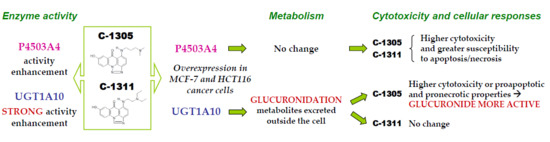
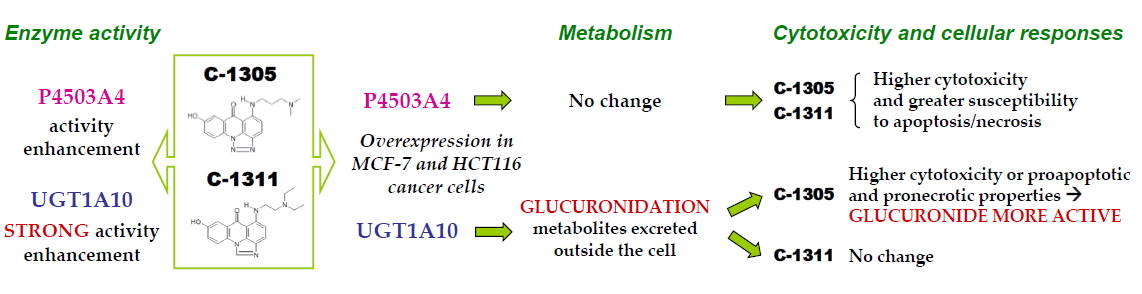
Enhanced Activity of P4503A4 and UGT1A10 Induced by Acridinone Derivatives C-1305 and C-1311 in MCF-7 and HCT116 Cancer Cells: Consequences for the Drugs’ Cytotoxicity, Metabolism and Cellular Response



Abstract
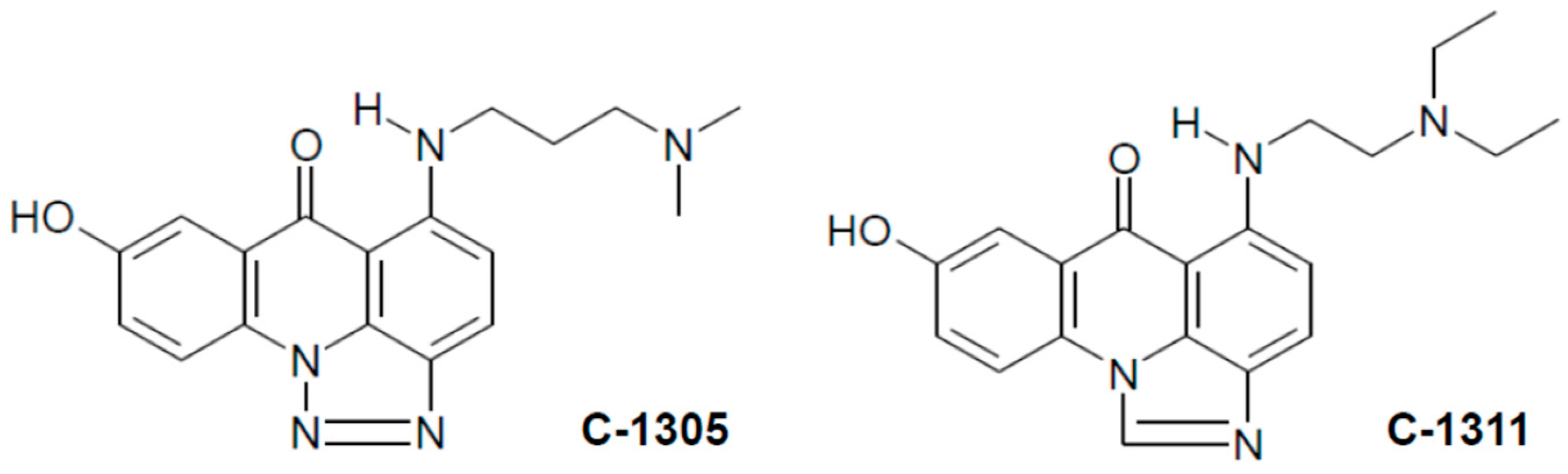
1. Introduction
2. Results and Discussion
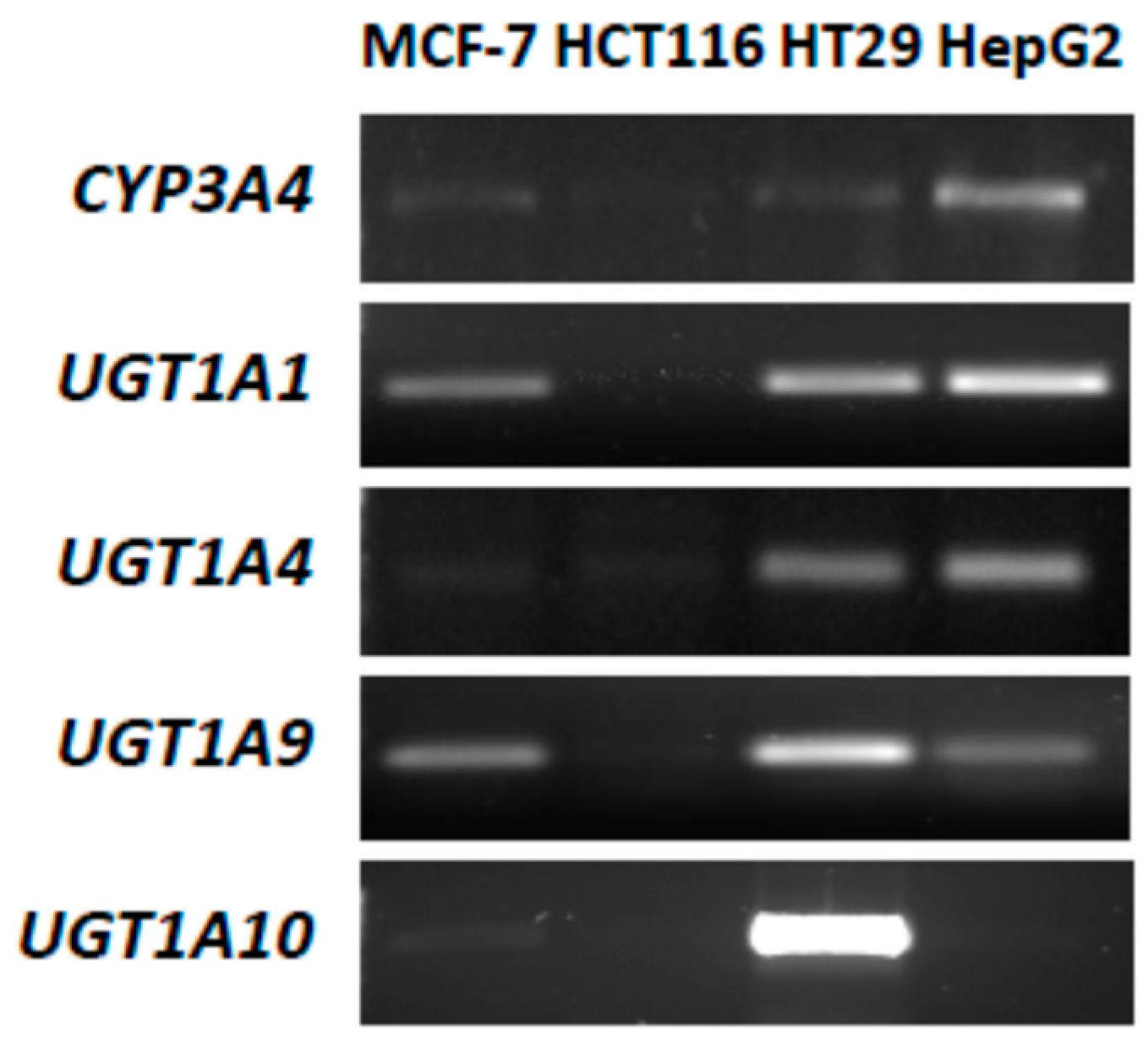
2.1. P4503A4 and UGT Expression in MCF-7 and HCT116 Cells
2.2. Cytotoxic Effects of Studied Compounds against Cancer Cells
2.3. Metabolic Transformation of C-1305 and C-1311 in MCF-7 and HCT116 Cells
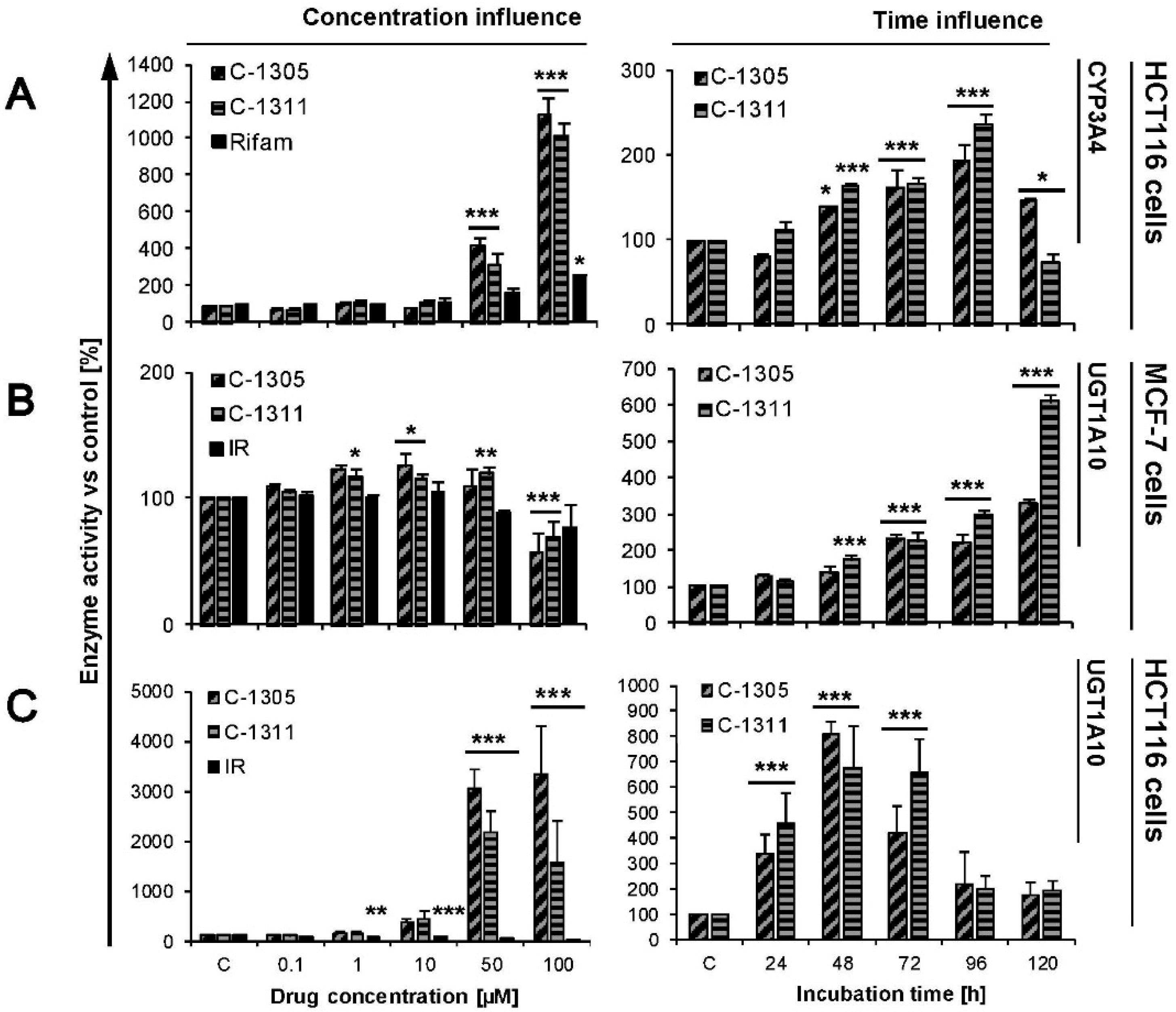
2.4. Modulation of P4503A4 and UGT1A10 Activity by C-1305 and C-1311
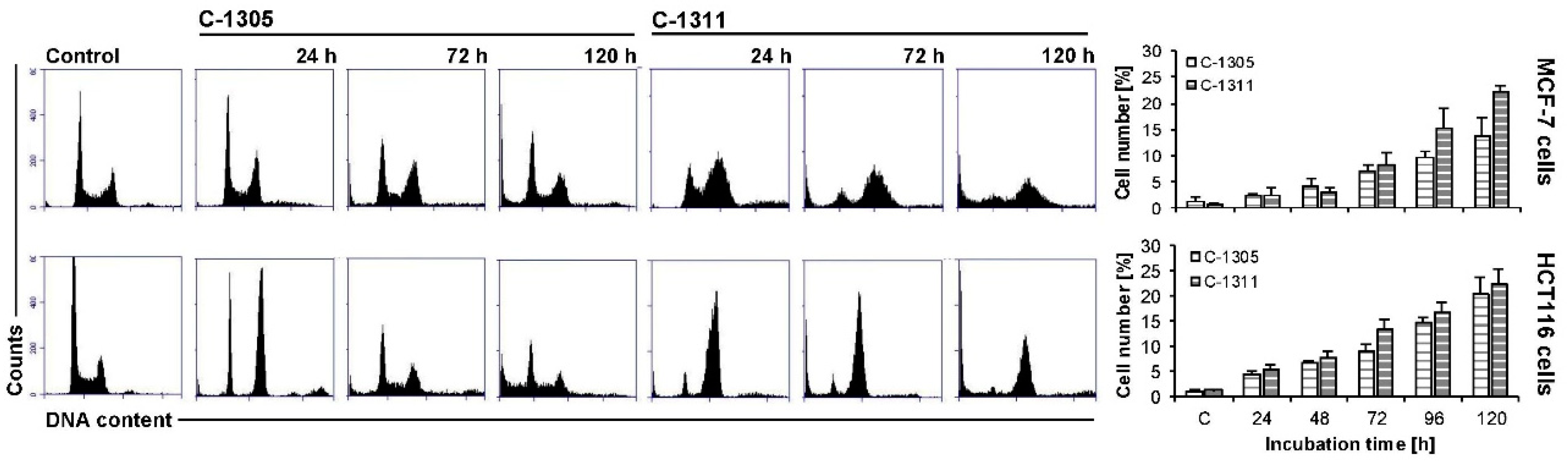
2.5. Effects of C-1305 and C-1311 on Cell Cycle Progression of Control MCF-7-EV and HCT116-EV Cells
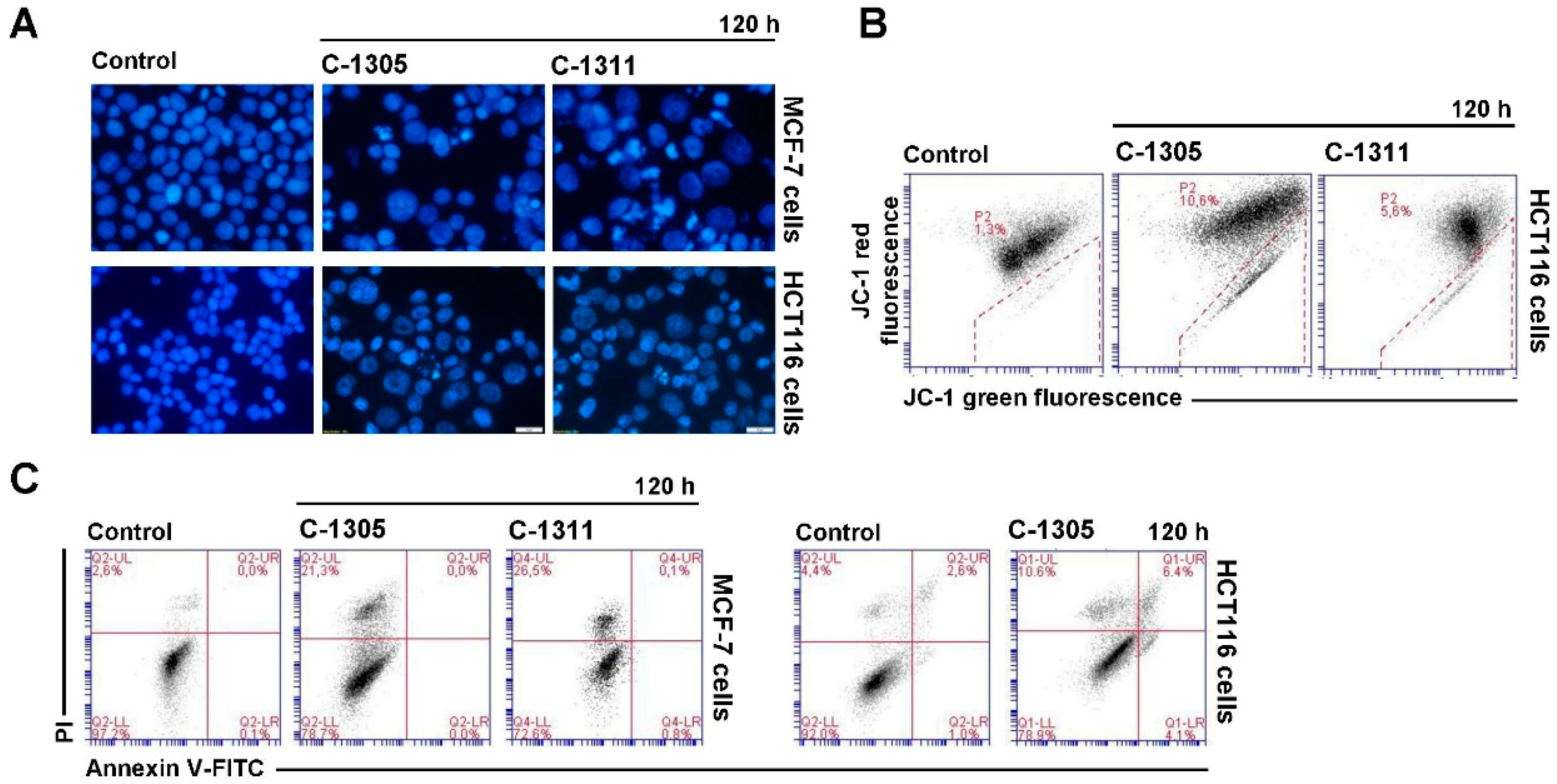
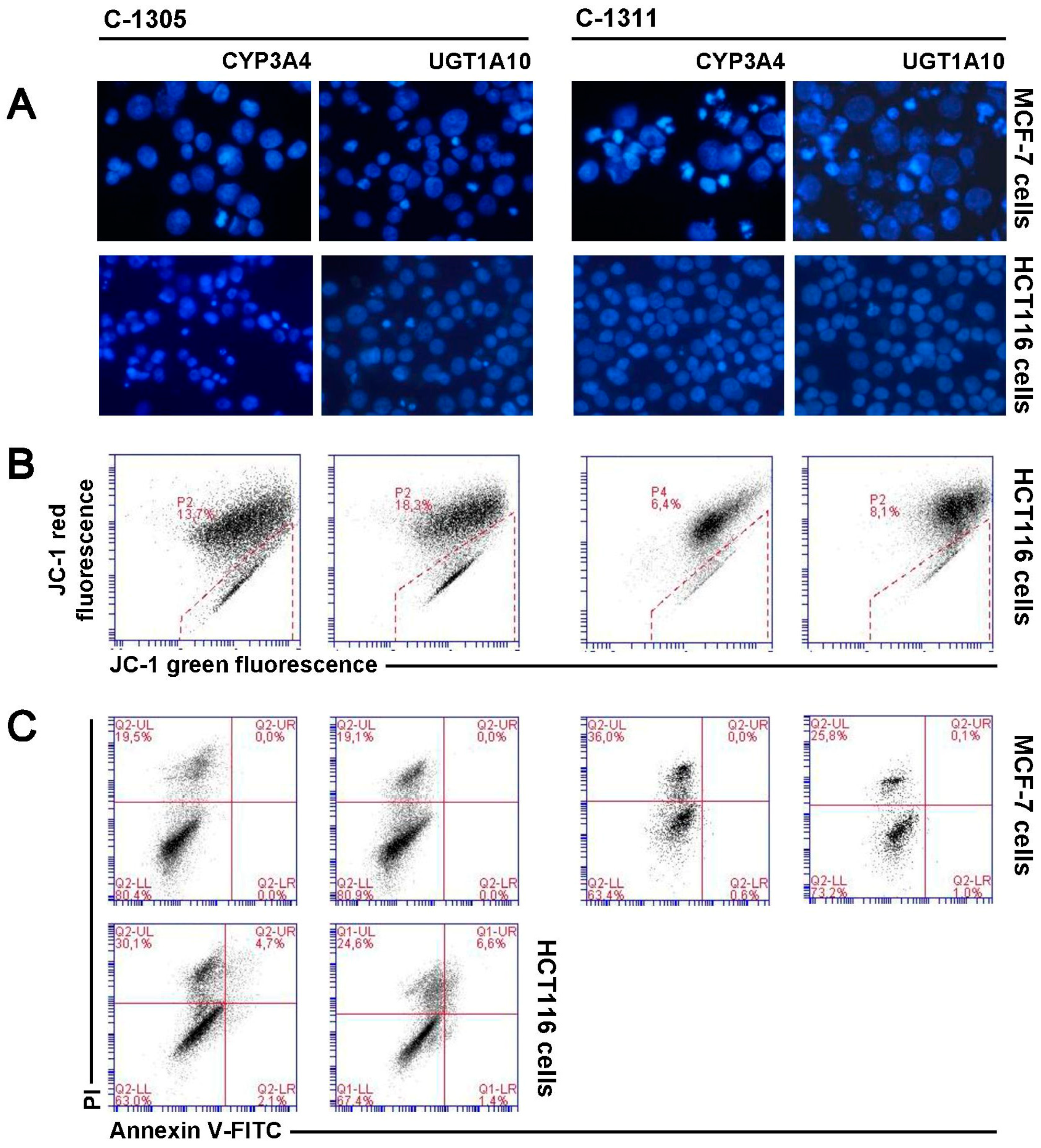
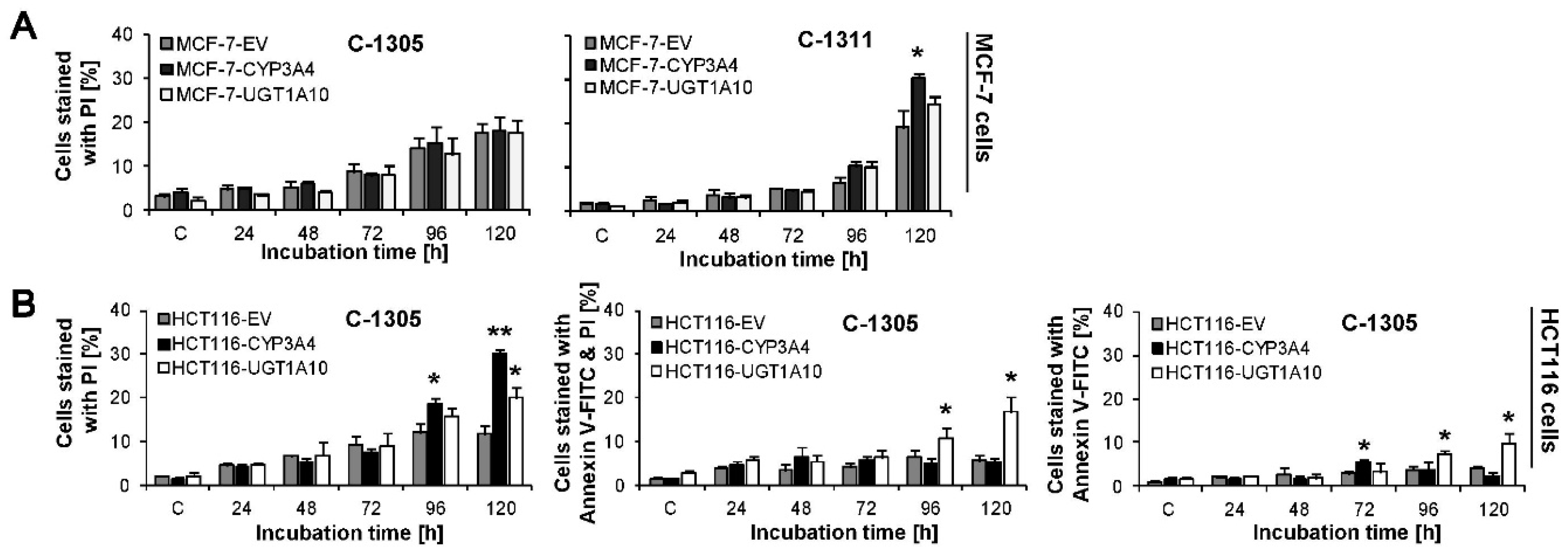
2.6. Induction of Cell Death in MCF-7-EV and HCT116-EV Cells
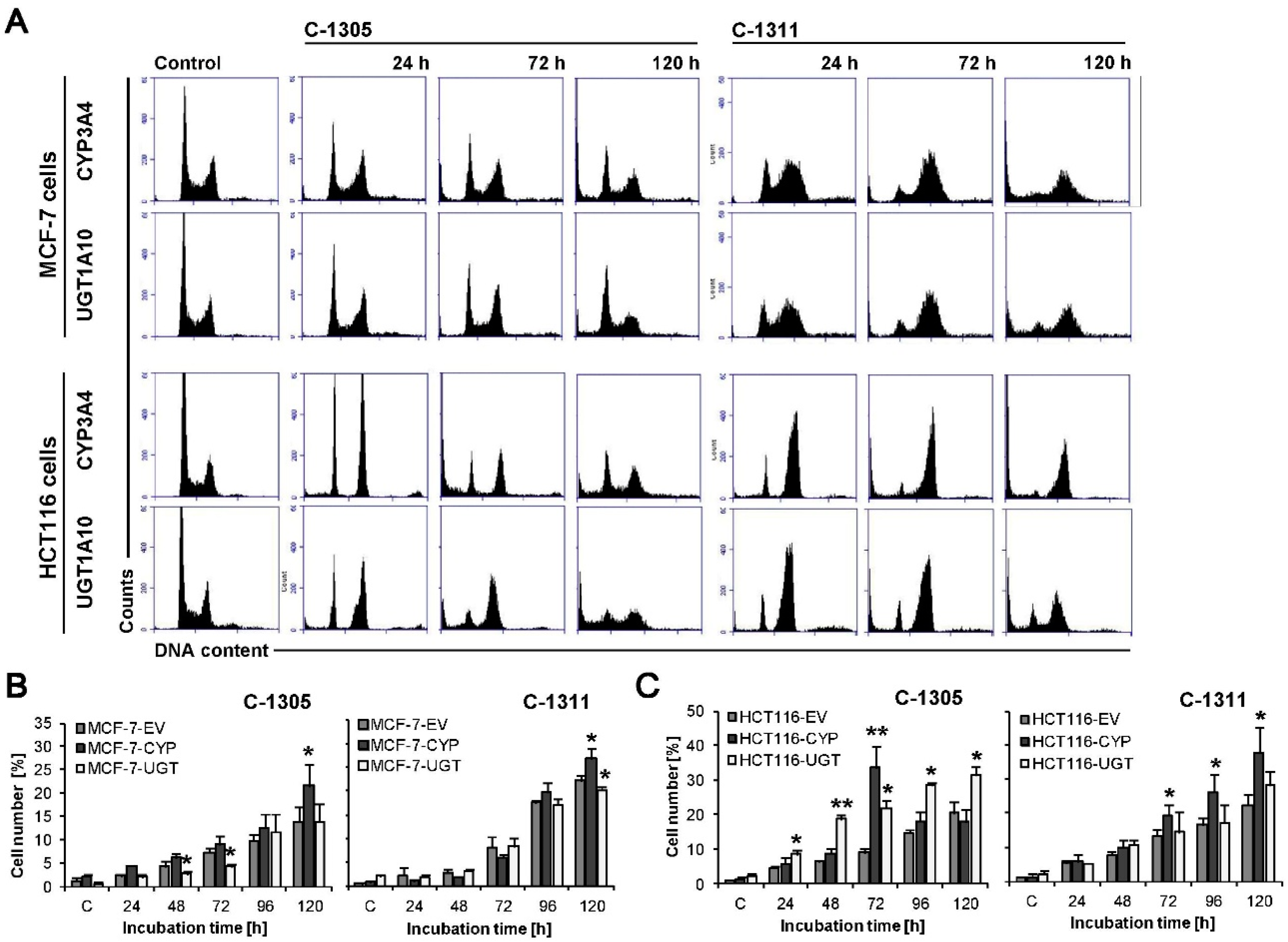
2.7. P4503A4 Influence on Cellular Response Induced by C-1305 and C-1311 in MCF-7 and HCT116 Cells
2.8. UGT1A10 Isoenzyme Influence on Cellular Response Induced by C-1305 and C-1311 in MCF-7 and HCT116 Cells
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Cell Lines, Cultures and Creation of Stable Cell Lines
3.3. Cell Growth Inhibition Assay
3.4. mRNA Isolation and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
3.5. P4503A4 and UGT Activity Modulation in Living Cells
3.6. Biotransformation of C-1305 and C-1311 in Alive Cells
3.7. High-Performance Liquid Chromatography (HPLC) Analysis
3.8. Cell Cycle Analysis
3.9. Assessment of Cell Morphology
3.10. Annexin V/PI Dual Staining
3.11. Mitochondrial Transmembrane Potential Measurement
3.12. Data Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 7-OH-TFC | 7-Hydroxy-4-trifluoromethylcoumarin (O-glucuronidation) standard substrate |
| ΔΨm | Mitochondrial membrane potential |
| DAPI | 4′,6-Diaminidino-2-phenylindole (fluorescent-DNA stain) |
| ESI-MS | Electrospray ionization mass spectroscopy |
| EV | Empty vector |
| FITC | Fluorescein isothiocyanate (fluorescent dye) |
