Flavin Adenine Dinucleotide Fluorescence as an Early Marker of Mitochondrial Impairment During Brain Hypoxia

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
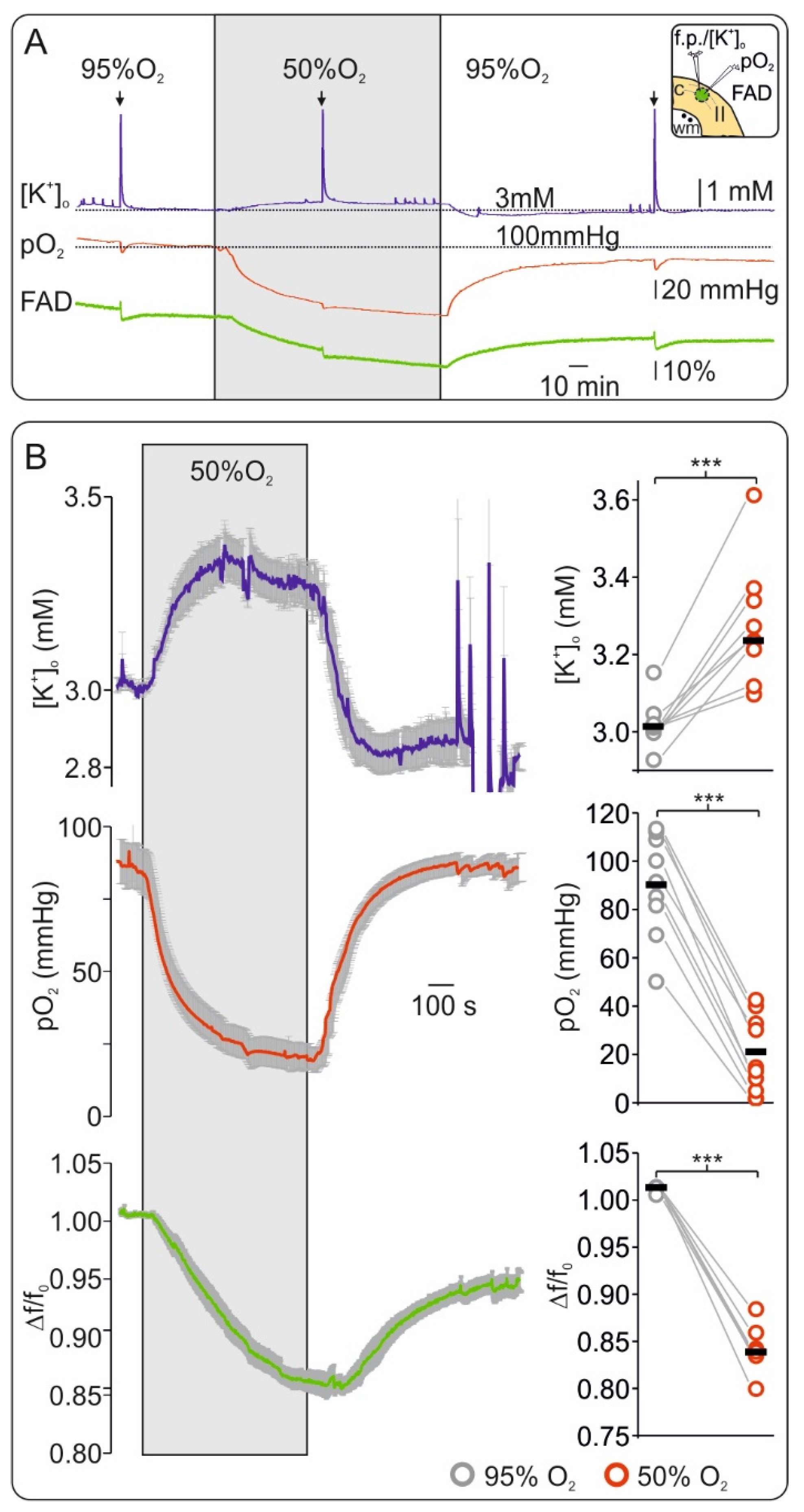
2.1. Oxygen Depletion Correlates with Changes in FAD Redox State and Impairment of Synaptic Transmission in Brain Slices
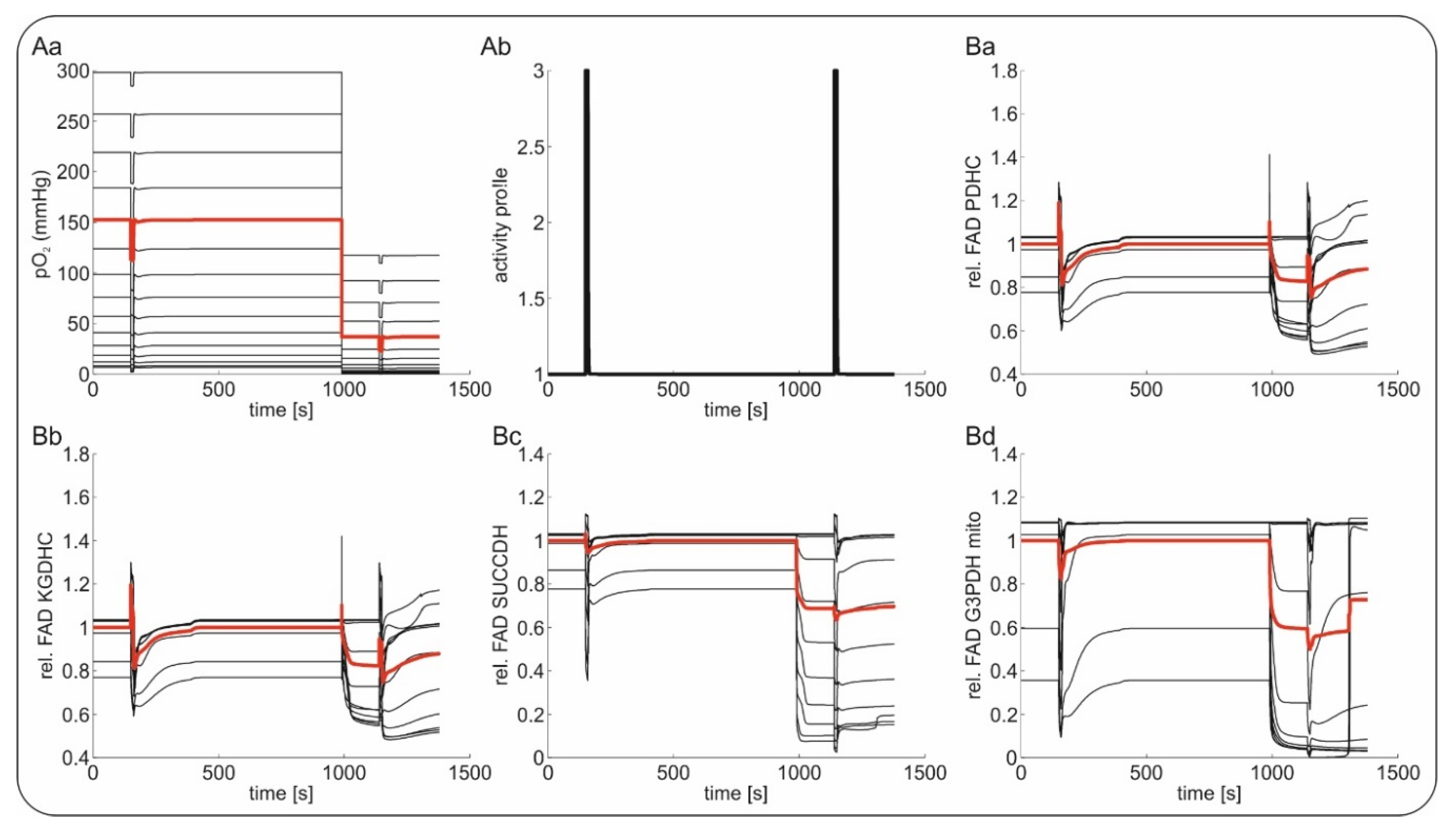
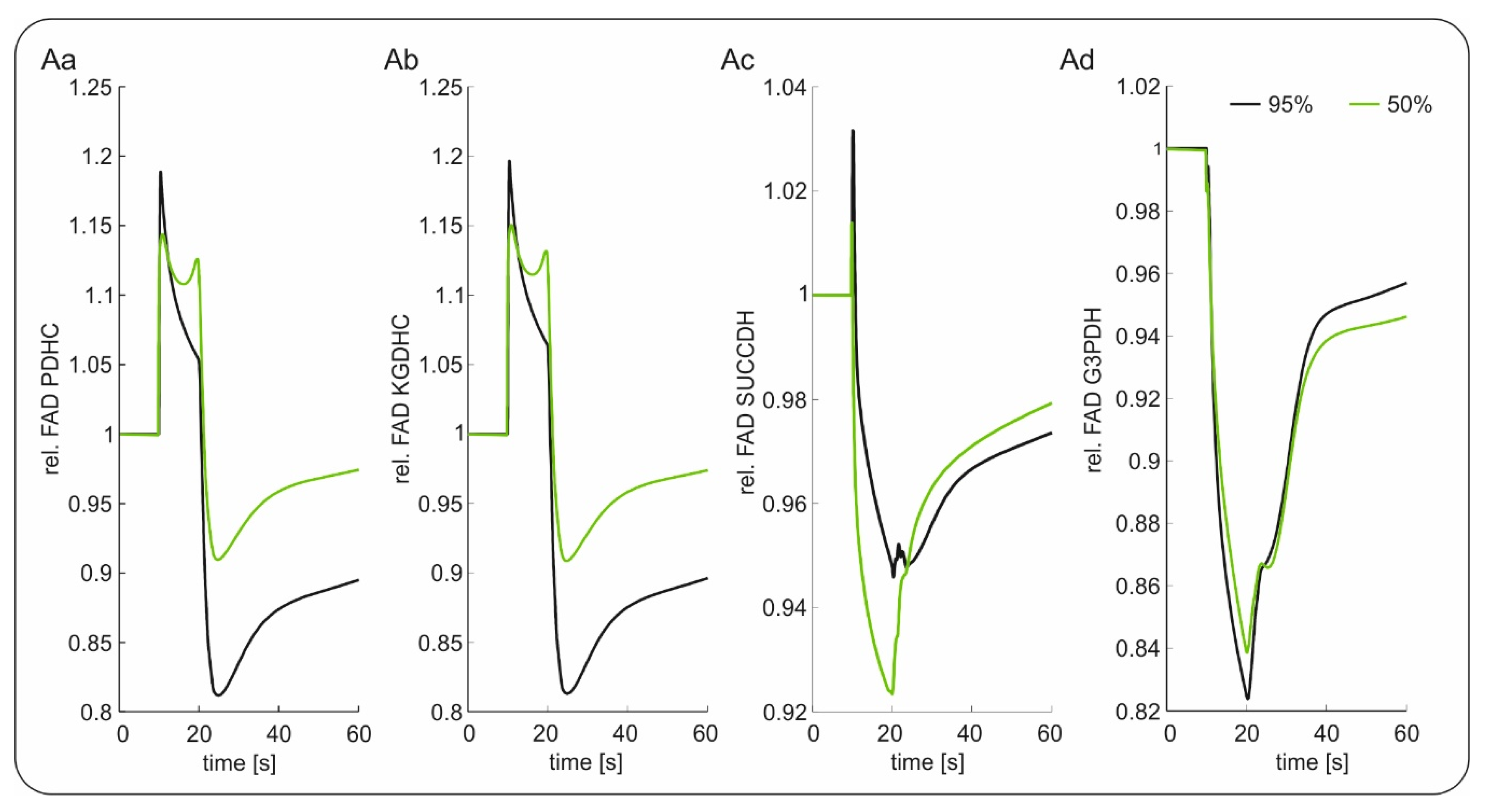
2.2. Slice Modeling: The Influence of Oxygen Supply on the Basal and the Activation-Induced FAD Response
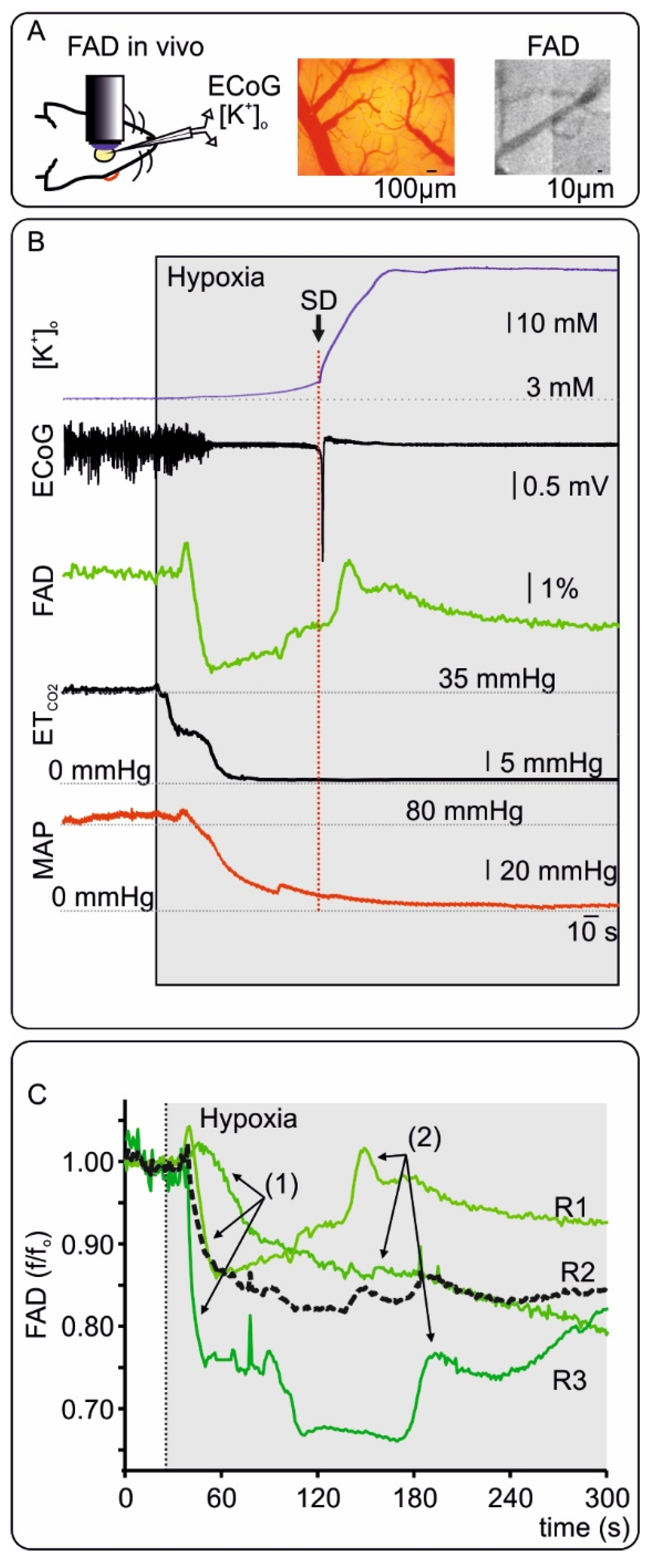
2.3. FAD Dynamics during Terminal Hypoxia In Vivo
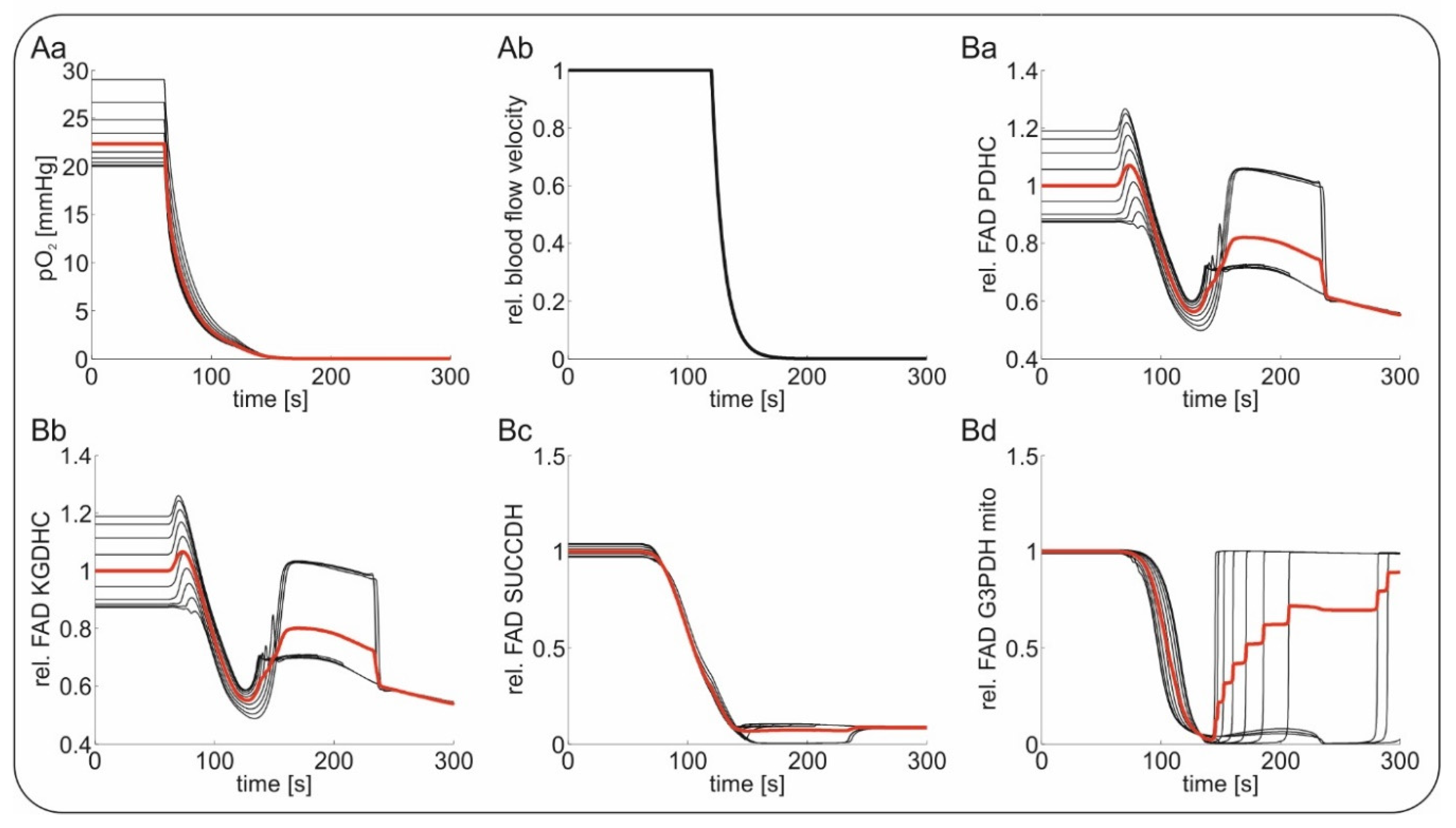
2.4. Computational Modeling of FAD Response to Hypoxia In Vivo
3. Discussion
3.1. A FAD Reductive Shift Is an Early Sign of Oxygen Depletion In Vitro
3.2. FAD Fluorescence during Hypoxia and Global Ischemia In Vivo
4. Materials and Methods
4.1. Slice Preparation and Maintenance
4.2. Electrophysiology, Oxygen Recordings and Fluorescence Recordings in Brain Slices
4.3. In Vivo FAD Recordings
4.4. Data Analysis of Experimental Data
4.5. Metabolic Model
4.6. In Vitro Slice Model
4.7. In Vivo Tissue Model
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harrison, T.R. Harrison’s Principles of Internal Medicine, 20th ed.; McGraw-Hill Education: New York, NY, USA, 2018; ISBN 978-1-259-64403-0. [Google Scholar]
- Frontera, J.; Ziai, W.; O’Phelan, K.; Leroux, P.D.; Kirkpatrick, P.J.; Diringer, M.N.; Suarez, J.I. Regional brain monitoring in the neurocritical care unit. Neurocrit. Care 2015, 22, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Reddy, U.; Robba, C.; Sharma, D.; Citerio, G. Acute ischaemic stroke: Challenges for the intensivist. Intensive Care Med. 2019, 45, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Reiffurth, C. The stroke-migraine depolarization continuum. Neuron 2015, 86, 902–922. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; Shuttleworth, C.W.; Kirov, S.A.; Ayata, C.; Hinzman, J.M.; Foreman, B.; Andrew, R.D.; Boutelle, M.G.; Brennan, K.C.; Carlson, A.P.; et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leão’s legacy. J. Cereb. Blood Flow Metab. 2017, 37, 1571–1594. [Google Scholar] [CrossRef]
