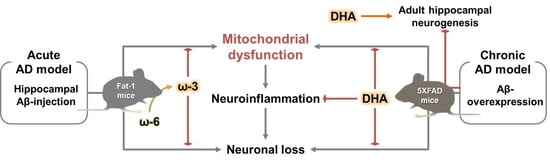
Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Aβ-Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model

 ,
,  ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
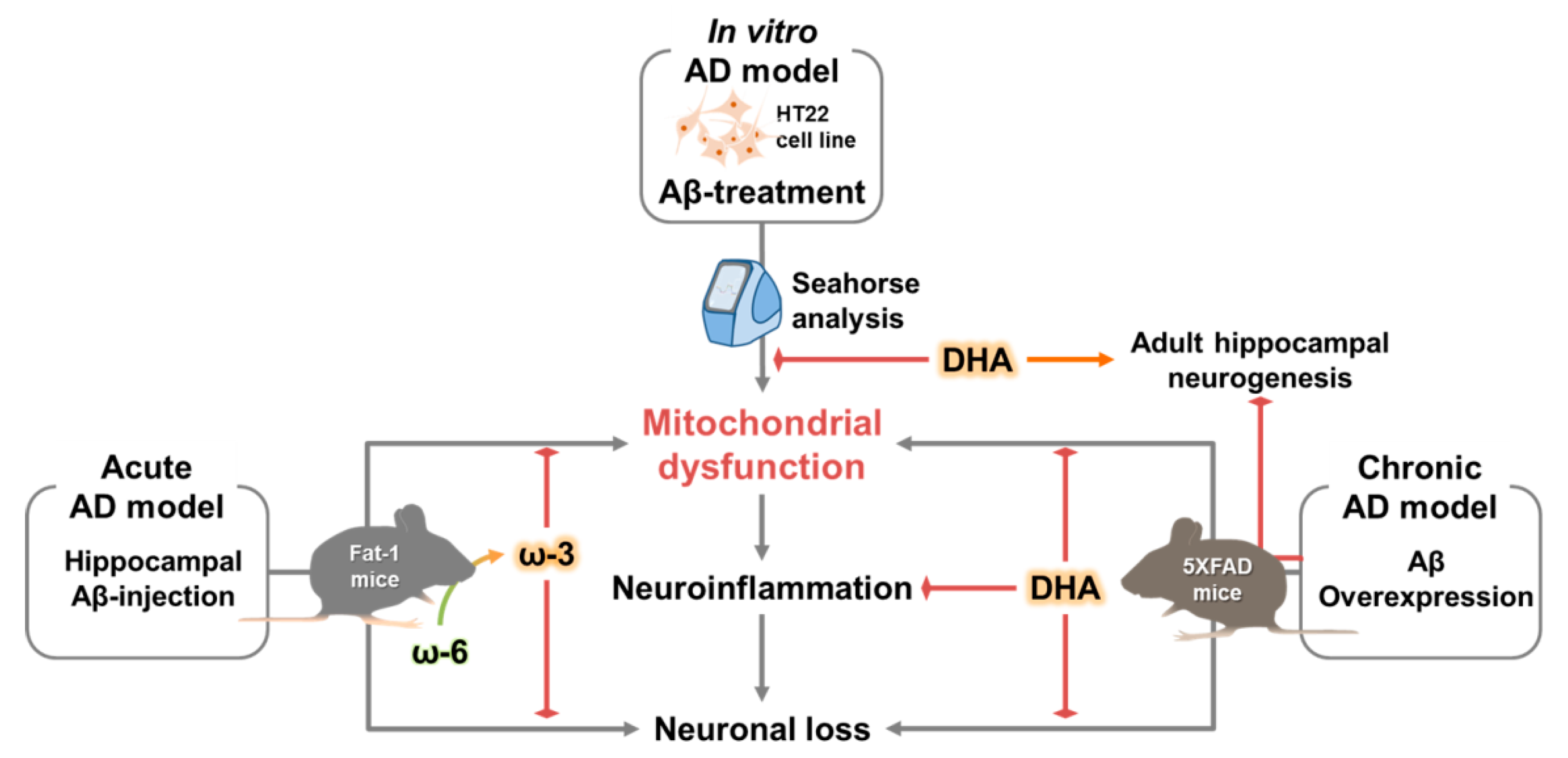{kind=link}
1. Introduction
2. Results
2.1. Cytotoxicity Evaluation of DHA in the HT22 Cell Line
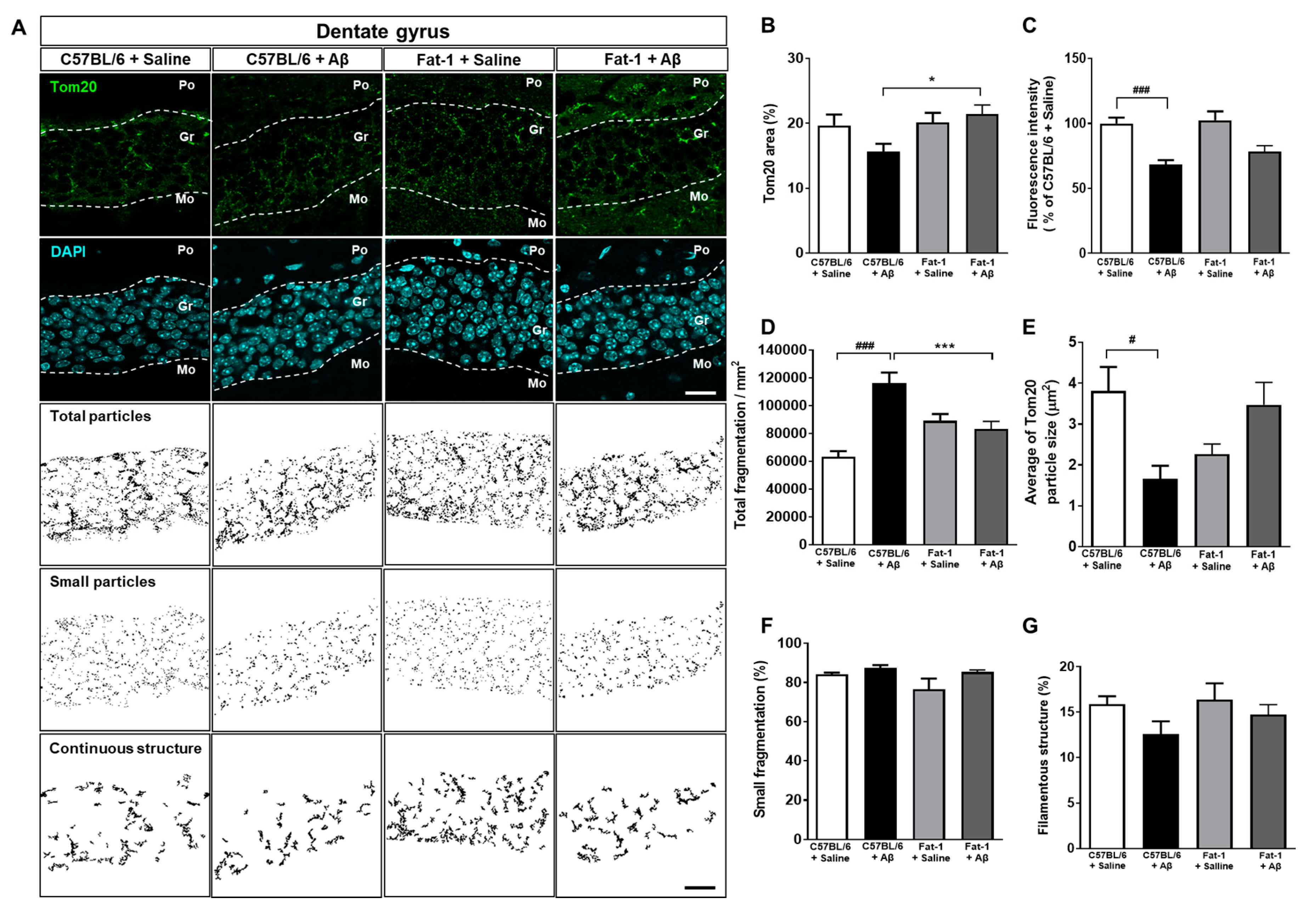
2.2. Endogenous Elevation of Omega-3 PUFAs Ameliorates Aβ-Induced Mitochondrial Dynamics Impairment
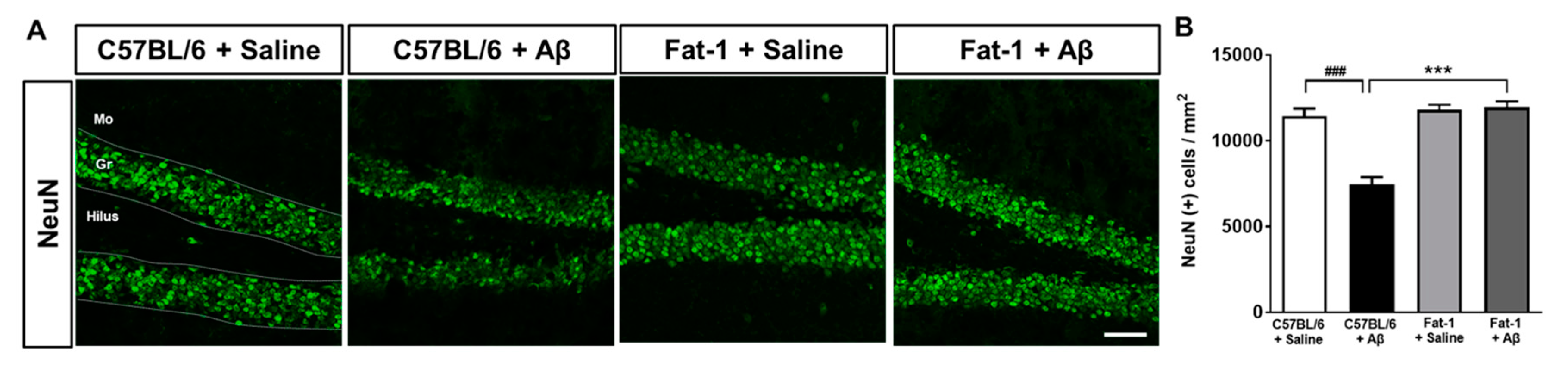
2.3. Endogenous Elevation of Omega-3 PUFAs Reduces Aβ-Induced Neuronal Loss
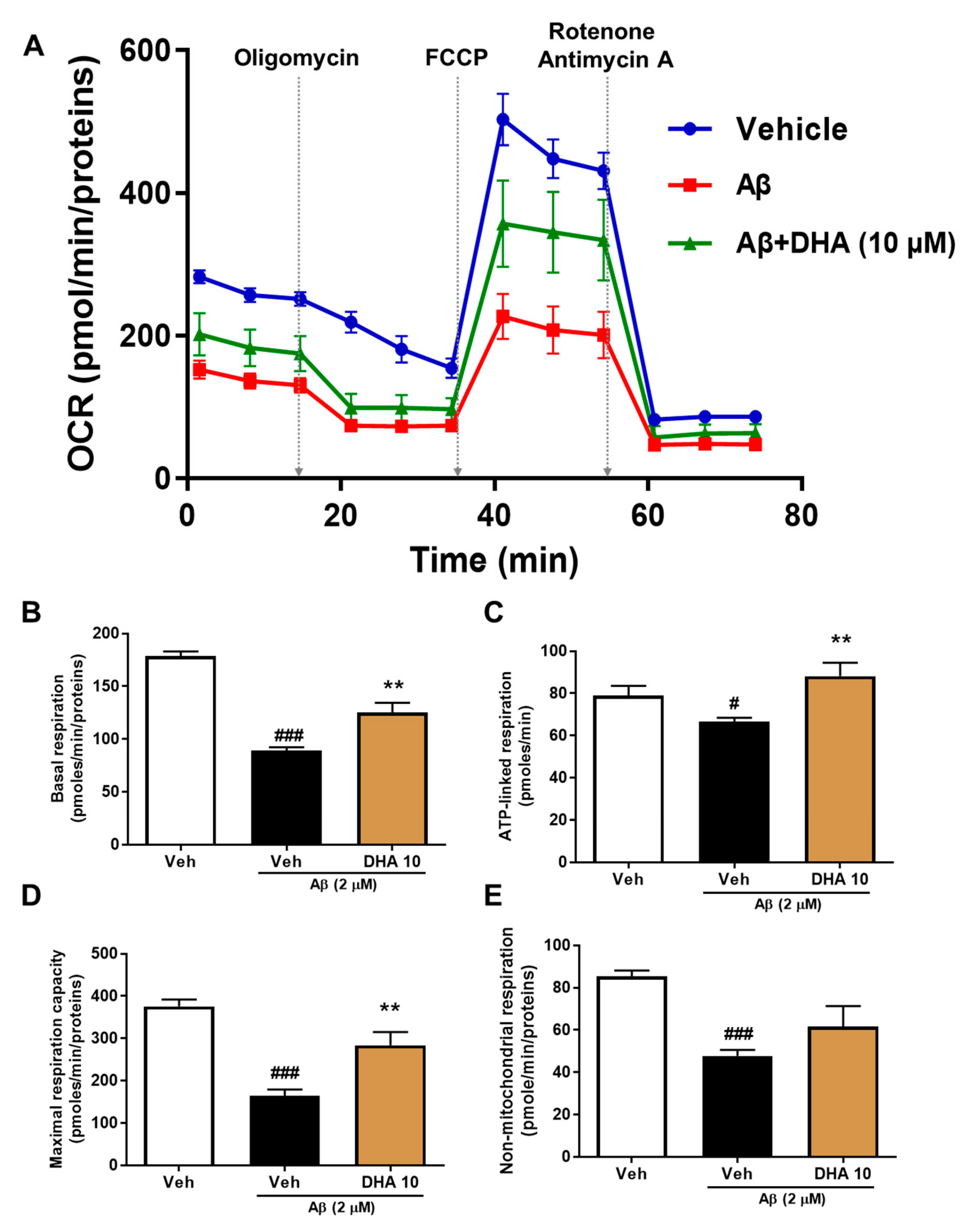
2.4. DHA Attenuates Aβ-Induced Mitochondrial Dysfunction in HT22 Cells
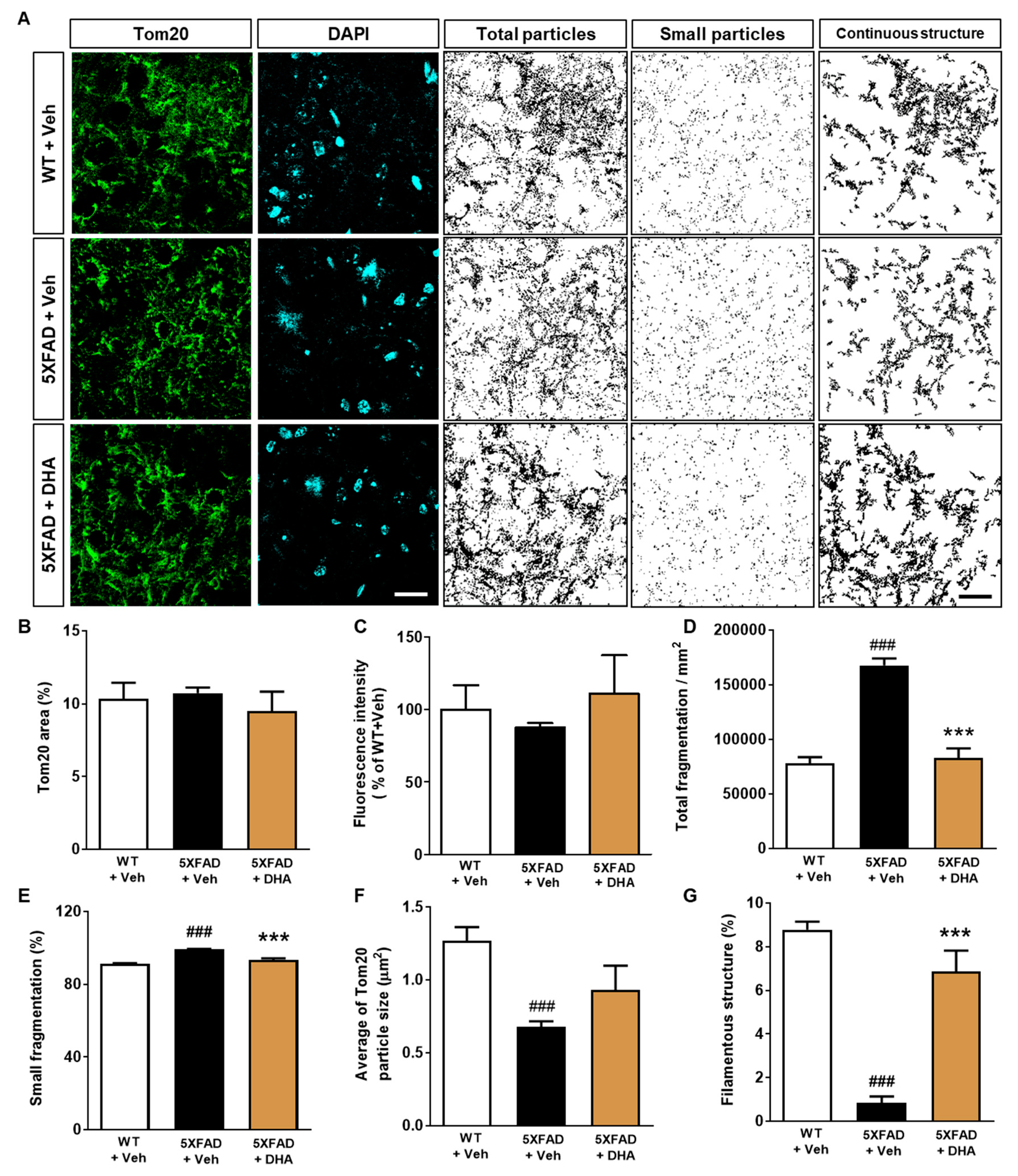
2.5. DHA Protects against the Alteration of Mitochondrial Dynamics in the Dorsal Subiculum of 5XFAD Mice
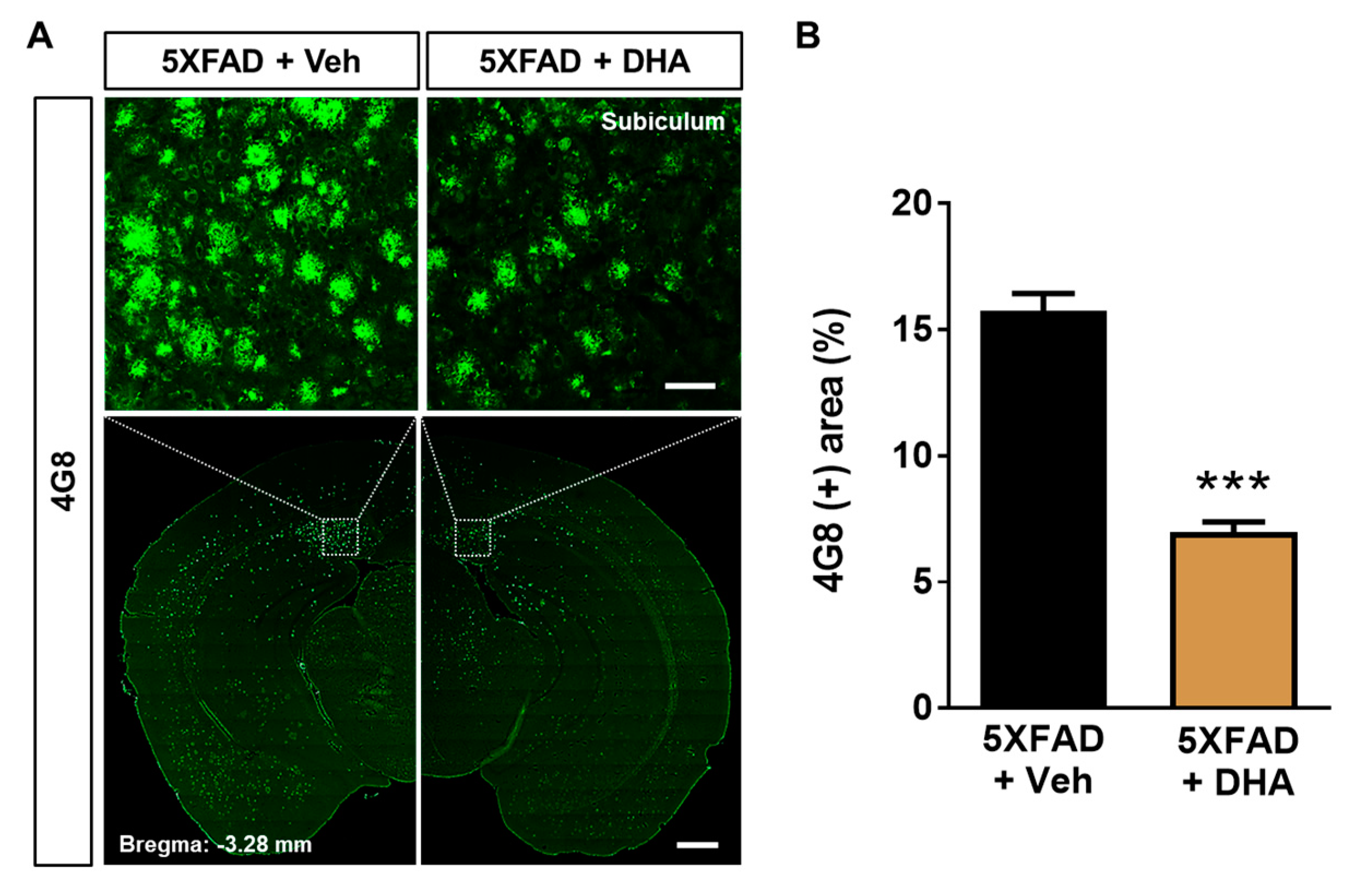
2.6. DHA Attenuates Aβ Burden in the Dorsal Subiculum of 5XFAD Mice
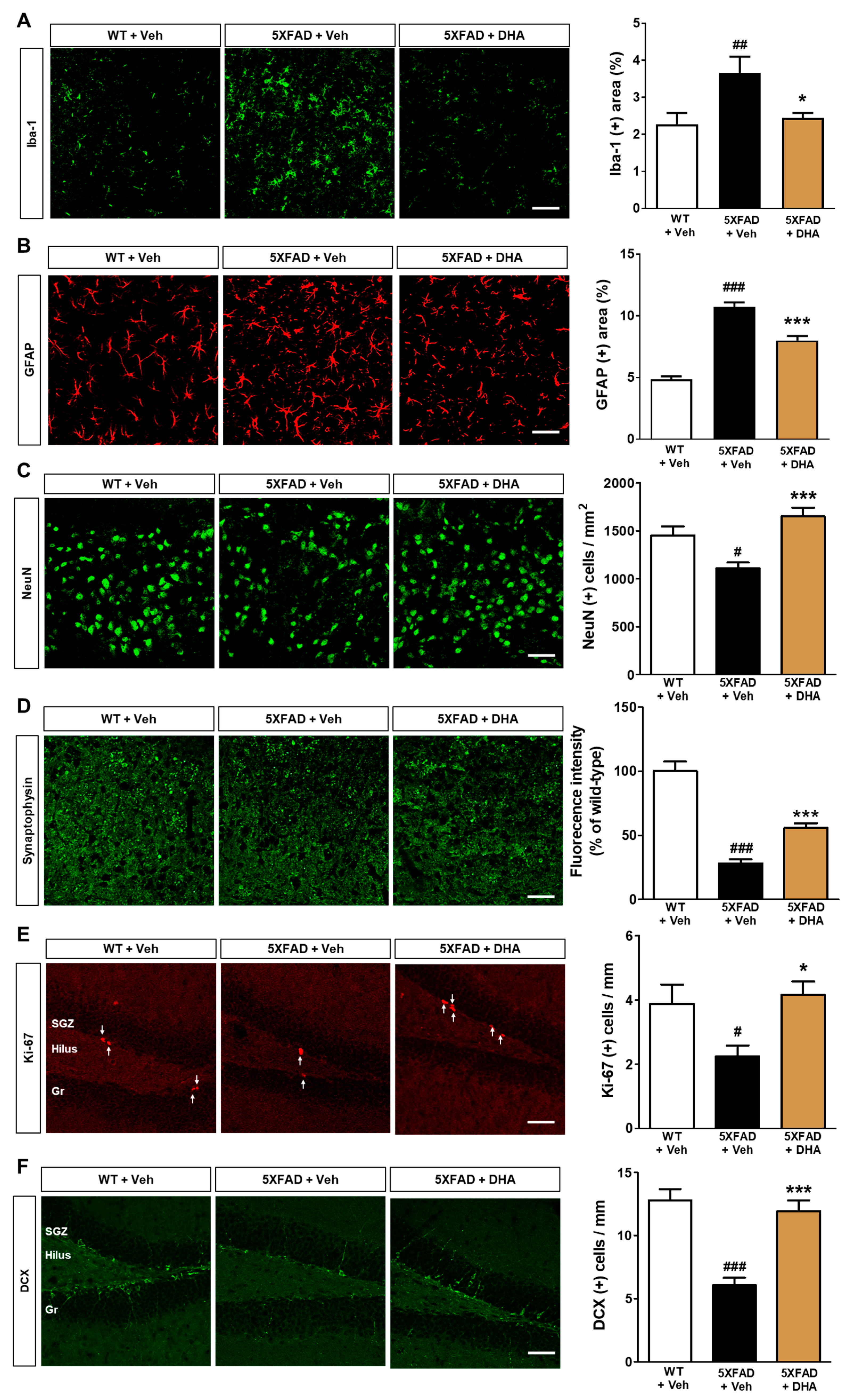
2.7. DHA Reduces Neuroinflammation and Neuronal Loss in the Dorsal Subiculum of 5XFAD Mice
2.8. DHA Significantly Alleviates Hippocampal Adult Neurogenesis Impairment in 5XFAD Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Polymerization Chain Reaction for Genotyping
4.3. Administration of DHA
4.4. Characterization of Aβ Aggregation
4.5. Stereotaxic Surgery
4.6. Preparation of Brain Tissue
4.7. Cell Culture and DHA Treatment
4.8. Cell Viability Assay
4.9. Measurements of Oxygen Consumption Rate
4.10. Immunohistochemistry
4.11. Image Acquisition and Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid-beta |
| DCX | Doublecortin |
| DG | Dentate gyrus |
| DHA | Docosahexaenoic acid |
| DPA | Docosapentaenoic acid |
| EPA | Eicosapentaenoic acid |
| GFAP | Glial fibrillary acidic protein for astrocyte |
| Iba-1 | Ionized calcium-binding adapter molecule 1 |
| NeuN | Neuronal nuclei |
| PUFAs | Polyunsaturated fatty acid |
| SGZ | Subgranular zone |
| SYN | Synaptophysin |
| Tom20 | Translocases of mitochondrial outer membrane 20 |
References
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of Abeta oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [PubMed]
- McGeer, E.G.; McGeer, P.L. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: A field in its infancy. J. Alzheimers Dis. 2010, 19, 355–361. [Google Scholar] [PubMed]
