SAK3 Administration Improves Spine Abnormalities and Cognitive Deficits in AppNL-G-F/NL-G-F Knock-in Mice by Increasing Proteasome Activity through CaMKII/Rpt6 Signaling


 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. SAK3 Administration Ameliorates the Reduction in Proteasome Activity in NL-G-F Mice
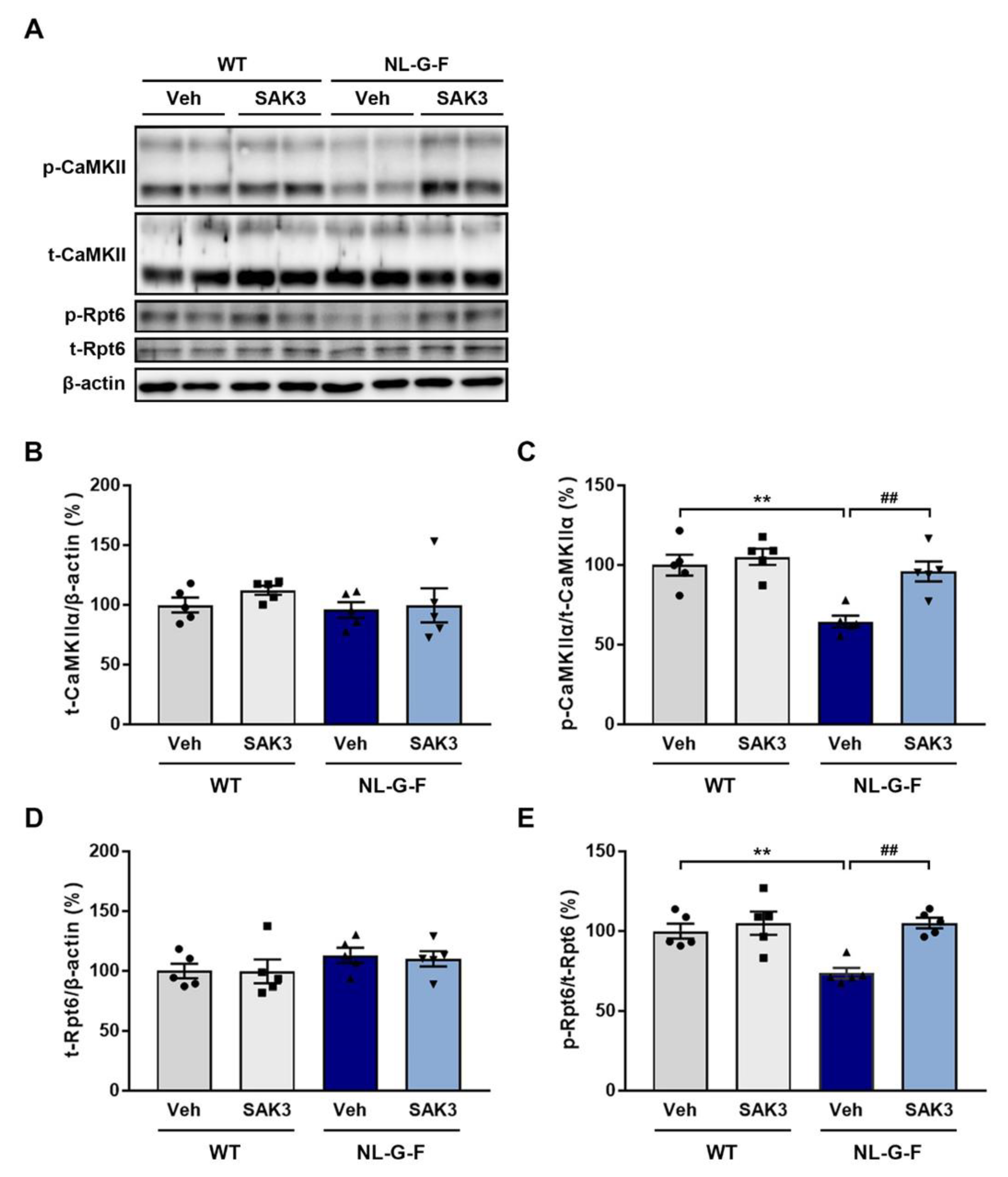
2.2. SAK3 Administration Improves the Reduction in CaMKII-Rpt6 Signaling in NL-G-F Mice
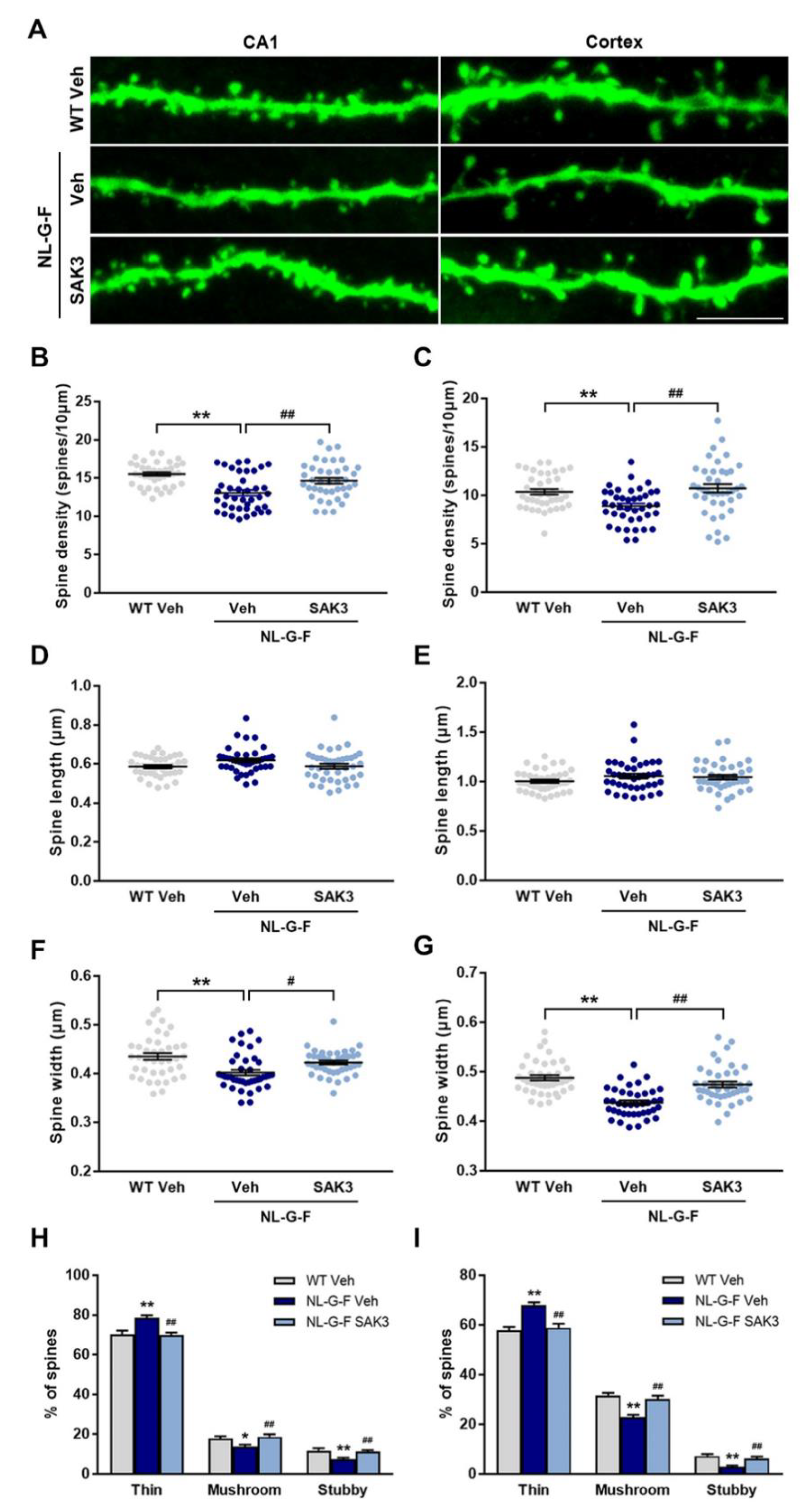
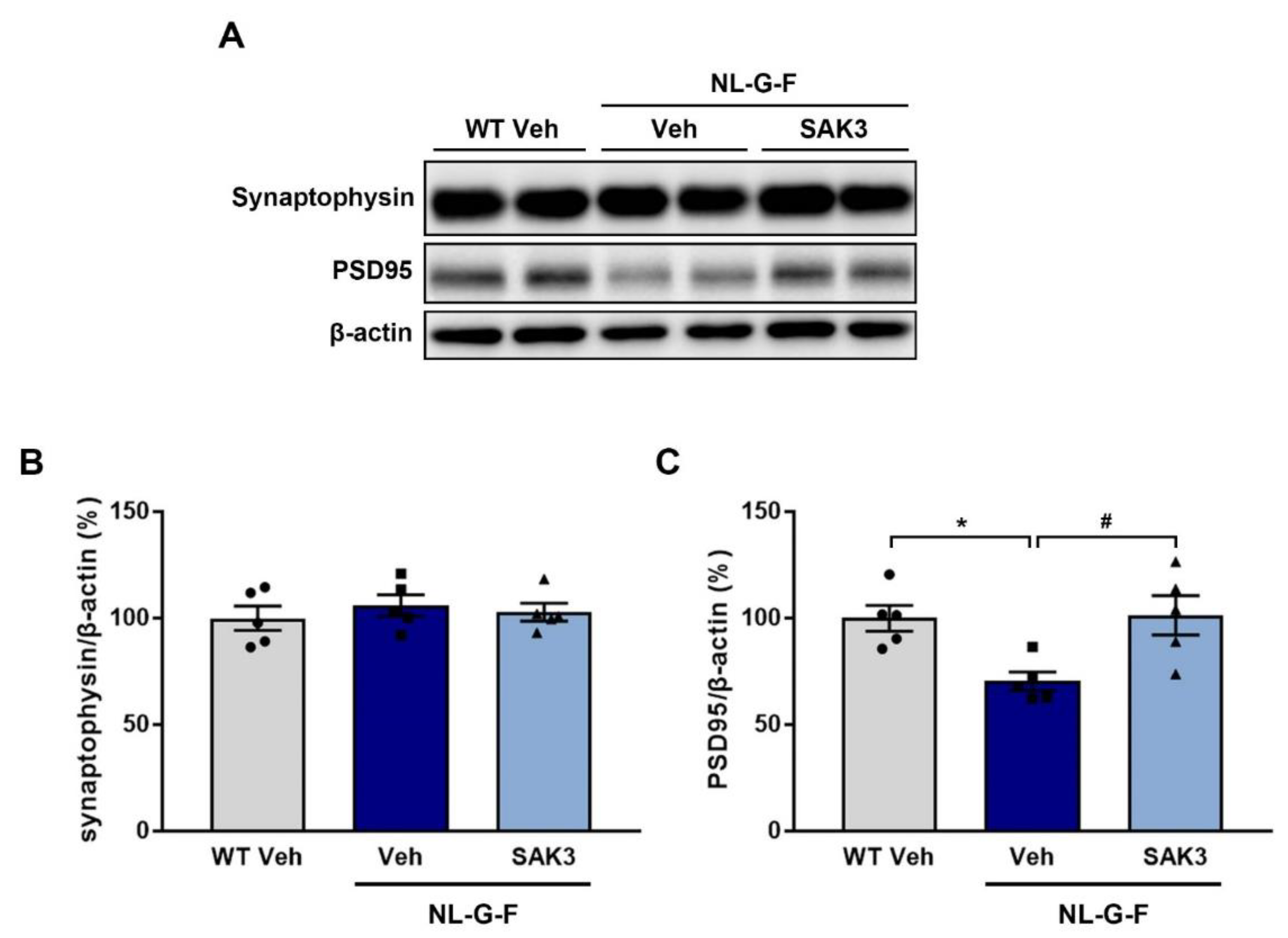
2.3. SAK3 Administration Rescues Dendritic Spine Abnormalities in NL-G-F Mice
2.4. SAK3 Administration Improves the Memory Impairments in NL-G-F Mice
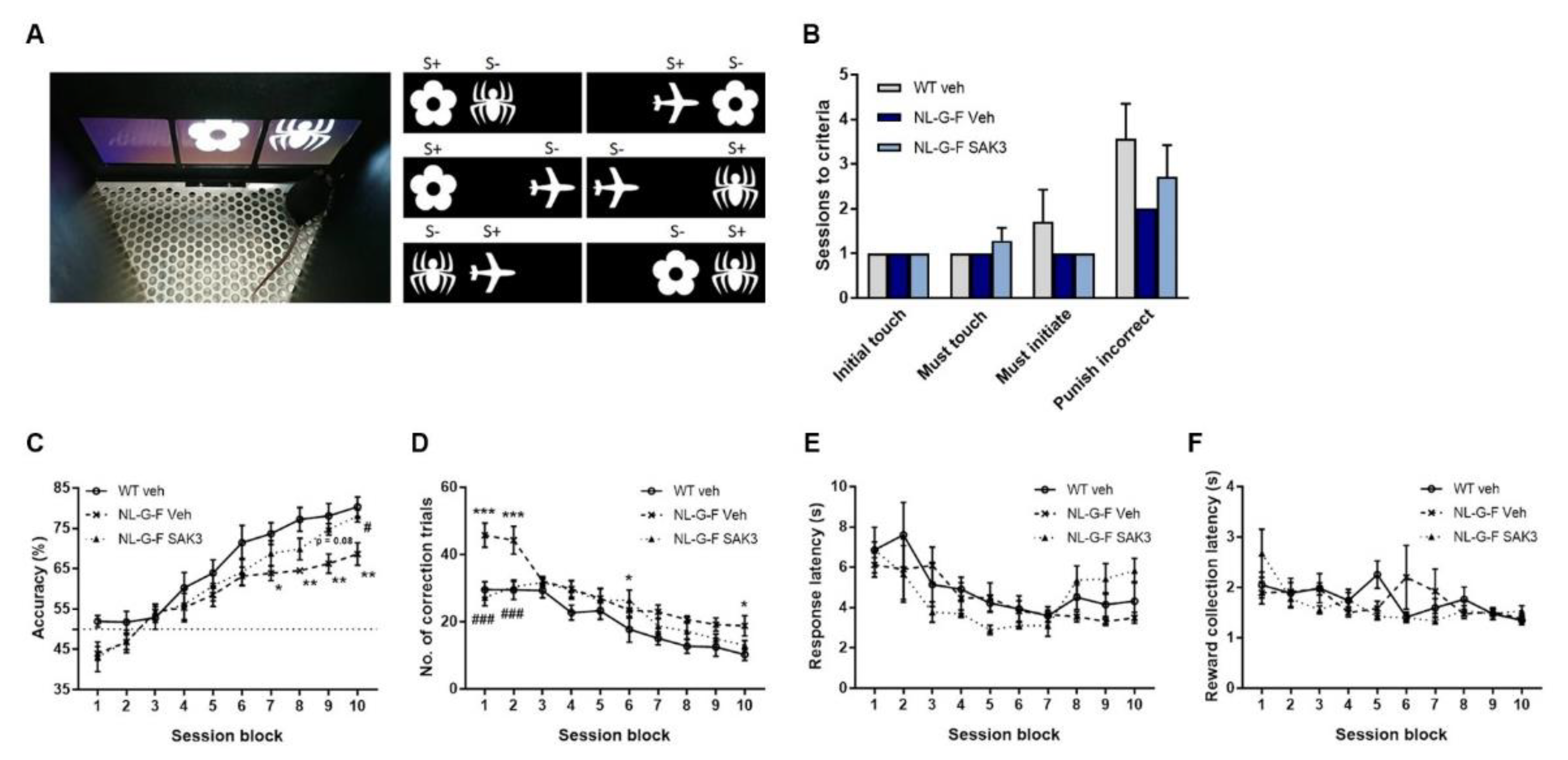
2.5. SAK3 Administration Improves Performance on the dPAL Task in NL-G-F Mice
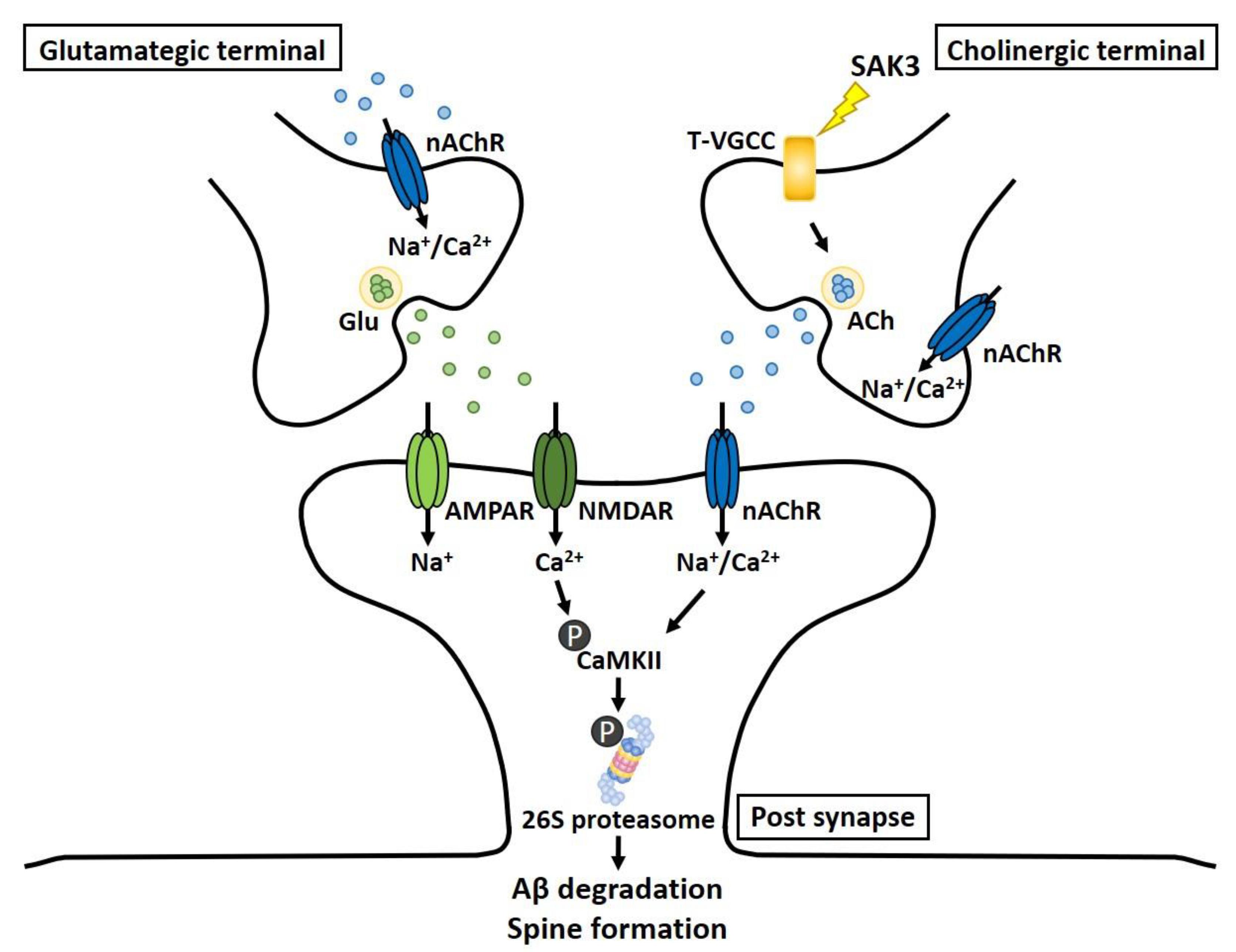
3. Discussion
4. Materials and Methods
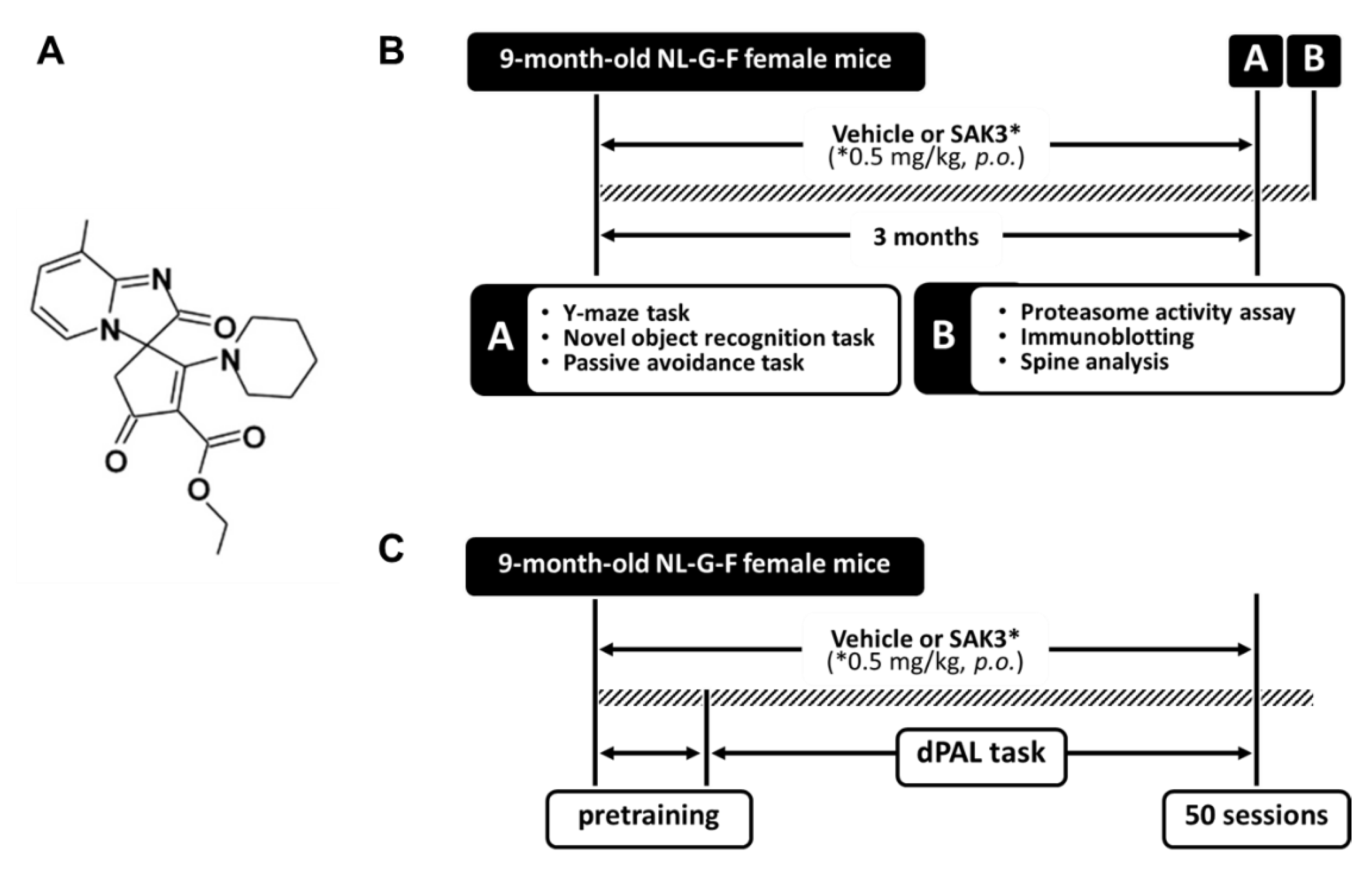
4.1. Animals and Experimental Design
4.2. Proteasome Activity Assay
4.3. Western Blot Analysis
4.4. Dendritic Spine Analysis Using Lucifer Yellow Labeling in Fixed Slices
4.5. Behavioral Tasks
4.5.1. Y-Maze Task
4.5.2. Novel Object Recognition Task
4.5.3. Step-Through Passive Avoidance Task
4.6. PAL Task
4.6.1. Apparatus for the PAL Task
4.6.2. Pretraining on the PAL Task
4.6.3. PAL Task
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | amyloid-β |
| CA1 | cornu ammonis 1 |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| LTP | long-term potentiation |
| NFT | neurofibrillary tangles |
| NL-F | AppNL-F/NL-F knock-in mice |
| NL-G-F | AppNL-F/NL-F knock-in mice |
| NNC | NNC 55-0396, a T-type calcium channel blocker |
| PAL | paired associate learning |
| PKA | protein kinase A/cAMP-dependent protein kinase |
| SAK3 | ethyl-8′-methyl-2′,4-dioxo-2-(piperidin-1-yl)-2′H-spiro[cyclopentane-1,3′- imidazo[1,2-a]pyridin] -2-ene-3-carboxylate |
