Abstract
Endoplasmic reticulum (ER) and mitochondrion are the key organelles in mammal cells and play crucial roles in a variety of biological functions in both physiological and pathological conditions. Valosin-containing protein (VCP), a newly identified calcium-associated ATPase protein, has been found to be involved in both ER and mitochondrial function. Impairment of VCP, caused by structural mutations or alterations of expressions, contributes to the development of various diseases, through an integrating effect on ER, mitochondria and the ubiquitin–proteasome system, by interfering with protein degradation, subcellular translocation and calcium homeostasis. Thus, understanding the role and the molecular mechanisms of VCP in these organelles brings new insights to the pathogenesis of the associated diseases, and leads to the discovery of new therapeutic strategies. In this review, we summarized the progress of studies on VCP, in terms of its regulation of ER and mitochondrial function and its implications for the associated diseases, focusing on the cancers, heart disease, and neurodegenerative disorders.
1. Introduction
The endoplasmic reticulum (ER) is one of the largest membrane organelles in cells, and plays an important role in protein synthesis, protein folding and quality control, lipid metabolism and Ca2+ homeostasis [1]. As ER is found in all cell types, the sarcoplasmic reticulum (SR), a morphologically distinct version of the ER, only exits in muscle cells that are specialized for Ca2+ release to fuel muscle contraction. SR consists of two spatial and functional organizations, termed longitudinal SR and junctional SR, tasked with the release and the uptake of Ca2+, ensuring delivery of Ca2+ for contraction to occur [2,3,4]. ER dysfunction has been associated with cellular dysfunction and cell death [5]. ER contains multiple domains, among which the biggest is a bilayer barrier around the cell nucleus, named the nuclear envelope. In addition, the peripheral ER membrane extends from the nuclear envelope to the whole cytoplasm, forming interactions with other organelles and contacting the plasma membrane [6]. One of the most essential organelles that the ER interacts with is the mitochondria, which are responsible for regulating Ca2+ homeostasis, apoptosis and ATP production. The dysfunction of mitochondria leads to impaired energy production and contributes to the pathogenesis of many metabolic diseases [7,8]. Most recent evidence has demonstrated that mitochondria communicate with the ER via mitochondria-associated ER membranes (MAMs) [8,9]. Although increasing evidence demonstrates that impaired function of the ER and mitochondria is associated with the pathogenesis of a variety of diseases, including cancers, neurodegeneration diseases and heart disease, as well as metabolic diseases such as obesity and diabetes [9,10,11], the underlying mechanisms remain largely unknown.
The valosin-containing protein (VCP), also known as p97 in mammals, Cdc48 in yeast and plants, CDC-48 in worms and Ter94 in flies, was initially recognized as one of the ER-associated proteins, and has been demonstrated as playing critical roles in regulating ER formation and morphology by participating in the ubiquitin–proteasome system (UPS) and other intracellular signaling pathways [12,13]. One of the key functions of VCP is to conjugate its substrates with the ubiquitin chain through interaction with a variety of ubiquitin adapters, and then transport the substrates to the 26S proteasome for subsequent degradation, such as ER-associated protein degradation (ERAD) [13]. The mutations of VCP are found to be associated with some human degenerative disorders, such as amyotrophic lateral sclerosis (ALS), inclusion body myopathy (IBM) associated with Paget disease of the bone (PDB), and frontotemporal dementia (FTD), also called IBMPFD, which mainly affect the brain and muscles [12]. In addition, the VCP expression level was also shown to be upregulated in some cancers, mainly in response to the increased burden of protein degradation, indicating that VCP inhibition could be a promising therapeutic approach to cancer management [13,14,15]. Furthermore, recent studies also found that VCP participates in cardiomyocyte growth and survival, and plays a protective role against the stress-induced pathogenesis in heart diseases by attenuating mitochondrial and ER stress through regulating calcium homeostasis [16,17,18,19].
In this review, we summarized the new progress achieved in studies of VCP, regarding its regulating effect on ER and mitochondria functions, and its implications for various diseases, focusing on cancer, heart disease and neurodegenerative disorders.
2. The Structure and Distribution of VCP in Mammal Cells
VCP belongs to the type II class of the AAA (ATPases Associated with various cellular Activities) ATPase family [20]. As shown in Figure 1, VCP possesses four structural domains, including a conserved N-terminal domain, two AAA ATPase domains (D1 and D2), and a C-terminal tail. D1 and D2 domains are stacked in a head-to-tail manner and connect with a short polypeptide linker, while the N-terminal domain is connected to the D1 domain by another short linker. In mammalian cells, VCP functions as a homohexamer [20]. The active form of VCP is a complex of a double ring structure, with the D1 and D2 domains sitting on top of each other.
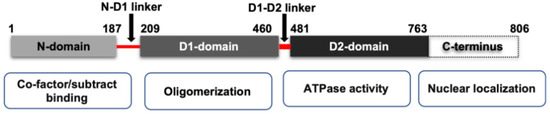
Figure 1.
The scheme of the mammalian isoform of VCP domains and their function. VCP is constituted by one binding domain N-domain, two ATPase domains (D1 domain and D2 domain) and a C-terminus. N-domains are responsible for substrate recognition and binding. The D2 domain contributes to the major ATPase activity of VCP, while the D1 domain is responsible for the assembly of VCP homohexamer. The N-domain and D1 domain are connected by an N-D1 linker, and the D1 and D2 domains are connected by D1–D2 linker.