- Schwab, C.; McGeer, P.L. Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J. Alzheimers Dis. 2008, 13, 359–369. [Google Scholar] [CrossRef]
- Cha, M.Y.; Han, S.H.; Son, S.M.; Hong, H.S.; Choi, Y.J.; Byun, J.; Mook-Jung, I. Mitochondria-specific accumulation of amyloid beta induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Shin, S.J.; Jeon, S.G.; Kim, J.I.; Jeong, Y.O.; Kim, S.; Park, Y.H.; Lee, S.K.; Park, H.H.; Hong, S.B.; Oh, S.; et al. Red Ginseng Attenuates Abeta-Induced Mitochondrial Dysfunction and Abeta-mediated Pathology in an Animal Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3030. [Google Scholar] [CrossRef]
- Sirk, D.; Zhu, Z.; Wadia, J.S.; Shulyakova, N.; Phan, N.; Fong, J.; Mills, L.R. Chronic exposure to sub-lethal beta-amyloid (Abeta) inhibits the import of nuclear-encoded proteins to mitochondria in differentiated PC12 cells. J. Neurochem. 2007, 103, 1989–2003. [Google Scholar] [CrossRef]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar]
- Khacho, M.; Clark, A.; Svoboda, D.S.; MacLaurin, J.G.; Lagace, D.C.; Park, D.S.; Slack, R.S. Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum. Mol. Genet. 2017, 26, 3327–3341. [Google Scholar] [CrossRef] [PubMed]
- Beckervordersandforth, R. Mitochondrial Metabolism-Mediated Regulation of Adult Neurogenesis. Brain Plast. 2017, 3, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and cell bioenergetics: Increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid. Redox Signal. 2012, 16, 1434–1455. [Google Scholar] [CrossRef]
- Surette, M.E. Dietary omega-3 PUFA and health: Stearidonic acid-containing seed oils as effective and sustainable alternatives to traditional marine oils. Mol. Nutr. Food Res. 2013, 57, 748–759. [Google Scholar] [CrossRef]
- Conquer, J.A.; Tierney, M.C.; Zecevic, J.; Bettger, W.J.; Fisher, R.H. Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids 2000, 35, 1305–1312. [Google Scholar] [CrossRef]
- Tixier-Vidal, A.; Picart, R.; Loudes, C.; Bauman, A.F. Effects of polyunsaturated fatty acids and hormones on synaptogenesis in serum-free medium cultures of mouse fetal hypothalamic cells. Neuroscience 1986, 17, 115–132. [Google Scholar] [CrossRef]
