Synthesis and Diels–Alder Reactivity of 4-Fluoro-4-Methyl-4H-Pyrazoles


Abstract
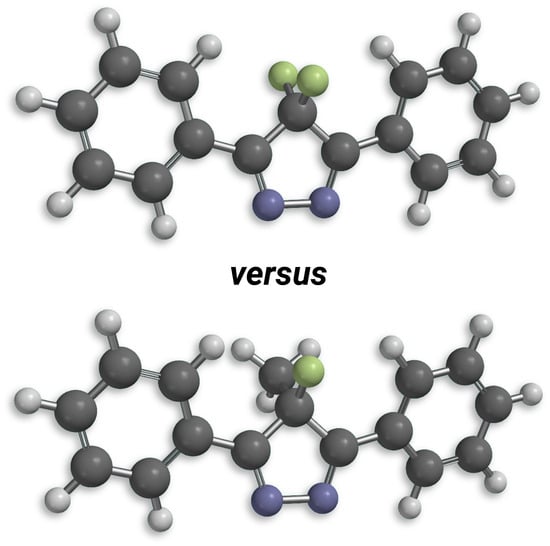
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.1.1. General
3.1.2. Synthesis of 2-Fluoro-1,3-diphenylpropane-1,3-dione (2)
3.1.3. Synthesis of 2-Fluoro-2-methyl-1,3-diphenylpropane-1,3-dione (3)
3.1.4. Synthesis of 4-Fluoro-4-methyl-3,5-diphenyl-4H-pyrazole (MFP) Using Method A
3.1.5. Synthesis of 2-Methyl-1,3-diphenylpropane-1,3-dione (4)
3.1.6. Synthesis of 4-Methyl-3,5-diphenyl-1H-pyrazole (5)
3.1.7. Synthesis of 4-Fluoro-4-methyl-3,5-diphenyl-4H-pyrazole (MFP) Using Method B
3.2. UV–vis Kinetics
3.3. Compound Stability Studies
3.3.1. Diene Stability in FBS
3.3.2. Diene Stability to Glutathione
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Oliveira, B.L.; Guo, Z.; Bernardes, G.J.L. Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef] [PubMed]
- Settle, A.E.; Berstis, L.; Rorrer, N.A.; Roman-Leshkov, Y.; Beckham, G.T.; Richards, R.M.; Vardon, D.R. Heterogeneous Diels–Alder catalysis for biomass-derived aromatic compounds. Green Chem. 2017, 19, 3468–3492. [Google Scholar] [CrossRef]
- Stocking, E.M.; Williams, R.M. Chemistry and biology of biosynthetic Diels–Alder reactions. Angew. Chem. Int. Ed. 2003, 42, 3078–3115. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Devaraj, N.K. Inverse electron-demand Diels–Alder bioorthogonal reactions. Top. Curr. Chem. 2016, 374, 3. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels–Alder reaction in total synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Adam, W.; Ammon, H.; Nau, W.M.; Peters, K. 4-Halo-4H-pyrazoles: Cycloaddition with cyclopentadiene to azoalkanes of the 2,3-diazabicyclo[2.2.1]hept-2-ene type versus electrophilic addition with cyclopentene. J. Org. Chem. 1994, 59, 7067–7071. [Google Scholar] [CrossRef]
- Adam, W.; Harrer, H.M.; Nau, W.M.; Peters, K. Electronic substituent effects on the acid-catalyzed [4 + 2] cycloaddition of isopyrazoles with cyclopentadiene and the photochemical and thermal denitrogenation of the resulting 1,4-diaryl-7,7-dimethyl-2,3-diazabicyclo[2.2.1]hept-2-ene azoalkanes to bicyclo[2.1.0]pentanes. J. Org. Chem. 1994, 59, 3786–3797. [Google Scholar] [CrossRef]
- Beck, K.; Höhn, A.; Hünig, S.; Prokschy, F. Azobrücken aus Azinen, I. Isopyrazole als elektronenarme Diene zur Synthese von 2,3-Diazabicyclo[2.2.1]heptenen. Chem. Ber. 1984, 117, 517–533. [Google Scholar] [CrossRef]
- Beck, K.; Hünig, S.; Klärner, F.-G.; Kraft, P.; Artschwager-Perl, U. Azobrücken aus Azinen, VII1) Diels-Alder-Reaktionen mit inversem Elektronenbedarf zwischen Isopyrazolen und Cycloalkenen sowie Cycloalkadienen—Ein Vergleich von Säure-Katalyse und Beschleunigung durch Druck. Chem. Ber. 1987, 120, 2041–2051. [Google Scholar] [CrossRef]
- Levandowski, B.J.; Abularrage, N.S.; Houk, K.N.; Raines, R.T. Hyperconjugative antiaromaticity activates 4H-pyrazoles as inverse-electron-demand Diels–Alder dienes. Org. Lett. 2019, 21, 8492–8495. [Google Scholar] [CrossRef] [PubMed]
- Fernández, I.; Wu, J.I.; Schleyer, P.V.R. Substituent effects on “hyperconjugative” aromaticity and antiaromaticity in planar cyclopolyenes. Org. Lett. 2013, 15, 2990–2993. [Google Scholar] [CrossRef] [PubMed]
- Nyulászi, L.; Schleyer, P.V.R. Hyperconjugative π-aromaticity: How to make cyclopentadiene aromatic. J. Am. Chem. Soc. 1999, 121, 6872–6875. [Google Scholar] [CrossRef]
- Levandowski, B.J.; Zou, L.; Houk, K.N. Schleyer hyperconjugative aromaticity and Diels–Alder reactivity of 5-substituted cyclopentadienes. J. Comput. Chem. 2016, 37, 117–123. [Google Scholar] [CrossRef]
- Levandowski, B.J.; Zou, L.; Houk, K.N. Hyperconjugative aromaticity and antiaromaticity control the reactivities and π-facial stereoselectivities of 5-substituted cyclopentadiene Diels–Alder cycloadditions. J. Org. Chem. 2018, 83, 14658–14666. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Sammes, M.P.; Katritzky, A.R. The 4H-pyrazoles. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: Cambridge, MA, USA, 1983; Volume 34, pp. 53–78. [Google Scholar]