While the N-terminal domain of VCP is involved in the substrate’s recognition and interaction with other cofactors, the C-terminal tail was shown to be involved in nuclear localization via interacting with other proteins. Meanwhile, the D2 domain undertakes the major ATPase activity, while the D1 domain mainly contributes to the assembly of the hexamers [20]. The linkers between the D1 and D2 domains, and the D1 and N terminal domains, are also reported to be critical to the functions of VCP. For instance, the linker region between the D1 and D2 domains is essential for the D2 ATPase activity [21]. Interestingly, most disease-associated VCP mutations are found in the N-domains and ATPase domains [22], and those mutations in VCP have been shown to impair mitochondrial function and protein homeostasis [23,24]. It also reported that the mutations in the D1–N terminal linker region of VCP may cause neurodegenerative disorders in humans [25].
VCP is one of the most broadly expressed proteins in human, and it can be detected in a variety of organs such as the brain, skeletal muscle, heart, kidney, liver, ovary, testis and lung [26]. In mammalian cells, VCP is found to be distributed in different subcellular organelles, such as cytoplasm, Golgi apparatus [27], nuclear envelope [28], ER and mitochondria [29]. It has been shown that VCP, an ATPase, participates in a diverse array of cellular functions, including DNA replication, protein folding and degradation, the ubiquitin–proteasome system (UPS), calcium homeostasis, chromatin remodeling, and the assembly of Golgi and nuclear membranes [30].
3. The Regulation of VCP in ER and Mitochondrial Function in Physiological Condition
The function of ER under normal conditions is responsible for regulating protein folding and synthesis, post-translational modification, and maintaining the transaction of different transmembrane proteins. It has been shown that VCP, as a ubiquitin-selective chaperone, plays an essential role in maintaining ER integrity through interacting with E3 ubiquitin ligases, such as Glycoprotein 78 (gp78, also known as AMFR) and ERAD-associated E3 ubiquitin-protein ligase HRD 1 (Hrd1) [31,32].
Under normal conditions, in order to restore ER homeostasis, accumulated misfolded proteins in the ER active the signal of the unfolded protein response (UPR), which initiates the ERAD process through activating transcription factor 6 (ATF6), inositol-requiring enzyme 1α (IRE-1 α) and the PKR-like ER kinase (PERK) signaling pathways [33], by which the misfolded proteins are exported from ER to cytoplasm, where those proteins are subject to proteasomal degradation. Studies have shown that VCP plays a critical role in maintaining the homeostasis of ER, by regulating ubiquitin-dependent processes through the ubiquitin–proteasome system (UPS). By interacting with various polyubiquitinated proteins, such as ubiquitin ligase gp78, ubiquitin fusion degradation 1L, UBX-domain containing protein 1 (UBXD1), p47, nuclear protein localization protein 4 (Npl4), or other ubiquitin-like proteins, VCP is involved in the regulation of the activity of these factors and the delivery of misfolded proteins for proteasomal degradation [29,32,34]. VCP also interacts and binds to the ubiquitinated substrates, and regulates the downstream of ubiquitylation [35]. During ERAD, VCP extracts ubiquitinated misfolded proteins from ER membranes to the 26S proteasome for degradation, preventing the misfolded proteins’ aggregation, which is the main cause of ER stress.
The ER physically directly interacts with mitochondria, and plays a critical role in maintaining mitochondrial function. For instance, VCP can extract misfolded proteins from mitochondria during the process of mitochondria-associated degradation with the VCP cofactors Ufd1-Np14. VCP has been shown to participate in mitophagy-related degradation, which depends on the E3 ligase Parkin [36]. VCP is also required for the retro-translocation of the anti-apoptotic protein myeloid cell leukemia sequence 1(Mcl1) from mitochondria to cytosol for the proteasomal degradation [29].
In addition, ER–mitochondria tethering plays a critical role in regulating calcium homeostasis through MAM. The inositol triphosphate receptor (IP3R), a Ca2+ channel on ER, is highly accumulated in MAM [37]. Through the interaction of the regulators and the Ca2+ channel regulation protein complex, Ca2+ transfer can be modulated between ER and mitochondria [38,39]. In mitochondria, different Ca2+ levels trigger different molecular activities. Increase of the mitochondrial Ca2+ stimulates electron activity and ATP generation [40]. However, excessive Ca2+ levels will trigger apoptosis, through inducing mitochondrial permeability transition pore (mPTP) opening [41]. Thus, the mechanism of VCP in regulating mitochondrial calcium homeostasis has attracted attention. Recent studies have shown that VCP participates in mitochondrial respiration, calcium intake and mPTP, through regulating the mitochondrial calcium uptake (MICU) proteins by regulating the degradation of these proteins [17].
In summary, as shown in Figure 2, VCP plays an essential role in maintaining cellular ER and mitochondrial function and calcium homeostasis in normal cells, and thus, impairment of the VCP activity, caused by deficiency of expression or the mutation of structure, will induce ER and mitochondrial dysfunctions, which subsequently induce cellular damage, resulting in the pathogenesis of the diseases.
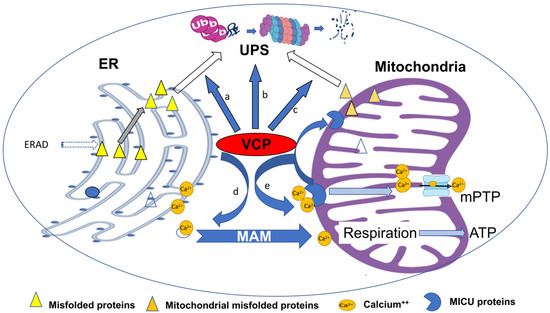
Figure 2.