| UPS | ubiquitin-proteasome system |
| WT | wild-type |
References
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- FukunagaHardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [PubMed]
- Blesneac, I.; Chemin, J.; Bidaud, I.; Huc-Brandt, S.; Vandermoere, F.; Lory, P. Phosphorylation of the Cav3.2 T-type calcium channel directly regulates its gating properties. Proc. Natl. Acad. Sci. USA 2015, 112, 13705–13710. [Google Scholar] [CrossRef]
- Powell, K.L.; Cain, S.M.; Snutch, T.P.; O’Brien, T.J. Low threshold T-type calcium channels as targets for novel epilepsy treatments. Br. J. Clin. Pharmacol. 2014, 77, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Lory, P.; Perez-Reyes, E. Role of voltage-gated calcium channels in epilepsy. Pflugers Arch. 2010, 460, 395–403. [Google Scholar] [CrossRef]
- Nelson, M.T.; Todorovic, S.M.; Perez-Reyes, E. The role of T-type calcium channels in epilepsy and pain. Curr. Pharm. Des. 2006, 12, 2189–2197. [Google Scholar] [CrossRef]
- McCormick, D.A.; Bal, T. Sleep and arousal: Thalamocortical mechanisms. Annu. Rev. Neurosci. 1997, 20, 185–215. [Google Scholar] [CrossRef]
- Altier, C.; Zamponi, G.W. Targeting Ca2+ channels to treat pain: T-type versus N-type. Trends Pharmacol. Sci. 2004, 25, 465–470. [Google Scholar] [CrossRef]
- Shin, H.S.; Cheong, E.J.; Choi, S.; Lee, J.; Na, H.S. T-type Ca2+ channels as therapeutic targets in the nervous system. Curr. Opin. Pharmacol. 2008, 8, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Gangarossa, G.; Laffray, S.; Bourinet, E.; Valjent, E. T-type calcium channel Cav3.2 deficient mice show elevated anxiety, impaired memory and reduced sensitivity to psychostimulants. Front. Behav. Neurosci. 