- Dreier, J.P.; Lemale, C.L.; Kola, V.; Friedman, A.; Schoknecht, K. Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology 2018, 134, 189–207. [Google Scholar] [CrossRef]
- Lückl, J.; Lemale, C.L.; Kola, V.; Horst, V.; Khojasteh, U.; Oliveira-Ferreira, A.I.; Major, S.; Winkler, M.K.L.; Kang, E.-J.; Schoknecht, K.; et al. The negative ultraslow potential, electrophysiological correlate of infarction in the human cortex. Brain 2018, 141, 1734–1752. [Google Scholar] [CrossRef]
- Dreier, J.P.; Major, S.; Foreman, B.; Winkler, M.K.L.; Kang, E.-J.; Milakara, D.; Lemale, C.L.; DiNapoli, V.; Hinzman, J.M.; Woitzik, J.; et al. Terminal spreading depolarization and electrical silence in death of human cerebral cortex. Ann. Neurol. 2018, 83, 295–310. [Google Scholar] [CrossRef]
- Somjen, G.G. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev. 2001, 81, 1065–1096. [Google Scholar] [CrossRef]
- Feuerstein, D.; Manning, A.; Hashemi, P.; Bhatia, R.; Fabricius, M.; Tolias, C.; Pahl, C.; Ervine, M.; Strong, A.J.; Boutelle, M.G. Dynamic metabolic response to multiple spreading depolarizations in patients with acute brain injury: An online microdialysis study. J. Cereb. Blood Flow Metab. 2010, 30, 1343–1355. [Google Scholar] [CrossRef]
- Selman, W.R.; Lust, W.D.; Pundik, S.; Zhou, Y.; Ratcheson, R.A. Compromised metabolic recovery following spontaneous spreading depression in the penumbra. Brain Res. 2004, 999, 167–174. [Google Scholar] [CrossRef]
- Takano, T.; Tian, G.-F.; Peng, W.; Lou, N.; Lovatt, D.; Hansen, A.J.; Kasischke, K.A.; Nedergaard, M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat. Neurosci. 2007, 10, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Piilgaard, H.; Witgen, B.M.; Rasmussen, P.; Lauritzen, M. Cyclosporine A, FK506, and NIM811 ameliorate prolonged CBF reduction and impaired neurovascular coupling after cortical spreading depression. J. Cereb. Blood Flow Metab. 