Summary of the integrating regulation of VCP on ER- and mitochondrial-associated degradation and calcium homeostasis. VCP plays an essential role in maintaining cellular function and calcium homeostasis in physiological conditions through multiple mechanisms. By interacting with other proteins, VCP participates in the delivery of misfolded proteins from the ER (a) and promotes the ubiquitin-dependent degradation processes through the ubiquitin–proteasome system (UPS) during ERAD (b). VCP is also involved in extracting the misfolded proteins from mitochondria during the process of mitochondria-associated degradation, and participates in mitophagy-related degradation (c). VCP participates in the regulation of calcium homeostasis through mitochondria-associated ER membranes (MAMs), which stimulates electron activity and ATP production (d). Reciprocally, VCP prevents excessive calcium entry in mitochondria by regulating the mitochondrial calcium intake through the degradation of mitochondrial calcium uptake (MICU) proteins (e), which subsequently inhibits mPTP opening, preventing cell death. Impairment of VCP activity induces ER and mitochondrial dysfunctions, which subsequently induce cellular damage, resulting in the pathogenesis of the diseases.
4. The Aberrant Expression of VCP Mediates ER and Mitochondria Dysfunctions in the Progress of Various Cancers
Accumulating evidence from clinical studies has revealed the prognostic value of VCP in a wide range of cancers (Table 1), and demonstrated an enhanced ERAD and upregulated VCP expression in cancer cells [42]. For example, VCP upregulation has been associated with unfavorable clinical outcomes in breast cancer patients, and was found to be involved in cell growth and survival in colorectal cancer [43]. In other malignancies in the lung, prostate and pancreas, the VCP expression level was correlated with recurrence rate and prognosis in the patients [44]. Interestingly, although the aberrant expression of VCP has been shown to contribute to cancer progression at different stages, the role of VCP in the pathogenesis of cancers appears controversial. For instance, VCP expression declines in osteosarcoma cell lines, however, VCP expression is continuously increased in its subline LM8 cells, which display higher metastatic potential [45]. Yamamoto et al. reported that in gastric carcinoma and in esophageal squamous cell carcinoma (ESCC), a higher expression of VCP indicates a higher rate of lymph node metastasis, deep tumor invasion, and poorer overall and disease-free survival [46,47]. However, in human papilloma virus (HPV)-negative oropharyngeal squamous cell carcinoma (OSCC), high expression of VCP was related to significantly better five-year disease-free survival. It has also been reported that in HPV-negative OSCC, patients who lose the chromosome region 9p13-p12, in which the VCP gene is located, display a poor survival rate [48].

Table 1.
Summary of VCP expression and prognostic value in different types of cancer.
VCP expression is upregulated in most types of cancer, and the level of VCP expression is genitively associated with survival rates in patients. However, the correlation varies in some types of cancers.
Several studies have revealed the potential mechanisms of VCP involved in cancer cell reprogramming. The VCP-interacting motif is regulated mainly by three proteins: E3 ubiquitin-protein ligase gp78, the recruitment factor for E3 ubiquitin-protein ligase sialoprotein S (VIMP), and the small VCP/p97-interacting protein (SVIP) [52,53]. While Gp78 and VIMP have been shown to promote ERAD in cancer cells, SVIP has been reported to play an opposing role, and exert an inhibitory effect on this process [54]. In many human tumors, SVIP is silenced [42], resulting in an upregulation of VCP, which in turn facilitates ERAD to avoid programmed cell death. These studies have provided the potential mechanism by which cancer cells avoid ER stress-induced programmed cell death, and become progressive in a VCP-dependent manner.
Evidence also indicates that VCP plays an important role in the metastatic process in cancer cells, by involvement in the ubiquitin-dependent proteasome degradation pathway via the Akt/NFκB pathway [55]. In addition, recently, ER stress has been reported to induce epithelial–mesenchymal transition (EMT) in cancers, which is critical for the progression and metastasis of the tumor. During the process of EMT, epithelial markers are decreased, while the mesenchymal markers are increased in the cells. ER stress level is the driving force of cell fate—either apoptosis or EMT [56]—and also induces abnormal cell differentiation and morphological changes [57]. Findings from the recent study suggest that, at the early stage of cell dysfunction, loss of VCP results in elevated ER stress, which may transform cells into an EMT-like state. However, prolonged deficiency of VCP will lead to programmed cell death [58].
Mitochondria have been also shown to be critical in cancer transformation, progression and metabolism [59]. The dysfunction of mitochondria has been related to many pathological processes in human cancers [60]. Mitochondrial quality control dysregulation causes mitochondria stress, and increases reactive oxygen species (ROS) production, which may be involved in cancer predisposition [61]. In the mitochondria, the quality control of the outer membrane (OMM) proteins plays an important role in the maintaining of mitochondria homeostasis. Many OMM proteins are regulated by the ubiquitin–proteasome system, in which VCP plays a pivotal role [29,62]. For example, VCP/Cdc48-associated mitochondrial stress-responsive 1(Vms1), a mitochondrial protein, translocates from the cytoplasm to mitochondria during mitochondria stress, and plays an important role in regulating mitochondria quality control in a complex with VCP, through the ubiquitin–proteasome system. The decrease of Vms1 impairs the degradation of the OMM protein and induces mitochondria stress [63]. Thus, as a major regulator of mitochondria, VCP could also play critical roles in the pathogenesis of cancers.
Based on these results, VCP is now considering as an important target for cancer treatment. It has been shown that the inhibition of VCP is able to induce proteotoxic stress and apoptosis in cancer cells, and improve radiation sensitivity in ESCC [64]. One of the VCP inhibitors, CB-5083, is the first selective VCP inhibitor to show preclinical effects [13]. However, the clinical trial of CB-5083 failed due to its toxicities. Some new compounds that may have better potential effects are being investigated, like the VCP inhibitor compound 35, in non-small cell lung cancer [44].
5. VCP Structural Mutations Induce ER and Mitochondrial Impairments Resulting in Neuro-Degenerative Disorders
The mutations of VCP have been associated with diverse myodegenerative and neurodegenerative disorders including inclusion body myopathy (IBM), with Paget disease of the bone, as well as frontotemporal dementia (IBMPFD) and amyotrophic lateral sclerosis (ALS).