2014, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, A.J.; Marks, W.N.; Liu, M.C.; Tahir, N.B.; Zabder, N.K.; Snutch, T.P.; Howland, J.G. Effects of the T-type calcium channel antagonist Z944 on paired associates learning and locomotor activity in rats treated with the NMDA receptor antagonist MK-801. Psychopharmacology 2018, 235, 3339–3350. [Google Scholar] [CrossRef]
- Zhao, Y.; Sivaji, S.; Chiang, M.C.; Ali, H.; Zukowski, M.; Ali, S.; Kennedy, B.; Sklyar, A.; Cheng, A.; Guo, Z.; et al. Amyloid Beta Peptides Block New Synapse Assembly by Nogo Receptor-Mediated Inhibition of T-Type Calcium Channels. Neuron 2017, 96, 355–372.e6. [Google Scholar] [CrossRef]
- Moriguchi, S.; Shioda, N.; Yamamoto, Y.; Tagashira, H.; Fukunaga, K. The T-type voltage-gated calcium channel as a molecular target of the novel cognitive enhancer ST101: Enhancement of long-term potentiation and CaMKII autophosphorylation in rat cortical slices. J. Neurochem. 2012, 121, 44–53. [Google Scholar] [CrossRef]
- Rice, R.A.; Berchtold, N.C.; Cotman, C.W.; Green, K.N. Age-related downregulation of the CaV3.1 T-type calcium channel as a mediator of amyloid beta production. Neurobiol. Aging 2014, 35, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Shinoda, Y.; Saito, T.; Saido, T.C.; Sato, K.; Yabuki, Y.; Matsumoto, Y.; Kanemitsu, Y.; Tomioka, Y.; Abolhassani, N.; et al. The Disease-modifying Drug Candidate, SAK3 Improves Cognitive Impairment and Inhibits Amyloid beta Deposition in App Knock-in Mice. Neuroscience 2018, 377, 87–97. [Google Scholar] [CrossRef]
- Schwartz, A.L.; Ciechanover, A. Targeting proteins for destruction by the ubiquitin system: Implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 73–96. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef]
- Lopez Salon, M.; Pasquini, L.; Besio Moreno, M.; Pasquini, J.M.; Soto, E. Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp. Neurol. 2003, 180, 131–143. [Google Scholar] [CrossRef]