- Greiner, R.S.; Moriguchi, T.; Hutton, A.; Slotnick, B.M.; Salem, N., Jr. Rats with low levels of brain docosahexaenoic acid show impaired performance in olfactory-based and spatial learning tasks. Lipids 1999, 34, S239–S243. [Google Scholar] [CrossRef]
- Echeverria, F.; Valenzuela, R.; Catalina Hernandez-Rodas, M.; Valenzuela, A. Docosahexaenoic acid (DHA), a fundamental fatty acid for the brain: New dietary sources. Prostaglandins Leukot. Essent. Fat. Acids 2017, 124, 1–10. [Google Scholar] [CrossRef]
- Kalmijn, S.; van Boxtel, M.P.J.; Ocké, M.; Verschuren, W.M.M.; Kromhout, D.; Launer, L.J. Dietary intake of fatty acids and fish in relation to cognitive performance at middle age. Neurology 2004, 62, 275–280. [Google Scholar] [CrossRef]
- Morris, M.C.; Evans, D.A.; Tangney, C.C.; Bienias, J.L.; Wilson, R.S. Fish Consumption and Cognitive Decline With Age in a Large Community Study. JAMA Neurol. 2005, 62, 1849–1853. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hossain, S.; Shimada, T.; Sugioka, K.; Yamasaki, H.; Fujii, Y.; Ishibashi, Y.; Oka, J.-I.; Shido, O. Docosahexaenoic acid provides protection from impairment of learning ability in Alzheimer’s disease model rats. J. Neurochem. 2002, 81, 1084–1091. [Google Scholar] [CrossRef]
- Hashimoto, M.; Tanabe, Y.; Fujii, Y.; Kikuta, T.; Shibata, H.; Shido, O. Chronic Administration of Docosahexaenoic Acid Ameliorates the Impairment of Spatial Cognition Learning Ability in Amyloid β–Infused Rats. J. Nutr. 2005, 135, 549–555. [Google Scholar] [CrossRef]
- Calon, F.; Lim, G.P.; Yang, F.; Morihara, T.; Teter, B.; Ubeda, O.; Rostaing, P.; Triller, A.; Salem, N.; Ashe, K.H.; et al. Docosahexaenoic Acid Protects from Dendritic Pathology in an Alzheimer’s Disease Mouse Model. Neuron 2004, 43, 633–645. [Google Scholar] [CrossRef]
- Green, K.N.; Martinez-Coria, H.; Khashwji, H.; Hall, E.B.; Yurko-Mauro, K.A.; Ellis, L.; LaFerla, F.M. Dietary Docosahexaenoic Acid and Docosapentaenoic Acid Ameliorate Amyloid-β and Tau Pathology via a Mechanism Involving Presenilin 1 Levels. J. Neurosci. 2007, 27, 4385–4395. [Google Scholar] [CrossRef]
- Avallone, R.; Vitale, G.; Bertolotti, M. Omega-3 Fatty Acids and Neurodegenerative Diseases: New Evidence in Clinical Trials. Int. J. Mol. Sci. 2019, 20, 4256. [Google Scholar] [CrossRef]