2011, 31, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Gerich, F.J.; Hepp, S.; Probst, I.; Müller, M. Mitochondrial inhibition prior to oxygen-withdrawal facilitates the occurrence of hypoxia-induced spreading depression in rat hippocampal slices. J. Neurophysiol. 2006, 96, 492–504. [Google Scholar] [CrossRef]
- Yang, J.-L.; Mukda, S.; Chen, S.-D. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018, 16, 263–275. [Google Scholar] [CrossRef]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef]
- Korbakis, G.; Vespa, P.M. Multimodal neurologic monitoring. Handb. Clin. Neurol. 2017, 140, 91–105. [Google Scholar] [CrossRef]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; Shuttleworth, C.W.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef]
- Kunz, W.S.; Kunz, W. Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria. Biochim. Biophys. Acta 1985, 841, 237–246. [Google Scholar] [CrossRef]
- Heikal, A.A. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark. Med. 2010, 4, 241–263. [Google Scholar] [CrossRef]
- Rösner, J.; Liotta, A.; Schmitz, D.; Heinemann, U.; Kovács, R. A LED-based method for monitoring NAD(P)H and FAD fluorescence in cell cultures and brain slices. J. Neurosci. Methods 2013, 212, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Cohen, P.; Jobsis, F.; Schoener, B. Intracellular oxidation-reduction states in vivo. Science 1962, 137, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Hassinen, I.E. Reflectance spectrophotometric and surface fluorometric methods for measuring the redox state of nicotinamide nucleotides and flavins in intact tissues. Meth. Enzymol. 1986, 123, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R.; Biscoe, T.J. Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. J. Physiol. (Lond.) 1992, 450, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.A.; Balaban, R.S. Direct imaging of dehydrogenase activity within living cells using enzyme-dependent fluorescence recovery after photobleaching (ED-FRAP). Biophys. J. 2001, 80, 2018–2028. [Google Scholar] [CrossRef][Green Version]