IBMPFD is a multisystem degenerative disorder caused by mutations in VCP, which includes IBM associated with Paget’s disease of the bone (PDB) and frontotemporal dementia (FTD) [65]. IBM is a hereditary disease with uncertain pathogenic mechanisms, and it is usually associated with progressive asymmetric muscle weakness and other pathological features, including invasion of myofibers by cytotoxic T cells, congophilic inclusions, cytoplasmic aggregates and rimmed vacuoles [66]. One of the characteristic hallmarks of this disorder is cytoplasmic protein aggregation, which is associated with a large range of proteins, such as ubiquitin, amyloid-β peptides, phosphorylated tau, RNA-binding protein, TAR-DNA binding protein 43 (TDP-43) and prion protein, which results in imbalanced protein homeostasis in those diseases [67,68]. Multiple mutations of VCP have been demonstrated to contribute to the pathogenesis of IBMPFD. For instance, multiple sites of mutations at R95G, R155P, R155C and R155H have been observed in the N-domain of VCP, while R155H is the most frequently identified mutation. Studies have shown that expressing VCP R155H in cultures cells results in an abundant accumulation of misfolded substrates, suggesting ERAD is severely disrupted in IBMPFD [69]. Other mutations were confirmed to be located in the N- to D1-domain linker and D1 domain, such as R191Q and A232E [65], which altered the structural orientation in N and/or D1 domains, consequently affecting its cofactor-binding features and ATPase activity [70,71].
Studies have postulated the potential molecular mechanisms by which VCP regulates IBMPFD pathogenesis. First, the IBMPFD-associated mutations in VCP disrupt the ubiquitin-conjugated protein turnover in the ER membrane and aggregate with other cofactors, ultimately impairing the UPS pathway. These phenomena may be attributed to the interruption of the protein–protein interactions in the ERAD pathway, which impairs the transferring of ubiquitinated proteins from ER to proteasome [69]. Second, overexpressing VCP mutants, VCP-R155H or VCP-A232E, results in defects in autophagy by impairing ubiquitin-containing autophagosome maturation, and leads to the accumulation of autophagosomes, which contribute to the pathogenesis of IBMPFD [65]. Third, VCP acts as an important part in the Shoc2– Ras–RAF-1 complex, in regulating the ERK1/2 signaling pathway [72]. VCP mutations disturb ERK1/2 phosphorylation and alter ERK1/2 activity, which may play an important role in IBMPFD pathogenesis. Finally, VCP, interacting with the endoplasmic reticulum (ER) morphology regulator Atlastin-1(ATL1), regulates dendritic spine formation in neurons by influencing tubular ER formation and protein synthesis [24].
ALS, a rapid progress disease caused by degeneration of motor neurons (MNs), is another hereditary disease associated with VCP mutations. Mislocalization and aggregation of TDP-43 in the cytoplasm has been observed in VCP-mutated MNs, accompanied with an increased Bip and p-eIF2alpha and a decreased ER calcium storage [73]. TDP-43 dislocation and aggregation have also been connected with mitochondrial quality control defects, which may disturb the ER–mitochondria contacts and prevent protein import to the mitochondria matrix [74]. These studies have suggested that aberrant VCP expression may be associated with the pathogenesis of this disorder.
Mitochondrial function is also found to be disturbed by VCP mutations in neurodegeneration diseases like ALS and IBMPFD, including the increased mitochondrial fragmentation, the loss of mitochondrial membrane potential and the increased reactive oxygen species (ROS). VCP, as the ubiquitin-dependent chaperone, maintains the mitochondrial function in neuronal cells through the UPS and the outer mitochondrial membrane-associated degradation (OMMAD) [75]. In ALS, VCP mutations disrupt the damaged mitochondria clearance through impairing the PINK1/Parkin pathway [62].
6. VCP Represents a New Regulator in Cardiac ER and Mitochondria Functions, and is Involved in Various Heart Diseases
Of note, VCP was identified to be expressed in the heart [18]. Moreover, studies in Drosophila have also revealed that the knockdown of TER94 (VCP homolog in Drosophila) with cardiac-restricted siRNA severely disrupts myofibrillar organization and heart function in adult flies [76]. Taken together, these findings highlight the importance of VCP in the heart.
Clinical observation found that dilated cardiomyopathy commonly existed in patients with IBMPFD [77]. Cardiac-specific overexpression of an enzymatically mutated VCP (VCPK524A) in transgenic mice resulted in an impairment of the ATPase activity within the D2 domain of VCP, and lead to the development of cardiomyopathy [78]. These mice displayed an ERAD dysfunction and an accumulation of ubiquitinated proteins in the heart.
In addition, recent studies have revealed that the reduction of VCP expression was associated with hypertensive heart diseases [16]. For example, VCP expression was found to be significantly decreased in the heart of spontaneously hypertensive rats (SHR), compared to their normotensive Wistar Kyoto (WKY) controls. VCP expression was also found to be progressively decreased in the mouse hearts, along with persistent pressure overload [16]. Reciprocally, cardiac-specific overexpression of VCP attenuated the pressure overload-induced cardiac pathogenesis [19]. These phenomena are linked to the inhibitory effects of VCP on the transduction of the AKT/mTOC1R/S6K signaling pathway [16]. It is also found that the disruption of VCP ATPase activity results in age-dependent cardiomyopathy, because of the defect in the ubiquitinated targeted proteins’ degradation, mediated by UPS [78]. This suggests the critical role of VCP in regulating ER function in aging- and pressure overload-related cardiac cardiomyopathy.