- Kang, J.X.; Wang, J.; Wu, L.; Kang, Z.B. Transgenic mice: Fat-1 mice convert n-6 to n-3 fatty acids. Nature 2004, 427, 504. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, F.; Leak, R.K.; Zhang, W.; Iwai, M.; Stetler, R.A.; Dai, Y.; Zhao, A.; Gao, Y.; Chen, J. Transgenic overproduction of omega-3 polyunsaturated fatty acids provides neuroprotection and enhances endogenous neurogenesis after stroke. Curr. Mol. Med. 2013, 13, 1465–1473. [Google Scholar] [CrossRef]
- He, C.; Qu, X.; Cui, L.; Wang, J.; Kang, J.X. Improved spatial learning performance of fat-1 mice is associated with enhanced neurogenesis and neuritogenesis by docosahexaenoic acid. Proc. Natl. Acad. Sci. USA 2009, 106, 11370–11375. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, R.; Dong, Y.; Tucker, D.; Zhao, N.; Ahmed, M.E.; Zhu, L.; Liu, T.C.; Cohen, R.M.; Zhang, Q. Low-level laser therapy for beta amyloid toxicity in rat hippocampus. Neurobiol. Aging 2017, 49, 165–182. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vassar, R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol. Neurodegener. 2013, 8, 2. [Google Scholar]
- Kornfeld, O.S.; Qvit, N.; Haileselassie, B.; Shamloo, M.; Bernardi, P.; Mochly-Rosen, D. Interaction of mitochondrial fission factor with dynamin related protein 1 governs physiological mitochondrial function in vivo. Sci. Rep. 2018, 8, 14034. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar]
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef]
- Feng, Z.; Zou, X.; Jia, H.; Li, X.; Zhu, Z.; Liu, X.; Bucheli, P.; Ballevre, O.; Hou, Y.; Zhang, W.; et al. Maternal docosahexaenoic acid feeding protects against impairment of learning and memory and oxidative stress in prenatally stressed rats: Possible role of neuronal mitochondria metabolism. Antioxid. Redox Signal. 2012, 16, 275–289. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef]
- Gu, M.; Li, Y.; Tang, H.; Zhang, C.; Li, W.; Zhang, Y.; Zhao, Y.; Song, C. Endogenous Omega (n)-3 Fatty Acids in Fat-1 Mice Attenuated Depression-Like Behavior, Imbalance between Microglial M1 and M2 Phenotypes, and Dysfunction of Neurotrophins Induced by Lipopolysaccharide Administration. Nutrients 2018, 10, 1351. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar]
- Hauptmann, S.; Scherping, I.; Drose, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Muller, W.E. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef]