| HPLC | High-performance liquid chromatography |
| IC | Inhibitory concentration |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| PI | Propidium iodide (fluorescent dye staining only dead cells) |
| UGT | UDP-glucuronosyltranspherase |
References
- Fleming, I. The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol. Rev. 2014, 66, 1106–1140. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Guengerich, F.P. Contributions of human enzymes in carcinogen metabolism. Chem. Res. Toxicol. 2012, 25, 1316–1383. [Google Scholar] [CrossRef] [PubMed]
- Konopa, J. Antitumor acridines with diaminoalkylo pharmacophoric group. Pure Appl. Chem. 2001, 73, 1421–1428. [Google Scholar] [CrossRef]
- Cholody, W.M.; Martelli, S.; Konopa, J. 8-Substituted 5-[(aminoalkyl)amino]-6H-v-triazolo[4,5,1-de]acridin-6-ones as potential antineoplastic agents. Synthesis and biological activity. J. Med. Chem. 1990, 33, 2852–2856. [Google Scholar] [CrossRef]
- Kuśnierczyk, H.; Chołody, W.M.; Paradziej-Łukowicz, J.; Radzikowski, C.; Konopa, J. Experimental antitumor activity and toxicity of the selected triazolo- and imidazoacridinones. Arch. Immunol. Ther. Exp. 1994, 42, 415–423. [Google Scholar]
- Cholody, W.M.; Martelli, S.; Paradziej-Łukowicz, J.; Konopa, J. 5-[(Aminoalkyl)amino]imidazo[4,5,1-de]acridin-6-ones as a novel class of antineoplastic agents. Synthesis and biological activity. J. Med. Chem. 1990, 33, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.M.; Double, J.A.; Konopa, J.; Bibby, M.C. Preclinical evaluation of novel imidazoacridinone derivatives with potent activity against experimental colorectal cancer. Br. J. Cancer. 1996, 74, 1369–1374. [Google Scholar] [CrossRef]
- Thomas, l.; Anthoney, A.; Scott, E.; Ahmed, S.; Lundberg, A.S.; Major, A.; Capizzi, R.L.; Twelves, C.J. C-1311, a novel inhibitor of FLT3 and topoisomerase II: A phase 1 trial of a once every three weeks schedule in patients with advanced solid tumors. J. Clin. Oncol. 2008, 26, 2576. [Google Scholar] [CrossRef]
- Isambert, N.; Campone, M.; Bourbouloux, E.; Drouin, M.; Major, A.; Yin, W.; Loadman, P.; Capizzi, R.; Grieshaber, C.; Fumoleau, P. Evaluation of the safety of C-1311 (SYMADEX) administered in a phase 1 dose escalation trial as a weekly infusion for 3 consecutive weeks in patients with advanced solid tumours. Eur. J. Cancer 2010, 46, 729–734. [Google Scholar] [CrossRef]