Furthermore, increasing evidence indicates that VCP plays an important role in regulating mitochondrial function in the heart. The heart is an organ with high energy demands, and approximately 95% of energy consumed by the heart is from mitochondria [79]. Mitochondrial dysfunction has been observed in a variety of heart diseases, particularly in conditions with reduced blood supply and oxygen, such as ischemic heart injury [80]. Recent studies have demonstrated that cardiac-specific overexpression of VCP can protect the heart against ischemia/reperfusion injury, and prevent cell death in an inducible nitric oxide (NO) synthase (iNOS)-dependent manner [17]. These protective effects of VCP are related to the preservation of the cardiac mitochondrial function and the inhibition of the opening of mitochondrial permeability transition pores, through inhibiting the stress-induced excessive calcium uptake by repressing MICU1, and activating MICU2, which may be associated with the selective degradation of the mitochondrial protein by VCP [81]. Moreover, VCP is also identified as a link between NF-κB-induced iNOS expression and the AKT signaling pathway, which is activated by the heat shock protein22 (HSP22) [18], which is also a key regulator of UPS [82] and mitochondrial function [83]. However, despite these significant findings, the role of VCP in the pathogenesis of cardiomyopathy, and the mechanisms involved, remain largely unknown.
7. Future Directions
In this review, we have summarized the involvement of VCP in a variety of pathological conditions, particularly in cancers, neurodegenerative disorders and heart diseases. Although the regulatory signaling pathways underlying the pathogenesis of these disorders are not fully understood, and may vary among the different diseases, there are some common features related to VCP in these disorders. For example, as shown in Figure 3, by interacting with different cofactors/adaptors, VCP could promote stress-induced misfolded protein degradation and maintain calcium homeostasis, which subsequently helps resist ER/mitochondrial stress-induced cell damage and death. Thus, the increase of VCP in cancer cells would protect cells against death, while the deficiency of VCP in neurodegenerative disorders and heart diseases would induce cell damage and death, due to the loss of these functions. This evidence has highlighted the role of VCP in mitochondrial and/or ER-related disorders.
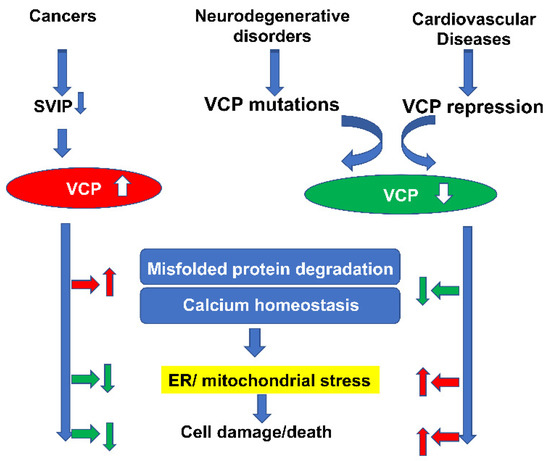
Figure 3.
Summary of the common features related to VCP in different diseases. Despite the fact that the cofactors and the regulatory signaling pathways underlying the pathogenesis of these disorders may vary, VCP could promote stress-induced misfolded protein degradation and maintain calcium homeostasis, which subsequently helps resist ER/mitochondrial stress-induced cell damage and death. As the increase of VCP in cancer cells would protect cells against death, the deficiency of VCP in neurodegenerative disorders and heart diseases would induce cell damage and death, due to the loss of these functions. (The red arrows represent the promotive effects and green arrows stand for the inhibitory/suppressive effects).
Therefore, future research could draw more attention to the definition of the detailed mechanism of VCP that is involved in the regulation of ER and mitochondrial interaction, in calcium metabolism and homeostasis. In addition, since VCP’s effects largely rely on its banded cofactors, exploring the potential interactions between VCP and other proteins, particularly the cofactors and substrates of the N-terminal domain of VCP, will bring new insight into the molecular mechanism in VCP-related diseases. Furthermore, VCP-mediated protein subcellular translocation is crucial in ER and mitochondrial function, and further understanding the mechanism by which VCP mediates other protein translocations will help to define the new effect of VCP in integrating homeostasis among multiple organelles. It is also necessary to explore the mechanisms that stimulate VCP in cancer cells, or their reduction under cardiac stress, which will lead to the discovery of the pathogenesis of the related diseases. Finally, it is important to design and develop VCP inhibitors for the treatment of various cancers, and activators of VCP to restore the VCP in stressed hearts, to prevent cardiomyopathy.
Author Contributions
X.S. and H.Q. wrote this manuscript and approved the submission. All authors have read and agreed to the published version of the manuscript.