- Rhein, V.; Baysang, G.; Rao, S.; Meier, F.; Bonert, A.; Muller-Spahn, F.; Eckert, A. Amyloid-beta leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell. Mol. Neurobiol. 2009, 29, 1063–1071. [Google Scholar] [CrossRef]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; Suire, C.; Maharana, C.; Jicha, G.A.; Liu, D.; et al. Extracellular Vesicle-Associated Abeta Mediates Trans-Neuronal Bioenergetic and Ca(2+)-Handling Deficits in Alzheimer’s Disease Models. NPJ Aging Mech. Dis. 2016, 2, 16019. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, S.; Shao, Q.; Ma, D.; Yang, Z.; Zhou, R. Mechanism by which DHA inhibits the aggregation of KLVFFA peptides: A molecular dynamics study. J. Chem. Phys. 2018, 148, 115102. [Google Scholar] [CrossRef]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a biological hydrotrope. Science 2017, 356, 753–756. [Google Scholar]
- Edison, P.; Brooks, D.J. Role of Neuroinflammation in the Trajectory of Alzheimer’s Disease and in vivo Quantification Using PET. J. Alzheimers Dis. 2018, 64, S339–S351. [Google Scholar] [CrossRef]
- Ising, C.; Heneka, M.T. Functional and structural damage of neurons by innate immune mechanisms during neurodegeneration. Cell Death Dis. 2018, 9, 120. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pedraza-Chaverri, J.; Perez-Rojas, J.M. Role of docosahexaenoic acid in the modulation of glial cells in Alzheimer’s disease. J. Neuroinflammation 2016, 13, 61. [Google Scholar]
- Wilkins, H.M.; Swerdlow, R.H. Relationships Between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Curr. Top. Med. Chem. 2016, 16, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Mediat. Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef] [PubMed]
- Donev, R.; Kolev, M.; Millet, B.; Thome, J. Neuronal death in Alzheimer’s disease and therapeutic opportunities. J. Cell Mol. Med. 2009, 13, 4329–4348. [Google Scholar]
- Marttinen, M.; Takalo, M.; Natunen, T.; Wittrahm, R.; Gabbouj, S.; Kemppainen, S.; Leinonen, V.; Tanila, H.; Haapasalo, A.; Hiltunen, M. Molecular Mechanisms of Synaptotoxicity and Neuroinflammation in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 963. [Google Scholar] [CrossRef]