- Oh, S.; Hong, H.S.; Hwang, E.; Sim, H.J.; Lee, W.; Shin, S.J.; Mook-Jung, I. Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech. Ageing Dev. 2005, 126, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Tseng, B.P.; Green, K.N.; Chan, J.L.; Blurton-Jones, M.; LaFerla, F.M. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 2008, 29, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Djakovic, S.N.; Schwarz, L.A.; Barylko, B.; DeMartino, G.N.; Patrick, G.N. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 2009, 284, 26655–26665. [Google Scholar] [CrossRef] [PubMed]
- Djakovic, S.N.; Marquez-Lona, E.M.; Jakawich, S.K.; Wright, R.; Chu, C.; Sutton, M.A.; Patrick, G.N. Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J. Neurosci. 2012, 32, 5126–5131. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.M.; Oh, W.C.; Vega-Ramirez, H.; Stein, I.S.; Hell, J.W.; Patrick, G.N.; Zito, K. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 2012, 74, 1023–1030. [Google Scholar] [CrossRef]
- Fonseca, R.; Vabulas, R.M.; Hartl, F.U.; Bonhoeffer, T.; Nagerl, U.V. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 2006, 52, 239–245. [Google Scholar] [CrossRef]
- Lopez-Salon, M.; Alonso, M.; Vianna, M.R.; Viola, H.; Mello e Souza, T.; Izquierdo, I.; Pasquini, J.M.; Medina, J.H. The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur. J. Neurosci. 2001, 14, 1820–1826. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef]
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The ubiquitin proteasomal system: A potential target for the management of Alzheimer’s disease. J. Cell Mol. Med. 2016, 20, 1392–1407. [Google Scholar] [CrossRef]