- Schuchmann, S.; Kovacs, R.; Kann, O.; Heinemann, U.; Buchheim, K. Monitoring NAD(P)H autofluorescence to assess mitochondrial metabolic functions in rat hippocampal-entorhinal cortex slices. Brain Res. Brain Res. Protoc. 2001, 7, 267–276. [Google Scholar] [CrossRef]
- Shibuki, K.; Hishida, R.; Murakami, H.; Kudoh, M.; Kawaguchi, T.; Watanabe, M.; Watanabe, S.; Kouuchi, T.; Tanaka, R. Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J. Physiol. (Lond.) 2003, 549, 919–927. [Google Scholar] [CrossRef]
- Shuttleworth, C.W.; Brennan, A.M.; Connor, J.A. NAD(P)H fluorescence imaging of postsynaptic neuronal activation in murine hippocampal slices. J. Neurosci. 2003, 23, 3196–3208. [Google Scholar] [CrossRef]
- Reinert, K.C.; Dunbar, R.L.; Gao, W.; Chen, G.; Ebner, T.J. Flavoprotein autofluorescence imaging of neuronal activation in the cerebellar cortex in vivo. J. Neurophysiol. 2004, 92, 199–211. [Google Scholar] [CrossRef]
- Foster, K.A.; Beaver, C.J.; Turner, D.A. Interaction between tissue oxygen tension and NADH imaging during synaptic stimulation and hypoxia in rat hippocampal slices. Neuroscience 2005, 132, 645–657. [Google Scholar] [CrossRef]
- Liotta, A.; Rösner, J.; Huchzermeyer, C.; Wojtowicz, A.; Kann, O.; Schmitz, D.; Heinemann, U.; Kovács, R. Energy demand of synaptic transmission at the hippocampal Schaffer-collateral synapse. J. Cereb. Blood Flow Metab. 2012, 32, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Rösner, J.; Liotta, A.; Angamo, E.A.; Spies, C.; Heinemann, U.; Kovács, R. Minimizing photodecomposition of flavin adenine dinucleotide fluorescence by the use of pulsed LEDs. J. Microsc. 2016, 264, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Mayevsky, A. Mitochondrial Function In Vivo Evaluated by NADH Fluorescence; Springer International Publishing: Cham, Switzerland, 2015; ISBN 978-3-319-16681-0. [Google Scholar]