- Kashyap, G.; Bapat, D.; Das, D.; Gowaikar, R.; Amritkar, R.E.; Rangarajan, G.; Ravindranath, V.; Ambika, G. Synapse loss and progress of Alzheimer’s disease—A network model. Sci. Rep. 2019, 9, 6555. [Google Scholar]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and Abeta. Mol. Neurodegener. 2009, 4, 48. [Google Scholar]
- Hajjar, T.; Goh, Y.M.; Rajion, M.A.; Vidyadaran, S.; Li, T.A.; Ebrahimi, M. Alterations in neuronal morphology and synaptophysin expression in the rat brain as a result of changes in dietary n-6: n-3 fatty acid ratios. Lipids Health Dis. 2013, 12, 113. [Google Scholar] [CrossRef]
- Petursdottir, A.L.; Farr, S.A.; Morley, J.E.; Banks, W.A.; Skuladottir, G.V. Effect of dietary n-3 polyunsaturated fatty acids on brain lipid fatty acid composition, learning ability, and memory of senescence-accelerated mouse. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 1153–1160. [Google Scholar]
- Bayer, T.A.; Wirths, O. Intracellular accumulation of amyloid-Beta—A predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front. Aging Neurosci. 2010, 2, 8. [Google Scholar]
- Han, X.J.; Hu, Y.Y.; Yang, Z.J.; Jiang, L.P.; Shi, S.L.; Li, Y.R.; Guo, M.Y.; Wu, H.L.; Wan, Y.Y. Amyloid beta-42 induces neuronal apoptosis by targeting mitochondria. Mol. Med. Rep. 2017, 16, 4521–4528. [Google Scholar] [CrossRef]
- Cai, Q.; Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1087–1103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wu, P.; Zhang, J.H.; Li, Y.; Xu, S.; Wang, C.; Wang, L.; Zhang, G.; Dai, J.; Zhu, S.; et al. Docosahexaenoic Acid Alleviates Oxidative Stress-Based Apoptosis via Improving Mitochondrial Dynamics in Early Brain Injury after Subarachnoid Hemorrhage. Cell Mol. Neurobiol. 2018, 38, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Oster, T.; Pillot, T. Docosahexaenoic acid and synaptic protection in Alzheimer’s disease mice. Biochim. Biophys. Acta 2010, 1801, 791–798. [Google Scholar] [PubMed]
- Moreno-Jimenez, E.P.; Flor-Garcia, M.; Terreros-Roncal, J.; Rabano, A.; Cafini, F.; Pallas-Bazarra, N.; Avila, J.; Llorens-Martin, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Beltz, B.S.; Tlusty, M.F.; Benton, J.L.; Sandeman, D.C. Omega-3 fatty acids upregulate adult neurogenesis. Neurosci. Lett. 2007, 415, 154–158. [Google Scholar] [CrossRef]
