermin, C., & Armant, O. (2020). Ionising Radiation Induces Promoter DNA Hypomethylation and Perturbs Transcriptional Activity of Genes Involved in Morphogenesis during Gastrulation in Zebrafish. International Journal of Molecular Sciences, 21(11), 4014. https://doi.org/10.3390/ijms21114014