Funding
This work is partially supported by NIH grants HL115195-01 (H.Q.), HL137962 (H.Q.), and HL 142291(H. Q &. G.Q).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wilkinson, S. ER-phagy: Shaping up and destressing the endoplasmic reticulum. FEBS J. 2019, 286, 2645–2663. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Crisosto, C.; Pennanen, C.; Vasquez-Trincado, C.; Morales, P.E.; Bravo-Sagua, R.; Quest, A.F.G.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 2017, 14, 342–360. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Opas, M. Endoplasmic and sarcoplasmic reticulum in the heart. Trends Cell Biol. 2009, 19, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Reddish, F.N.; Miller, C.L.; Gorkhali, R.; Yang, J.J. Calcium Dynamics Mediated by the Endoplasmic/Sarcoplasmic Reticulum and Related Diseases. Int. J. Mol. Sci. 2017, 18, 1024. [Google Scholar] [CrossRef]
- Welihinda, A.A.; Tirasophon, W.; Kaufman, R.J. The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr. 1999, 7, 293–300. [Google Scholar]
- Okeke, E.; Dingsdale, H.; Parker, T.; Voronina, S.; Tepikin, A.V. Endoplasmic reticulum-plasma membrane junctions: Structure, function and dynamics. J. Physiol. 2016, 594, 2837–2847. [Google Scholar] [CrossRef]
- Thoudam, T.; Jeon, J.H.; Ha, C.M.; Lee, I.K. Role of Mitochondria-Associated Endoplasmic Reticulum Membrane in Inflammation-Mediated Metabolic Diseases. Mediat. Inflamm. 2016, 2016, 1851420. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders —A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Ann. Rev. Pharm. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.T.; Hsueh, Y.P. The involvement of endoplasmic reticulum formation and protein synthesis efficiency in VCP- and ATL1-related neurological disorders. J. Biomed. Sci. 2018, 25, 2. [Google Scholar] [CrossRef] [PubMed]
- Lan, B.; Chai, S.; Wang, P.; Wang, K. VCP/p97/Cdc48, A Linking of Protein Homeostasis and Cancer Therapy. Curr. Mol. Med. 2017, 17, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tomita, Y.; Uruno, T.; Hoshida, Y.; Qiu, Y.; Iizuka, N.; Nakamichi, I.; Miyauchi, A.; Aozasa, K. Increased expression of valosin-containing protein (p97) is correlated with disease recurrence in follicular thyroid cancer. Ann. Surg. Oncol. 2005, 12, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tomita, Y.; Hoshida, Y.; Iizuka, N.; Monden, M.; Yamamoto, S.; Iuchi, K.; Aozasa, K. Expression level of valosin-containing protein (p97) is correlated with progression and prognosis of non-small-cell lung carcinoma. Ann. Surg. Oncol. 2004, 11, 697–704. [Google Scholar] [CrossRef]
- Zhou, N.; Ma, B.; Stoll, S.; Hays, T.T.; Qiu, H. The valosin-containing protein is a novel repressor of cardiomyocyte hypertrophy induced by pressure overload. Aging Cell 2017, 16, 1168–1179. [Google Scholar] [CrossRef]
- Lizano, P.; Rashed, E.; Stoll, S.; Zhou, N.; Wen, H.; Hays, T.T.; Qin, G.; Xie, L.H.; Depre, C.; Qiu, H. The valosin-containing protein is a novel mediator of mitochondrial respiration and cell survival in the heart in vivo. Sci. Rep. 2017, 7, 46324. [Google Scholar] [CrossRef]
- Lizano, P.; Rashed, E.; Kang, H.; Dai, H.; Sui, X.; Yan, L.; Qiu, H.; Depre, C. The valosin-containing protein promotes cardiac survival through the inducible isoform of nitric oxide synthase. Cardiovasc. Res. 2013, 99, 685–693. [Google Scholar] [CrossRef]
- Zhou, N.; Stoll, S.; Qiu, H. VCP represses pathological cardiac hypertrophy. Aging 2017, 9, 2469–2470. [Google Scholar] [CrossRef]
- Xia, D.; Tang, W.K.; Ye, Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 2016, 583, 64–77. [Google Scholar] [CrossRef]
- Tang, W.K.; Xia, D. Role of the D1-D2 Linker of Human VCP/p97 in the Asymmetry and ATPase Activity of the D1-domain. Sci. Rep. 2016, 6, 20037. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.K.; Xia, D. Mutations in the Human AAA(+) Chaperone p97 and Related Diseases. Front. Mol. Biosci. 2016, 3, 79. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Wu, H.C.; Burchell, V.S.; Preza, E.; Wray, S.; Mahoney, C.J.; Fox, N.C.; Calvo, A.; Canosa, A.; Moglia, C.; et al. Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 2013, 78, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.T.; Hsueh, Y.P. VCP and ATL1 regulate endoplasmic reticulum and protein synthesis for dendritic spine formation. Nat. Commun. 2016, 7, 11020. [Google Scholar] [CrossRef]
- Gonzalez-Perez, P.; Cirulli, E.T.; Drory, V.E.; Dabby, R.; Nisipeanu, P.; Carasso, R.L.; Sadeh, M.; Fox, A.; Festoff, B.W.; Sapp, P.C.; et al. Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology 2012, 79, 2201–2208. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef]
- Rabouille, C.; Levine, T.P.; Peters, J.M.; Warren, G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell 1995, 82, 905–914. [Google Scholar] [CrossRef]
- Hetzer, M.; Meyer, H.H.; Walther, T.C.; Bilbao-Cortes, D.; Warren, G.; Mattaj, I.W. Distinct AAA-ATPase p97 complexes function in discrete steps of nuclear assembly. Nat. Cell. Biol. 2001, 3, 1086–1091. [Google Scholar] [CrossRef]
- Guo, X.; Qi, X. VCP cooperates with UBXD1 to degrade mitochondrial outer membrane protein MCL1 in model of Huntington’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 552–559. [Google Scholar] [CrossRef]
- Stolz, A.; Hilt, W.; Buchberger, A.; Wolf, D.H. Cdc48: A power machine in protein degradation. Trends Biochem. Sci. 2011, 36, 515–523. [Google Scholar] [CrossRef]