- Zelenin, K.N.; Tugusheva, A.R.; Yakimovich, S.I.; Alekseev, V.V.; Zerova, E.V. 5-Hydroxy-2-pyrazolines and some of their 1-substituted analogs. Chem. Heterocycl. Compd. 2002, 38, 668–676. [Google Scholar] [CrossRef]
- Breen, J.R.; Sandford, G.; Patel, B.; Fray, J. Synthesis of 4,4-difluoro-1H-pyrazole derivatives. Synlett 2015, 26, 51–54. [Google Scholar] [CrossRef][Green Version]
- Walton, L.; Duplain, H.R.; Knapp, A.L.; Eidell, C.K.; Bacsa, J.; Stephens, C.E. Synthesis and charge density determination of 4,4-difluoro-3,5-diphenyl-4H-pyrazole and a hydrate derivative. J. Fluor. Chem. 2015, 173, 12–17. [Google Scholar] [CrossRef]
- Yang, K.; Song, M.; Ali, A.I.M.; Mudassir, S.M.; Ge, H. Recent advances in the application of Selectfluor as a “fluorine-free” functional reagent in organic synthesis. Chem.—Asian J. 2020, 15, 729–741. [Google Scholar] [CrossRef]
- Nyffeler, P.T.; Durón, S.G.; Burkhart, M.D.; Vincent, S.P.; Wong, C.-H. Selectfluor: Mechanistic insight and applications. Angew. Chem. Int. Ed. 2004, 44, 192–212. [Google Scholar] [CrossRef] [PubMed]
- Rozatian, N.; Ashworth, I.W.; Sandford, G.; Hodgson, D.R.W. A quantitative reactivity scale for electrophilic fluorinating reagents. Chem. Sci. 2018, 9, 8692–8702. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Stanger, A. Can substituted cyclopentadiene become aromatic or antiaromatic? Chem. Eur. J. 2006, 12, 2745–2751. [Google Scholar] [CrossRef]
- Levandowski, B.J.; Houk, K.N. Hyperconjugative, secondary orbital, electrostatic, and steric effects on the reactivities and endo and exo stereoselectivities of cyclopropene Diels–Alder reactions. J. Am. Chem. Soc. 2016, 138, 16731–16736. [Google Scholar] [CrossRef]
- Sloop, J.C.; Jackson, J.L.; Schmidt, R.D. Microwave-mediated pyrazole fluorinations using Selectfluor®. Heteroat. Chem. 2009, 20, 341–345. [Google Scholar] [CrossRef]
- Lyles, M.M.; Gilbert, H.F. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: Dependence of the rate on the composition of the redox buffer. Biochemistry 1991, 30, 613–619. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Guo, F.; Li, Q.; Zhou, C. Synthesis and biological applications of fluoro-modified nucleic acids. Org. Biomol. Chem. 2017, 22, 9552–9565. [Google Scholar] [CrossRef]
- Buer, B.C.; Marsh, E.N. Fluorine: A new element in protein design. Protein Sci. 2012, 21, 453–462. [Google Scholar] [CrossRef]
- Odar, C.; Winkler, M.; Wiltschi, B. Fluoro amino acids: A rarity in nature, yet a prospect for protein engineering. Biotechnol. J. 2015, 10, 427–446. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of fluorine-containing amino acids for drug design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef] [PubMed]
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D.; et al. XSEDE: Accelerating scientific discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abularrage, N.S.; Levandowski, B.J.; Raines, R.T. Synthesis and Diels–Alder Reactivity of 4-Fluoro-4-Methyl-4H-Pyrazoles. Int. J. Mol. Sci. 2020, 21, 3964. https://doi.org/10.3390/ijms21113964
Abularrage NS, Levandowski BJ, Raines RT. Synthesis and Diels–Alder Reactivity of 4-Fluoro-4-Methyl-4H-Pyrazoles. International Journal of Molecular Sciences. 2020; 21(11):3964. https://doi.org/10.3390/ijms21113964
Chicago/Turabian StyleAbularrage, Nile S., Brian J. Levandowski, and Ronald T. Raines. 2020. "Synthesis and Diels–Alder Reactivity of 4-Fluoro-4-Methyl-4H-Pyrazoles" International Journal of Molecular Sciences 21, no. 11: 3964. https://doi.org/10.3390/ijms21113964
APA StyleAbularrage, N. S., Levandowski, B. J., & Raines, R. T. (2020). Synthesis and Diels–Alder Reactivity of 4-Fluoro-4-Methyl-4H-Pyrazoles. International Journal of Molecular Sciences, 21(11), 3964. https://doi.org/10.3390/ijms21113964