- Jarome, T.J.; Kwapis, J.L.; Ruenzel, W.L.; Helmstetter, F.J. CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front. Behav. Neurosci. 2013, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Gregori, L.; Fuchs, C.; Figueiredo-Pereira, M.E.; Van Nostrand, W.E.; Goldgaber, D. Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitro. J. Biol. Chem. 1995, 270, 19702–19708. [Google Scholar] [CrossRef]
- Yabuki, Y.; Matsuo, K.; Izumi, H.; Haga, H.; Yoshida, T.; Wakamori, M.; Kakei, A.; Sakimura, K.; Fukuda, T.; Fukunaga, K. Pharmacological properties of SAK3, a novel T-type voltage-gated Ca(2+) channel enhancer. Neuropharmacology 2017, 117, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yabuki, Y.; Yu, M.; Fukunaga, K. T-type calcium channel enhancer SAK3 produces anti-depressant-like effects by promoting adult hippocampal neurogenesis in olfactory bulbectomized mice. J. Pharmacol. Sci. 2018, 137, 333–341. [Google Scholar] [CrossRef] [PubMed]
- De Vrij, F.M.; Sluijs, J.A.; Gregori, L.; Fischer, D.F.; Hermens, W.T.; Goldgaber, D.; Verhaagen, J.; Van Leeuwen, F.W.; Hol, E.M. Mutant ubiquitin expressed in Alzheimer’s disease causes neuronal death. FASEB J. 2001, 15, 2680–2688. [Google Scholar] [CrossRef] [PubMed]
- Hope, A.D.; de Silva, R.; Fischer, D.F.; Hol, E.M.; van Leeuwen, F.W.; Lees, A.J. Alzheimer’s associated variant ubiquitin causes inhibition of the 26S proteasome and chaperone expression. J. Neurochem. 2003, 86, 394–404. [Google Scholar] [CrossRef]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Kawahata, I.; Tokuoka, H.; Parvez, H.; Ichinose, H. Accumulation of phosphorylated tyrosine hydroxylase into insoluble protein aggregates by inhibition of an ubiquitin-proteasome system in PC12D cells. J. Neural Transm. 2009, 116, 1571–1578. [Google Scholar] [CrossRef]
- Urushitani, M.; Kurisu, J.; Tsukita, K.; Takahashi, R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem. 2002, 83, 1030–1042. [Google Scholar] [CrossRef]