- Capizzi, R.; Roman, L.; Tjulandin, S.; Smirnova, I.; Manikhas, A.; Paterson, J.; Major, A.; Lundberg, A.; Fumoleau, P. Phase II trial of C1311, a novel inhibitor of topoisomerase II in advanced breast cancer. J. Clin. Oncol. 2008, 26, 1055. [Google Scholar] [CrossRef]
- Smith, S.C.; Havaleshko, D.M.; Moon, K.; Lee, J.; Bekiranov, S.; Burke, D.J.; Theodorescu, D. Use of yeast chemigenomics and COXEN informatics in preclinical evaluation of anticancer agents. Neoplasia 2011, 13, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Mazerska, Z.; Dzięgielewski, J.; Konopa, J. Enzymatic activation of a new antitumour drug, 5-diethylaminoethylamino-8-hydroxyimidazoacridinone, C-1311, observed after its intercalation into DNA. Biochem. Pharmacol. 2001, 61, 685–694. [Google Scholar] [CrossRef]
- Mazerska, Z.; Sowiński, P.; Konopa, J. Molecular mechanism of the enzymatic oxidation investigated for imidazoacridinone antitumor drug, C-1311. Biochem. Pharmacol. 2003, 66, 1727–1736. [Google Scholar] [CrossRef]
- Augustin, E.; Moś-Rompa, A.; Skwarska, A.; Witkowski, J.M.; Konopa, J. Induction of G2/M phase arrest and apoptosis of human leukemia cells by potent antitumor triazoloacridinone C-1305. Biochem. Pharmacol. 2006, 72, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- Skwarska, A.; Augustin, E.; Konopa, J. Sequential induction of mitotic catastrophe followed by apoptosis in human leukemia MOLT4 cells by imidazoacridinone C-1311. Apoptosis 2007, 12, 2245–2257. [Google Scholar] [CrossRef] [PubMed]
- Dzięgielewski, J.; Ślusarski, B.; Konitz, A.; Składanowski, A.; Konopa, J. Intercalation of imidazoacridinones to DNA and its relevance to cytotoxic and antitumor activity. Biochem. Pharmacol. 2002, 63, 1653–1662. [Google Scholar] [CrossRef]
- Skałdanowski, A.; Plisov, S.Y.; Konopa, J.; Larsen, A.K. Inhibition of DNA topoisomerase II by imidazoacridinones, new antineoplastic agents with strong activity against solid tumors. Mol. Pharmacol. 1996, 49, 772–780. [Google Scholar]