- Schulz, J.; Avci, D.; Queisser, M.A.; Gutschmidt, A.; Dreher, L.S.; Fenech, E.J.; Volkmar, N.; Hayashi, Y.; Hoppe, T.; Christianson, J.C. Conserved cytoplasmic domains promote Hrd1 ubiquitin ligase complex formation for ER-associated degradation (ERAD). J. Cell Sci. 2017, 130, 3322–3335. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.; Upadhyay, A.; Kumar, A.; Mishra, A. Gp78 E3 Ubiquitin Ligase: Essential Functions and Contributions in Proteostasis. Front. Cell. Neurosci. 2017, 11, 259. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef] [PubMed]
- Byrne, D.J.; Harmon, M.J.; Simpson, J.C.; Blackstone, C.; O’Sullivan, N.C. Roles for the VCP co-factors Npl4 and Ufd1 in neuronal function in Drosophila melanogaster. J. Genet. Genom. 2017, 44, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, N.; Rapoport, T. Toward an understanding of the Cdc48/p97 ATPase. F1000Res 2017, 6, 1318. [Google Scholar] [CrossRef]
- Guo, X.; Sun, X.; Hu, D.; Wang, Y.J.; Fujioka, H.; Vyas, R.; Chakrapani, S.; Joshi, A.U.; Luo, Y.; Mochly-Rosen, D.; et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat. Commun. 2016, 7, 12646. [Google Scholar] [CrossRef]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef]
- Honrath, B.; Metz, I.; Bendridi, N.; Rieusset, J.; Culmsee, C.; Dolga, A.M. Glucose-regulated protein 75 determines ER-mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 2017, 3, 17076. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Hansford, R.G.; Zorov, D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol. Cell. Biochem. 1998, 184, 359–369. [Google Scholar] [CrossRef]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef] [PubMed]
- Llinas-Arias, P.; Rossello-Tortella, M.; Lopez-Serra, P.; Perez-Salvia, M.; Setien, F.; Marin, S.; Munoz, J.P.; Junza, A.; Capellades, J.; Calleja-Cervantes, M.E.; et al. Epigenetic loss of the endoplasmic reticulum-associated degradation inhibitor SVIP induces cancer cell metabolic reprogramming. JCI Insight 2019, 5, e125888. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Jiang, Y.; Zhang, D.; Liu, X.; Guo, J.; Zhao, J. Valosin-containing protein (VCP) promotes the growth, invasion, and metastasis of colorectal cancer through activation of STAT3 signaling. Mol. Cell. Biochem. 2016, 418, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bai, E.; Zhou, H.; Sha, S.; Miao, H.; Qin, Y.; Liu, Z.; Wang, J.; Zhang, H.; Lei, M.; et al. Discovery of a new class of valosine containing protein (VCP/P97) inhibitors for the treatment of non-small cell lung cancer. Bioorg. Med. Chem. 2019, 27, 533–544. [Google Scholar] [CrossRef]
- Asai, T.; Tomita, Y.; Nakatsuka, S.; Hoshida, Y.; Myoui, A.; Yoshikawa, H.; Aozasa, K. VCP (p97) regulates NFkappaB signaling pathway, which is important for metastasis of osteosarcoma cell line. Jpn. J. Cancer Res. 2002, 93, 296–304. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tomita, Y.; Hoshida, Y.; Takiguchi, S.; Fujiwara, Y.; Yasuda, T.; Yano, M.; Nakamori, S.; Sakon, M.; Monden, M.; et al. Expression level of valosin-containing protein is strongly associated with progression and prognosis of gastric carcinoma. J. Clin. Oncol. 2003, 21, 2537–2544. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tomita, Y.; Hoshida, Y.; Iizuka, N.; Kidogami, S.; Miyata, H.; Takiguchi, S.; Fujiwara, Y.; Yasuda, T.; Yano, M.; et al. Expression level of valosin-containing protein (p97) is associated with prognosis of esophageal carcinoma. Clin. Cancer Res. 2004, 10, 5558–5565. [Google Scholar] [CrossRef][Green Version]
- Meyer, M.F.; Seuthe, I.M.; Drebber, U.; Siefer, O.; Kreppel, M.; Klein, M.O.; Mikolajczak, S.; Klussmann, J.P.; Preuss, S.F.; Huebbers, C.U. Valosin-containing protein (VCP/p97)-expression correlates with prognosis of HPV- negative oropharyngeal squamous cell carcinoma (OSCC). PLoS ONE 2014, 9, e114170. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tomita, Y.; Hoshida, Y.; Toyosawa, S.; Inohara, H.; Kishino, M.; Kogo, M.; Nakazawa, M.; Murakami, S.; Iizuka, N.; et al. Expression level of valosin-containing protein (VCP) as a prognostic marker for gingival squamous cell carcinoma. Ann. Oncol. 2004, 15, 1432–1438. [Google Scholar] [CrossRef]
- Cui, Y.; Niu, M.; Zhang, X.; Zhong, Z.; Wang, J.; Pang, D. High expression of valosin-containing protein predicts poor prognosis in patients with breast carcinoma. Tumor Biol. 2015, 36, 9919–9927. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tomita, Y.; Nakamori, S.; Hoshida, Y.; Nagano, H.; Dono, K.; Umeshita, K.; Sakon, M.; Monden, M.; Aozasa, K. Elevated expression of valosin-containing protein (p97) in hepatocellular carcinoma is correlated with increased incidence of tumor recurrence. J. Clin. Oncol. 2003, 21, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef]
- Tang, W.K.; Zhang, T.; Ye, Y.; Xia, D. Structural basis for nucleotide-modulated p97 association with the ER membrane. Cell Discov. 2017, 3, 17045. [Google Scholar] [CrossRef] [PubMed]
- Ballar, P.; Zhong, Y.; Nagahama, M.; Tagaya, M.; Shen, Y.; Fang, S. Identification of SVIP as an endogenous inhibitor of endoplasmic reticulum-associated degradation. J. Biol. Chem 2007, 282, 33908–33914. [Google Scholar] [CrossRef]
- Long, X.H.; Zhang, Z.H.; Liu, Z.L.; Huang, S.H.; Luo, Q.F. Inhibiting valosin-containing protein suppresses osteosarcoma cell metastasis via AKT/nuclear factor of kappa B signaling pathway in vitro. Indian J. Pathol. Microbiol. 2013, 56, 190–195. [Google Scholar] [PubMed]