- Hishikawa, N.; Niwa, J.; Doyu, M.; Ito, T.; Ishigaki, S.; Hashizume, Y.; Sobue, G. Dorfin localizes to the ubiquitylated inclusions in Parkinson’s disease, dementia with Lewy bodies, multiple system atrophy, and amyotrophic lateral sclerosis. Am. J. Pathol. 2003, 163, 609–619. [Google Scholar] [CrossRef]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef]
- Perry, G.; Friedman, R.; Shaw, G.; Chau, V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc. Natl. Acad. Sci. USA 1987, 84, 3033–3036. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Kondo, J.; Ihara, Y. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 1987, 235, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Satoh, K.; Sasajima, H.; Nyoumura, K.I.; Yokosawa, H.; Sawada, H. Assembly of the 26S proteasome is regulated by phosphorylation of the p45/Rpt6 ATPase subunit. Biochemistry 2001, 40, 314–319. [Google Scholar] [CrossRef]
- Zhang, F.; Hu, Y.; Huang, P.; Toleman, C.A.; Paterson, A.J.; Kudlow, J.E. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J. Biol. Chem. 2007, 282, 22460–22471. [Google Scholar] [CrossRef]
- Alghamdi, A.; Vallortigara, J.; Howlett, D.R.; Broadstock, M.; Hortobagyi, T.; Ballard, C.; Thomas, A.J.; O’Brien, J.T.; Aarsland, D.; Attems, J.; et al. Reduction of RPT6/S8 (a Proteasome Component) and Proteasome Activity in the Cortex is Associated with Cognitive Impairment in Lewy Body Dementia. J. Alzheimers Dis. 2017, 57, 373–386. [Google Scholar] [CrossRef]
- Wang, H.; Saunders, A.J. The role of ubiquitin-proteasome in the metabolism of amyloid precursor protein (APP): Implications for novel therapeutic strategies for Alzheimer’s disease. Discov. Med. 2014, 18, 41–50. [Google Scholar]