- Clavijo, J.A.; van Bastelaar, J.; Pinsky, M.R.; Puyana, J.C.; Mayevsky, A. Minimally invasive real time monitoring of mitochondrial NADH and tissue blood flow in the urethral wall during hemorrhage and resuscitation. Med. Sci. Monit. 2008, 14, BR175-82. [Google Scholar] [PubMed]
- Mayevsky, A.; Sonn, J.; Barbiro-Michaely, E. Physiological Mapping of Brain Functions In Vivo: Surface Monitoring of Hemodynamic Metabolic Ionic and Electrical Activities in Real-Time. J. Neurosci. Neuroeng. 2013, 2, 150–177. [Google Scholar] [CrossRef]
- Mayevsky, A.; Rogatsky, G.G. Mitochondrial function in vivo evaluated by NADH fluorescence: From animal models to human studies. Am. J. Physiol. Cell Physiol. 2007, 292, C615-40. [Google Scholar] [CrossRef]
- Rampil, I.J.; Litt, L.; Mayevsky, A. Correlated, simultaneous, multiple-wavelength optical monitoring in vivo of localized cerebrocortical NADH and brain microvessel hemoglobin oxygen saturation. J. Clin. Monit. 1992, 8, 216–225. [Google Scholar] [CrossRef]
- Berndt, N.; Kann, O.; Holzhütter, H.-G. Physiology-based kinetic modeling of neuronal energy metabolism unravels the molecular basis of NAD(P)H fluorescence transients. J. Cereb. Blood Flow Metab. 2015, 35, 1494–1506. [Google Scholar] [CrossRef]
- Berndt, N.; Rösner, J.; Haq, R.U.; Kann, O.; Kovács, R.; Holzhütter, H.-G.; Spies, C.; Liotta, A. Possible neurotoxicity of the anesthetic propofol: Evidence for the inhibition of complex II of the respiratory chain in area CA3 of rat hippocampal slices. Arch. Toxicol. 2018, 92, 3191–3205. [Google Scholar] [CrossRef]
- Schoknecht, K.; Berndt, N.; Rösner, J.; Heinemann, U.; Dreier, J.P.; Kovács, R.; Friedman, A.; Liotta, A. Event-Associated Oxygen Consumption Rate Increases ca. Five-Fold When Interictal Activity Transforms into Seizure-Like Events In Vitro. Int. J. Mol. Sci. 2017, 18, 1925. [Google Scholar] [CrossRef]
- Lu, H.-H.; Wu, Y.-M.; Chang, W.-T.; Luo, T.; Yang, Y.-C.; Cho, H.-D.; Liau, I. Molecular imaging of ischemia and reperfusion in vivo with mitochondrial autofluorescence. Anal. Chem. 2014, 86, 5024–5031. [Google Scholar] [CrossRef]