- Lemke, K.; Poindessous, V.; Składanowski, A.; Larsen, A.K. The antitumor triazoloacridone C-1305 is a topoisomerase II poison with unusual properties. Mol. Pharmacol. 2004, 66, 1035–1042. [Google Scholar] [CrossRef]
- Dzięgielewski, J.; Konopa, J. Interstrand crosslinking of DNA induced in tumor cells by a new group of antitumor imidazoacridinones. Proc. Am. Assoc. Cancer Res. 1996, 37, 410. [Google Scholar]
- Koba, M.; Konopa, J. Interactions of antitumor triazoloacridinones with DNA. Acta Biochim. Pol. 2007, 54, 297–306. [Google Scholar] [CrossRef]
- Paradziej-Łukowicz, J.; Skwarska, A.; Peszyńska-Sularz, G.; Brillowska-Dąbrowska, A.; Konopa, J. Anticancer imidazoacridinone C-1311 inhibits hypoxia-inducible factor-1α (HIF-1α), vascular endothelial growth factor (VEGF) and angiogenesis. Cancer Biol. Ther. 2011, 12, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.A.; Dunstan, M.S.; Caraher, M.C.; Scott, K.A.; Leys, D.; Stradford, I.J. In silico screening reveals structurally diverse, nanomolar inhibitors of NQO2 that are functionally active in cells and can modulate NF-κB signaling. Mol. Cancer Ther. 2012, 11, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Augustin, E.; Skwarska, A.; Weryszko, A.; Pelikant, I.; Sankowska, E.; Borowa-Mazgaj, B. The antitumor compound triazoloacridinone C-1305 inhibits FLT3 kinase activity and potentiates apoptosis in mutant FLT3-ITD leukemia cells. Acta Pharmacol. Sin. 2015, 36, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Skwarska, A.; Augustin, E.; Beffinger, M.; Wojtczyk, A.; Konicz, S.; Laskowska, K.; Polewska, J. Targeting of FLT3-ITD kinase contributes to high selectivity of imidazoacridinone C-1311 against FLT3-activated leukemia cells. Biochem. Pharmacol. 2015, 95, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Potęga, A.; Fedejko-Kap, B.; Mazerska, Z. Mechanism-based inactivation of human cytochrome P450 1A2 and 3A4 isoenzymes by anti-tumor triazoloacridinone C-1305. Xenobiotica 2016, 46, 1056–1065. [Google Scholar] [CrossRef]
- Potęga, A.; Fedejko-Kap, B.; Mazerska, Z. Imidazoacridinone antitumor agent C-1311 as a selective mechanism-based inactivator of human cytochrome P450 1A2 and 3A4 isoenzymes. Pharmacol. Rep. 2016, 68, 663–670. [Google Scholar] [CrossRef]