- Fukunaga, K.; Izumi, H.; Yabuki, Y.; Shinoda, Y.; Shioda, N.; Han, F. Alzheimer’s disease therapeutic candidate SAK3 is an enhancer of T-type calcium channels. J. Pharmacolo. Sci. 2019, 139, 51–58. [Google Scholar] [CrossRef]
- Chow, W.N.; Ngo, J.C.; Li, W.; Chen, Y.W.; Tam, K.M.; Chan, H.Y.; Miller, C.C.; Lau, K.F. phosphorylation of FE65 Ser 610 by serum- and glucocoricoid-induced kinase i modulates Alzheimer’s disease amyloid precursor processing. Biochem. J. 2015, 470, 303–317. [Google Scholar] [CrossRef]
- Mo, J.S.; Yoon, J.H.; Hong, J.A.; Kim, M.Y.; Ann, E.J.; Ahn, J.S.; Kim, S.M.; Baek, H.J.; Lang, F.; Choi, E.J.; et al. Phosphorylation of nicastrin by SGK1 leads to ist degradation through lysocomal and proteasomal pathways. PLoS ONE 2012, 7, e37111. [Google Scholar]
- Lian, B.; Liu, M.; Lan, Z.; Meng, Z.; Cahng, Q.; Liu, Z.; Zhang, J.; Zhao, C. Hippocampal overexpression of SGK1 ameliorates saptial memory, resuces Ab pathology and actin cytoskeleton polymerization in milde-aged APP/PS mice. Behav. Brain Res. 2020, 383, 112503. [Google Scholar]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Freund, R.K.; Gibson, E.S.; Potter, H.; Dell’Acqua, M.L. Inhibition of the Motor Protein Eg5/Kinesin-5 in Amyloid beta-Mediated Impairment of Hippocampal Long-Term Potentiation and Dendritic Spine Loss. Mol. Pharmacol. 2016, 89, 552–559. [Google Scholar] [CrossRef]
- Smith, D.L.; Pozueta, J.; Gong, B.; Arancio, O.; Shelanski, M. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc. Natl. Acad. Sci. USA 2009, 106, 16877–16882. [Google Scholar] [CrossRef]
- Bittner, T.; Fuhrmann, M.; Burgold, S.; Ochs, S.M.; Hoffmann, N.; Mitteregger, G.; Kretzschmar, H.; LaFerla, F.M.; Herms, J. Multiple events lead to dendritic spine loss in triple transgenic Alzheimer’s disease mice. PLoS ONE 2010, 5, e15477. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Sun, M.K.; Alkon, D.L. PKC epsilon activation prevents synaptic loss, Abeta elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J. Neurosci. 2011, 31, 630–643. [Google Scholar] [CrossRef]