- Ibrahim, B.A.; Wang, H.; Lesicko, A.M.H.; Bucci, B.; Paul, K.; Llano, D.A. Effect of temperature on FAD and NADH-derived signals and neurometabolic coupling in the mouse auditory and motor cortex. Pflugers Arch. 2017, 469, 1631–1649. [Google Scholar] [CrossRef] [PubMed]
- Frostig, R.D. In Vivo Optical Imaging of Brain Function, 2nd ed.; Taylor and Francis Group: Boca Raton, FL, USA, 2010; ISBN 978-1-4200-7684-4. [Google Scholar]
- Schneider, J.; Berndt, N.; Papageorgiou, I.E.; Maurer, J.; Bulik, S.; Both, M.; Draguhn, A.; Holzhütter, H.-G.; Kann, O. Local oxygen homeostasis during various neuronal network activity states in the mouse hippocampus. J. Cereb. Blood Flow Metab. 2019, 39, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Káradóttir, R.; Attwell, D. A preferential role for glycolysis in preventing the anoxic depolarization of rat hippocampal area CA1 pyramidal cells. J. Neurosci. 2005, 25, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Isele, T.; Reiffurth, C.; Offenhauser, N.; Kirov, S.A.; Dahlem, M.A.; Herreras, O. Is spreading depolarization characterized by an abrupt, massive release of gibbs free energy from the human brain cortex? Neuroscientist 2013, 19, 25–42. [Google Scholar] [CrossRef]
- Tasaki, I.; Byrne, P.M. Demonstration of heat production associated with spreading depression in the amphibian retina. Biochem. Biophys. Res. Commun. 1991, 174, 293–297. [Google Scholar] [CrossRef]
- Hashemi, P.; Bhatia, R.; Nakamura, H.; Dreier, J.P.; Graf, R.; Strong, A.J.; Boutelle, M.G. Persisting depletion of brain glucose following cortical spreading depression, despite apparent hyperaemia: Evidence for risk of an adverse effect of Leão’s spreading depression. J. Cereb. Blood Flow Metab. 2009, 29, 166–175. [Google Scholar] [CrossRef]