- Fedejko-Kap, B.; Niemira, M.; Radominska-Pandya, A.; Mazerska, Z. Flavin monooxygenases, FMO1 and FMO3, not cytochrome P450 isoenzymes, contribute to metabolism of anti-tumour triazoloacridinone, C-1305, in liver microsomes and HepG2 cells. Xenobiotica 2011, 41, 1044–1055. [Google Scholar] [CrossRef]
- Potęga, A.; Dąbrowska, E.; Niemira, M.; Kot-Wasik, A.; Ronseaux, S.; Henderson, C.J.; Wolf, R.; Mazerska, Z. The imidazoacridinone antitumor drug, C-1311, is metabolized by flavin monooxygenases but not by cytochrome P450s. Drug Metab. Dispos. 2011, 39, 1423–1432. [Google Scholar] [CrossRef]
- Augustin, E.; Niemira, M.; Hołownia, A.; Mazerska, Z. CYP3A4-dependent cellular response does not relate to CYP3A4-catalysed metabolites of C-1748 and C-1305 acridine antitumor agents in HepG2 cells. Cell Biol. Int. 2014, 38, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Augustin, E.; Borowa-Mazgaj, B.; Kikulska, A.; Kordalewska, M.; Pawłowska, M. CYP3A4 overexpression enhances the cytotoxicity of the antitumor triazoloacridinone derivative C-1305 in CHO cells. Acta Pharmacol. Sin. 2013, 34, 146–156. [Google Scholar] [CrossRef]
- Augustin, E.; Pawłowska, M.; Polewska, J.; Potęga, A.; Mazerska, Z. Modulation of CYP3A4 activity and induction of apoptosis, necrosis and senescence by the anti-tumour imidazoacridinone C-1311 in human hepatoma cells. Cell Biol. Int. 2013, 37, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Pawłowska, M.; Augustin, E.; Mazerska, Z. CYP3A4 overexpression enhances apoptosis induced by anticancer agent imidazoacridinone C-1311, but does not change the metabolism of C-1311 in CHO cells. Acta Pharmacol. Sin. 2014, 35, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Fedejko-Kap, B.; Bratton, S.M.; Finel, M.; Radominska-Pandya, A.; Mazerska, Z. Role of human UDP-glucuronosyltransferases in the biotransformation of the triazoloacridinone and imidazoacridinone antitumor agents C-1305 and C-1311: Highly selective substrates for UGT1A10. Drug Metab. Dispos. 2012, 40, 1736–1743. [Google Scholar] [CrossRef]
- Pawłowska, M.; Chu, R.; Fedejko-Kap, B.; Augustin, E.; Mazerska, Z.; Radomińska-Pandya, A.; Chambers, T. Metabolic transformation of antitumor acridinone C-1305 but not C-1311 via selective cellular expression of UGT1A10 increases cytotoxic response: Implications for clinical use. Drug Metab. Dispos. 2013, 41, 414–421. [Google Scholar] [CrossRef]