- Mayurasakorn, K.; Niatsetskaya, Z.V.; Sosunov, S.A.; Williams, J.J.; Zirpoli, H.; Vlasakov, I.; Deckelbaum, R.J.; Ten, V.S. DHA but Not EPA Emulsions Preserve Neurological and Mitochondrial Function after Brain Hypoxia-Ischemia in Neonatal Mice. PLoS ONE 2016, 11, e0160870. [Google Scholar] [CrossRef]
- Maarouf, C.L.; Kokjohn, T.A.; Whiteside, C.M.; Macias, M.P.; Kalback, W.M.; Sabbagh, M.N.; Beach, T.G.; Vassar, R.; Roher, A.E. Molecular Differences and Similarities between Alzheimer’s Disease and the 5XFAD Transgenic Mouse Model of Amyloidosis. Biochem. Insights 2013, 6, 1–10. [Google Scholar]
- Mead, R. The Design of Experiments: Statistical Principles for Practical Applications; Cambridge University Press: New York, NY, USA, 1988. [Google Scholar]
- Arifin, W.N.; Zahiruddin, W.M. Sample Size Calculation in Animal Studies Using Resource Equation Approach. Malays. J. Med Sci. (MJMS) 2017, 24, 101–105. [Google Scholar]
- Charan, J.; Kantharia, N.D. How to calculate sample size in animal studies? J. Pharmacol. Pharmacother. 2013, 4, 303–306. [Google Scholar] [CrossRef]
- Truett, G.E.; Heeger, P.; Mynatt, R.L.; Truett, A.A.; Walker, J.A.; Warman, M.L. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 2000, 29, 52–54. [Google Scholar]
- Ren, H.; Luo, C.; Feng, Y.; Yao, X.; Shi, Z.; Liang, F.; Kang, J.X.; Wan, J.B.; Pei, Z.; Su, H. Omega-3 polyunsaturated fatty acids promote amyloid-beta clearance from the brain through mediating the function of the glymphatic system. FASEB J. 2017, 31, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Casali, B.T.; Corona, A.W.; Mariani, M.M.; Karlo, J.C.; Ghosal, K.; Landreth, G.E. Omega-3 Fatty Acids Augment the Actions of Nuclear Receptor Agonists in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 9173–9181. [Google Scholar] [CrossRef] [PubMed]
- Noor, H.; Cao, P.; Raleigh, D.P. Morin hydrate inhibits amyloid formation by islet amyloid polypeptide and disaggregates amyloid fibers. Protein Sci. 2012, 21, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Monteleone, D.; Piras, A.; Valsecchi, V.; Tropiano, M.; Ariano, S.; Fornaro, M.; Vercelli, A.; Puyal, J.; Arancio, O.; et al. Abeta1–42 monomers or oligomers have different effects on autophagy and apoptosis. Autophagy 2014, 10, 1827–1843. [Google Scholar] [CrossRef]