- Rogers, M.L.; Feuerstein, D.; Leong, C.L.; Takagaki, M.; Niu, X.; Graf, R.; Boutelle, M.G. Continuous online microdialysis using microfluidic sensors: Dynamic neurometabolic changes during spreading depolarization. ACS Chem. Neurosci. 2013, 4, 799–807. [Google Scholar] [CrossRef]
- Huchzermeyer, C.; Berndt, N.; Holzhütter, H.-G.; Kann, O. Oxygen consumption rates during three different neuronal activity states in the hippocampal CA3 network. J. Cereb. Blood Flow Metab. 2013, 33, 263–271. [Google Scholar] [CrossRef]
- Mironov, S.L.; Richter, D.W. Oscillations and hypoxic changes of mitochondrial variables in neurons of the brainstem respiratory centre of mice. J. Physiol. (Lond.) 2001, 533, 227–236. [Google Scholar] [CrossRef]
- Engl, E.; Attwell, D. Non-signalling energy use in the brain. J. Physiol. (Lond.) 2015, 593, 3417–3429. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Fleidervish, I.A.; Gebhardt, C.; Astman, N.; Gutnick, M.J.; Heinemann, U. Enhanced Spontaneous Transmitter Release Is the Earliest Consequence of Neocortical Hypoxia That Can Explain the Disruption of Normal Circuit Function. J. Neurosci. 2001, 21, 4600–4608. [Google Scholar] [CrossRef] [PubMed]
- Revah, O.; Lasser-Katz, E.; Fleidervish, I.A.; Gutnick, M.J. The earliest neuronal responses to hypoxia in the neocortical circuit are glutamate-dependent. Neurobiol. Dis. 2016, 95, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Erdemli, G.; Xu, Y.Z.; Krnjević, K. Potassium conductance causing hyperpolarization of CA1 hippocampal neurons during hypoxia. J. Neurophysiol. 1998, 80, 2378–2390. [Google Scholar] [CrossRef]
- Müller, M.; Somjen, G.G. Na(+) and K(+) concentrations, extra- and intracellular voltages, and the effect of TTX in hypoxic rat hippocampal slices. J. Neurophysiol. 2000, 83, 735–745. [Google Scholar] [CrossRef]
- Canals, S.; Larrosa, B.; Pintor, J.; Mena, M.A.; Herreras, O. Metabolic challenge to glia activates an adenosine-mediated safety mechanism that promotes neuronal survival by delaying the onset of spreading depression waves. J. Cereb. Blood Flow Metab. 2008, 28, 1835–1844. [Google Scholar] [CrossRef]