- Iyer, L.; Das, S.; Janisch, L.; Wen, M.; Ramírez, J.; Karrison, T.; Fleming, G.F.; Vokes, E.E.; Schilsky, R.L.; Ratain, M.J. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002, 2, 43–47. [Google Scholar] [CrossRef]
- Goetz, M.P.; Rae, J.M.; Suman, V.J.; Safgren, S.L.; Ames, M.M.; Visscher, D.W.; Reynolds, C.; Couch, F.J.; Lingle, W.L.; Flockhart, D.A.; et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J. Clin. Oncol. 2005, 23, 9312–9318. [Google Scholar] [CrossRef]
- Hirth, J.; Watkins, P.B.; Strawderman, M.; Schott, A.; Bruno, M.; Baker, L.H. The effect of an individual’s cytochrome CYP3A4 activity on docetaxel clearance. Clin. Cancer Res. 2000, 6, 1255–1258. [Google Scholar]
- Saini, S.; Hirata, H.; Majid, S.; Dahiya, R. Functional significance of cytochrome P450 1B1 in endometrial carcinogenesis. Cancer Res. 2009, 69, 7038–7045. [Google Scholar] [CrossRef]
- Mitra, R.; Guo, Z.; Milani, M.; Mesaros, C.; Rodriguez, M.; Nguyen, J.; Luo, X.; Clarke, D.; Lamba, J.; Schuetz, E.; et al. CYP3A4 mediates growth of estrogen receptor-positive breast cancer cells in part by inducing nuclear translocation of phospho-Stat3 through biosynthesis of (±)-14,15-epoxyeicosatrienoic acid (EET). J. Biol. Chem. 2011, 286, 17543–17559. [Google Scholar] [CrossRef]
- Jiang, J.G.; Ning, Y.G.; Chen, C.; Ma, D.; Liu, Z.J.; Yang, S.; Zhou, J.; Xiao, X.; Zhang, X.A.; Edin, M.L.; et al. Cytochrome p450 epoxygenase promotes human cancer metastasis. Cancer Res. 2007, 67, 6665–6674. [Google Scholar] [CrossRef]
- Hu, D.G.; Selth, L.A.; Tarulli, G.A.; Meech, R.; Wijayakumara, D.; Chanawong, A.; Russell, R.; Caldas, C.; Robinson, J.L.; Carroll, J.S.; et al. Androgen and Estrogen Receptors in Breast Cancer Coregulate Human UDP-Glucuronosyltransferases 2B15 and 2B17. Cancer Res. 2016, 76, 5881–5893. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Gelbart, T.; Demina, A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: A balanced polymorphism for regulation of bilirubin metabolism? Proc. Natl. Acad. Sci. USA 1998, 95, 8170–8174. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-glucuronosyltransferases: Their role in drug metabolism and detoxification. Int. J. Bioch. Cell Biol. 2013, 45, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Rahikainen, T.; Häkkinen, M.R.; Finel, M.; Pasanen, M.; Juvonen, R.O. A high throughput assay for the glucuronidation of 7-hydroxy-4-trifluoromethylcoumarin by recombinant human UDP-glucuronosyltransferases and liver microsomes. Xenobiotica 2013, 43, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Mróz, A.; Mazerska, Z. Glucuronidation of antitumour therapeutics—detoxification, mechanism of resistance or prodrug formation? Postepy Hig. Med. Dosw. 2015, 69, 1462–1477. [Google Scholar]
- Guillemette, C.; Lévesque, É.; Rouleau, M. Pharmacogenomics of human uridine diphospho-glucuronosyltransferases and clinical implications. Clin. Pharmacol. Ther. 2014, 96, 324–339. [Google Scholar] [CrossRef]
- Pawłowska, M.; (Gdańsk University of Technology, Gdańsk, Poland). Personal communication, 2017.
- Pawłowska, M.; Mazerska, Z.; (Gdańsk University of Technology, Gdańsk, Poland). Personal communication, 2017.
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Wilson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Invest. 1998, 102, 1016–1023. [Google Scholar] [CrossRef]
- Niemira, M.; Dastych, J.; Mazerska, Z. Pregnane X receptor dependent up-regulation of CYP2C9 and CYP3A4 in tumor cells by antitumor acridine agents, C-1748 and C-1305, selectively diminished under hypoxia. Biochem. Pharmacol. 2013, 86, 231–241. [Google Scholar] [CrossRef]
- Bejrowska, A.; Pawłowska, M.; Mróz, A.; Mazerska, Z. Modulation of UDP-glucuronidation by acridinone antitumor agents C-1305 and C-1311 in HepG2 and HT29 cell lines, despite slight impact in noncellular systems. Pharmacol. Rep. 2018, 70, 470–475. [Google Scholar] [CrossRef]
- Jänicke, R.U. MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res. Treat. 2009, 117, 119–221. [Google Scholar] [CrossRef]
- Fischer, U.; Jänicke, R.U.; Schulze-Osthoff, K. Many cuts to ruin: A comprehensive update of caspase substrates. Cell Death Differ. 2003, 10, 76–100. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2001, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Pawłowska, M.; (Gdańsk University of Technology, Gdańsk, Poland). Personal communication, 2013.