- Zhong, Q.; Zhou, B.; Ann, D.K.; Minoo, P.; Liu, Y.; Banfalvi, A.; Krishnaveni, M.S.; Dubourd, M.; Demaio, L.; Willis, B.C.; et al. Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: Effects of misfolded surfactant protein. Am. J. Respir. Cell Mol. Biol. 2011, 45, 498–509. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Shah, P.P.; Beverly, L.J. Regulation of VCP/p97 demonstrates the critical balance between cell death and epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 2015, 6, 17725–17737. [Google Scholar] [CrossRef]
- Rogalinska, M. The Role of Mitochondria in Cancer Induction, Progression and Changes in Metabolism. Mini Rev. Med. Chem. 2016, 16, 524–530. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Ann. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.C.; Tresse, E.; Kolaitis, R.M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 2013, 78, 65–80. [Google Scholar] [CrossRef]
- Heo, J.-M.; Livnat-Levanon, N.; Taylor, E.B.; Jones, K.T.; Dephoure, N.; Ring, J.; Xie, J.; Brodsky, J.L.; Madeo, F.; Gygi, S.P.; et al. A Stress-Responsive System for Mitochondrial Protein Degradation. Mol. Cell 2010, 40, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Song, H.; Mao, R.; Gao, Q.; Feng, Z.; Wang, N.; Song, S.; Jiao, R.; Ni, P.; Ge, H. Targeting valosin-containing protein enhances the efficacy of radiation therapy in esophageal squamous cell carcinoma. Cancer Sci. 2019, 110, 3464–3475. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, O.; Oral, O.; Kocaturk, N.M.; Akkoc, Y.; Eberhart, K.; Kosar, A.; Gozuacik, D. IBMPFD Disease-Causing Mutant VCP/p97 Proteins Are Targets of Autophagic-Lysosomal Degradation. PLoS ONE 2016, 11, e0164864. [Google Scholar] [CrossRef]
- Greenberg, S.A. Inclusion body myositis: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 2019, 15, 257–272. [Google Scholar] [CrossRef]
- Salajegheh, M.; Pinkus, J.L.; Taylor, J.P.; Amato, A.A.; Nazareno, R.; Baloh, R.H.; Greenberg, S.A. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve 2009, 40, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Zanusso, G.; Vattemi, G.; Ferrari, S.; Tabaton, M.; Pecini, E.; Cavallaro, T.; Tomelleri, G.; Filosto, M.; Tonin, P.; Nardelli, E.; et al. Increased expression of the normal cellular isoform of prion protein in inclusion-body myositis, inflammatory myopathies and denervation atrophy. Brain Pathol. 2001, 11, 182–189. [Google Scholar] [CrossRef]
- Weihl, C.C.; Dalal, S.; Pestronk, A.; Hanson, P.I. Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet. 2006, 15, 189–199. [Google Scholar] [CrossRef]
- Tang, W.K.; Li, D.; Li, C.C.; Esser, L.; Dai, R.; Guo, L.; Xia, D. A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants. EMBO J. 2010, 29, 2217–2229. [Google Scholar] [CrossRef]
- Fernandez-Saiz, V.; Buchberger, A. Imbalances in p97 co-factor interactions in human proteinopathy. EMBO Rep. 2010, 11, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Jang, E.R.; Wilson, P.G.; Anderson, D.; Galperin, E. VCP/p97 controls signals of the ERK1/2 pathway transmitted via the Shoc2 scaffolding complex: Novel insights into IBMPFD pathology. Mol. Biol. Cell 2019, 30, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.E.; Yao, Z.; Choi, M.; Tyzack, G.E.; Serio, A.; Luisier, R.; Harley, J.; Preza, E.; Arber, C.; Crisp, S.J.; et al. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. 2017, 19, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.; Lievens, J.C. Mitochondrial quality control in amyotrophic lateral sclerosis: Towards a common pathway? Neural Regen. Res. 2017, 12, 1052–1061. [Google Scholar] [CrossRef]
- Fang, L.; Hemion, C.; Pinho Ferreira Bento, A.C.; Bippes, C.C.; Flammer, J.; Neutzner, A. Mitochondrial function in neuronal cells depends on p97/VCP/Cdc48-mediated quality control. Front. Cell Neurosci. 2015, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Blice-Baum, A.C.; Sang, T.K.; Cammarato, A. Cardiac-Restricted Expression of VCP/TER94 RNAi or Disease Alleles Perturbs Drosophila Heart Structure and Impairs Function. J. Cardiovasc. Dev. Dis. 2016, 3, 19. [Google Scholar] [CrossRef]
- Kimonis, V. Inclusion Body Myopathy with Paget Disease of Bone and/or Frontotemporal Dementia. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews®: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1476/ (accessed on 25 May 2020).
- Brody, M.J.; Vanhoutte, D.; Bakshi, C.V.; Liu, R.; Correll, R.N.; Sargent, M.A.; Molkentin, J.D. Disruption of valosin-containing protein activity causes cardiomyopathy and reveals pleiotropic functions in cardiac homeostasis. J. Biol. Chem. 2019, 294, 8918–8929. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Stoll, S.; Xi, J.; Ma, B.; Leimena, C.; Behringer, E.J.; Qin, G.; Qiu, H. The valosin-containing protein protects the heart against pathological Ca2+ overload by modulating Ca2+ uptake proteins. Toxicol. Sci. 2019, 171, 473–484. [Google Scholar] [CrossRef]
- Hedhli, N.; Wang, L.; Wang, Q.; Rashed, E.; Tian, Y.; Sui, X.; Madura, K.; Depre, C. Proteasome activation during cardiac hypertrophy by the chaperone H11 Kinase/Hsp22. Cardiovasc. Res. 2008, 77, 497–505. [Google Scholar] [CrossRef]
- Morrow, G.; Kim, H.J.; Pellerito, O.; Bourrelle-Langlois, M.; Le Pecheur, M.; Groebe, K.; Tanguay, R.M. Changes in Drosophila mitochondrial proteins following chaperone-mediated lifespan extension confirm a role of Hsp22 in mitochondrial UPR and reveal a mitochondrial localization for cathepsin D. Mech. Ageing Dev. 2016, 155, 36–47. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).