- Mayevsky, A. Brain energy metabolism of the conscious rat exposed to various physiological and pathological situations. Brain Res. 1976, 113, 327–338. [Google Scholar] [CrossRef]
- Mayevsky, A.; Chance, B. Metabolic responses of the awake cerebral cortex to anoxia hypoxia spreading depression and epileptiform activity. Brain Res. 1975, 98, 149–165. [Google Scholar] [CrossRef]
- Warden, M.R.; Cardin, J.A.; Deisseroth, K. Optical neural interfaces. Annu. Rev. Biomed. Eng. 2014, 16, 103–129. [Google Scholar] [CrossRef]
- Liotta, A.; Caliskan, G.; ul Haq, R.; Hollnagel, J.O.; Rösler, A.; Heinemann, U.; Behrens, C.J. Partial disinhibition is required for transition of stimulus-induced sharp wave-ripple complexes into recurrent epileptiform discharges in rat hippocampal slices. J. Neurophysiol. 2011, 105, 172–187. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berndt, N.; Kovács, R.; Rösner, J.; Wallach, I.; Dreier, J.P.; Liotta, A. Flavin Adenine Dinucleotide Fluorescence as an Early Marker of Mitochondrial Impairment During Brain Hypoxia. Int. J. Mol. Sci. 2020, 21, 3977. https://doi.org/10.3390/ijms21113977
Berndt N, Kovács R, Rösner J, Wallach I, Dreier JP, Liotta A. Flavin Adenine Dinucleotide Fluorescence as an Early Marker of Mitochondrial Impairment During Brain Hypoxia. International Journal of Molecular Sciences. 2020; 21(11):3977. https://doi.org/10.3390/ijms21113977
Chicago/Turabian StyleBerndt, Nikolaus, Richard Kovács, Jörg Rösner, Iwona Wallach, Jens P. Dreier, and Agustin Liotta. 2020. "Flavin Adenine Dinucleotide Fluorescence as an Early Marker of Mitochondrial Impairment During Brain Hypoxia" International Journal of Molecular Sciences 21, no. 11: 3977. https://doi.org/10.3390/ijms21113977
APA StyleBerndt, N., Kovács, R., Rösner, J., Wallach, I., Dreier, J. P., & Liotta, A. (2020). Flavin Adenine Dinucleotide Fluorescence as an Early Marker of Mitochondrial Impairment During Brain Hypoxia. International Journal of Molecular Sciences, 21(11), 3977. https://doi.org/10.3390/ijms21113977