- Floriano-Sanchez, E.; Rodriguez, N.C.; Bandala, C.; Coballase-Urrutia, E.; Lopez-Cruz, J. CYP3A4 expression in breast cancer and its association with risk factors in Mexican women. Asian Pac. J. Cancer Prev. 2014, 15, 3805–3809. [Google Scholar] [CrossRef] [PubMed]
- Hlaváč, V.; Brynychová, V.; Václavíková, R.; Ehrlichová, M.; Vrána, D.; Pecha, V.; Trnková, M.; Kodet, R.; Mrhalová, M.; Kubáčková, K.; et al. The role of cytochromes p450 and aldo-keto reductases in prognosis of breast carcinoma patients. Medicine 2014, 93, e255. [Google Scholar] [CrossRef]
- Thuy Phuong, N.T.; Kim, J.W.; Kim, J.A.; Jeon, J.S.; Lee, J.Y.; Xu, W.J.; Yang, J.W.; Kim, S.K.; Kang, K.W. Role of the CYP3A4-mediated 11,12-epoxyeicosatrienoic acid pathway in the development of tamoxifen-resistant breast cancer. Oncotarget 2017, 8, 71054–71069. [Google Scholar] [CrossRef]
- Pawłowska, M.; (Gdańsk University of Technology, Gdańsk, Poland). Personal communication, 2014.
- Hołownia, A.; Braszko, J.J. Tamoxifen cytotoxicity in hepatoblastoma cells stably transfected with human CYP3A4. Biochem. Pharmacol. 2004, 67, 1057–1064. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Ando, A.; Takamura, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Prediction of response to docetaxel by CYP3A4 mRNA expression in breast cancer tissues. Int. J Cancer 2002, 97, 129–132. [Google Scholar] [CrossRef]
- Gut, I.; Danielova, V.; Holuboa, J.; Soucek, P.; Kluckova, H. Cytotoxicity of cyclophosphamide, paclitaxel, and docetaxel for tumor cell lines in vitro effects of concentration, time and cytochrome P450-catalyzed metabolism. Mol. Toxicol. 2002, 74, 437–446. [Google Scholar] [CrossRef]
- Saba, N.; Seal, A. Identification of a less toxic vinca alkaloid derivative for use as a chemotherapeutic agent, based on in silico structural insights and metabolic interactions with CYP3A4 and CYP3A5. J. Mol. Model. 2018, 24, 82. [Google Scholar] [CrossRef]
- Peng, B.; Lloyd, P.; Schran, H. Clinical pharmacokinetics of imatinib. Clin. Pharmacokinet. 2005, 44, 879–894. [Google Scholar] [CrossRef]
- Tian, D.; Hu, Z. CYP3A4-mediated pharmacokinetic interactions in cancer therapy. Curr. Drug Metab. 2014, 15, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Starlard-Davenport, A.; Xiong, Y.; Bratton, S.; Gallus-Zawada, A.; Finel, M.; Radomińska-Pandya, A. Phenylalanine(90) and phenylalanine(93) are crucial amino acids within the estrogen binding site of the human UDP-glucuronosyltransferase 1A10. Steroids 2007, 72, 85–94. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activity of Enzyme (% Conversion of Standard Substrate) | Cell Line | |||||
|---|---|---|---|---|---|---|
| MCF-7 | MCF-7 3A4/1A10 | HCT116 | HCT116 3A4/1A10 | HT29 | HepG2 | |
| P4503A4 | 0.08 ± 0.04 | 0.28 ± 0.04 | 0.10 ± 0.03 | 0.86 ± 0.35 | no data | 1.38 ± 0.50 |
| UGT (O-glucuronidation) | 0.09 ± 0.04 | 1.94 ± 0.62 | 0.01 ± 0.01 | 3.42 ± 0.64 | 59.13 ± 8.61 | 2.28 ± 0.53 |
| Drug Dose (µM) | C-1305 | C-1311 | ||||
|---|---|---|---|---|---|---|
| MCF-7 Cells | MCF-7 Cells | |||||
| EV | CYP3A4 | UGT1A10 | EV | CYP3A4 | UGT1A10 | |
| IC50 | 1.87 ± 0.05 | 1.55 ± 0.23 * | 1.57 ± 0.08 * | 0.36 ± 0.08 | 0.23 ± 0.01 * | 0.33 ± 0.04 |
| IC80 | 9.19 ± 1.45 | 6.28 ± 0.73 * | 6.37 ± 0.73 * | 1.04 ± 0.12 | 0.70 ± 0.02 * | 1.00 ± 0.06 |
| HCT116 Cells | HCT116 Cells | |||||
| EV | CYP3A4 | UGT1A10 | EV | CYP3A4 | UGT1A10 | |
| IC50 | 1.09 ± 0.32 | 1.36 ± 0.37 | 1.34 ± 0.15 | 0.96 ± 0.13 | 0.77 ± 0.11 | 1.38 ± 0.10 ** |
| IC80 | 5.75 ± 0.89 | 6.53 ± 2.37 | 6.23 ± 0.71 | 5.37 ± 1.08 | 4.93 ± 0.82 | 9.31 ± 2.08 ** |
| IC90 | 10.39 ± 1.67 | 26.35 ± 3.63 *** | 10.77 ± 1.43 | 11.19 ± 1.86 | 9.79 ± 1.69 | 19.37 ± 5.86 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawłowska, M.; Kwaśniewska, A.; Mazerska, Z.; Augustin, E. Enhanced Activity of P4503A4 and UGT1A10 Induced by Acridinone Derivatives C-1305 and C-1311 in MCF-7 and HCT116 Cancer Cells: Consequences for the Drugs’ Cytotoxicity, Metabolism and Cellular Response. Int. J. Mol. Sci. 2020, 21, 3954. https://doi.org/10.3390/ijms21113954
Pawłowska M, Kwaśniewska A, Mazerska Z, Augustin E. Enhanced Activity of P4503A4 and UGT1A10 Induced by Acridinone Derivatives C-1305 and C-1311 in MCF-7 and HCT116 Cancer Cells: Consequences for the Drugs’ Cytotoxicity, Metabolism and Cellular Response. International Journal of Molecular Sciences. 2020; 21(11):3954. https://doi.org/10.3390/ijms21113954
Chicago/Turabian StylePawłowska, Monika, Anna Kwaśniewska, Zofia Mazerska, and Ewa Augustin. 2020. "Enhanced Activity of P4503A4 and UGT1A10 Induced by Acridinone Derivatives C-1305 and C-1311 in MCF-7 and HCT116 Cancer Cells: Consequences for the Drugs’ Cytotoxicity, Metabolism and Cellular Response" International Journal of Molecular Sciences 21, no. 11: 3954. https://doi.org/10.3390/ijms21113954
APA StylePawłowska, M., Kwaśniewska, A., Mazerska, Z., & Augustin, E. (2020). Enhanced Activity of P4503A4 and UGT1A10 Induced by Acridinone Derivatives C-1305 and C-1311 in MCF-7 and HCT116 Cancer Cells: Consequences for the Drugs’ Cytotoxicity, Metabolism and Cellular Response. International Journal of Molecular Sciences, 21(11), 3954. https://doi.org/10.3390/ijms21113954