- Moon, M.; Choi, J.G.; Nam, D.W.; Hong, H.S.; Choi, Y.J.; Oh, M.S.; Mook-Jung, I. Ghrelin ameliorates cognitive dysfunction and neurodegeneration in intrahippocampal amyloid-beta1–42 oligomer-injected mice. J. Alzheimers Dis. 2011, 23, 147–159. [Google Scholar] [CrossRef]
- Moon, M.; Choi, J.G.; Kim, S.Y.; Oh, M.S. Bombycis excrementum reduces amyloid-beta oligomer-induced memory impairments, neurodegeneration, and neuroinflammation in mice. J. Alzheimers Dis. 2014, 41, 599–613. [Google Scholar] [CrossRef]
- McIntee, F.L.; Giannoni, P.; Blais, S.; Sommer, G.; Neubert, T.A.; Rostagno, A.; Ghiso, J. In vivo Differential Brain Clearance and Catabolism of Monomeric and Oligomeric Alzheimer’s Abeta protein. Front. Aging Neurosci. 2016, 8, 223. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B. Paxinos and Franklin’s The Mouse Brain in Stereotaxic Coordinates, 4th ed.; Elsevier Academic Press: San Diego, CA, USA, 2013. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.H.; Shin, S.J.; Kim, H.s.; Hong, S.B.; Kim, S.; Nam, Y.; Kim, J.-J.; Lim, K.; Kim, J.-S.; Kim, J.-i.; et al. Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Aβ-Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model. Int. J. Mol. Sci. 2020, 21, 3879. https://doi.org/10.3390/ijms21113879
Park YH, Shin SJ, Kim Hs, Hong SB, Kim S, Nam Y, Kim J-J, Lim K, Kim J-S, Kim J-i, et al. Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Aβ-Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model. International Journal of Molecular Sciences. 2020; 21(11):3879. https://doi.org/10.3390/ijms21113879
Chicago/Turabian StylePark, Yong Ho, Soo Jung Shin, Hyeon soo Kim, Sang Bum Hong, Sujin Kim, Yunkwon Nam, Jwa-Jin Kim, Kyu Lim, Jong-Seok Kim, Jin-il Kim, and et al. 2020. "Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Aβ-Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model" International Journal of Molecular Sciences 21, no. 11: 3879. https://doi.org/10.3390/ijms21113879
APA StylePark, Y. H., Shin, S. J., Kim, H. s., Hong, S. B., Kim, S., Nam, Y., Kim, J.-J., Lim, K., Kim, J.-S., Kim, J.-i., Jeon, S. G., & Moon, M. (2020). Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Aβ-Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model. International Journal of Molecular Sciences, 21(11), 3879. https://doi.org/10.3390/ijms21113879