- Lee, H.G.; Won, S.M.; Gwag, B.J.; Lee, Y.B. Microglial P2X(7) receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Exp. Mol. Med. 2011, 43, 7–14. [Google Scholar] [CrossRef]
- Yang, E.J.; Ahn, S.; Ryu, J.; Choi, M.S.; Choi, S.; Chong, Y.H.; Hyun, J.W.; Chang, M.J.; Kim, H.S. Phloroglucinol Attenuates the Cognitive Deficits of the 5XFAD Mouse Model of Alzheimer’s Disease. PLoS ONE 2015, 10, e0135686. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Liang, Y.; Chen, H.; Xu, B.; Chai, C.; Xing, P. The Role of Fluoxetine in Activating Wnt/beta-Catenin Signaling and Repressing beta-Amyloid Production in an Alzheimer Mouse Model. Front. Aging Neurosci. 2018, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef] [PubMed]
- Mahmmoud, R.R.; Sase, S.; Aher, Y.D.; Sase, A.; Groger, M.; Mokhtar, M.; Hoger, H.; Lubec, G. Spatial and Working Memory Is Linked to Spine Density and Mushroom Spines. PLoS ONE 2015, 10, e0139739. [Google Scholar] [CrossRef] [PubMed]
- McCann, R.F.; Ross, D.A. A Fragile Balance: Dendritic Spines, Learning, and Memory. Biol. Psychiatry 2017, 82, e11–e13. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Wang, C.F.; Arnott, D.; Cheng, D.; Peng, J.; Sheng, M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 2010, 140, 567–578. [Google Scholar] [CrossRef]
- Lokireddy, S.; Kukushkin, N.V.; Goldberg, A.L. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. USA 2015, 112, E7176–E7185. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Tanahashi, N.; Murakami, Y.; Minami, Y.; Shimbara, N.; Hendil, K.B.; Tanaka, K. Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J. Biol. Chem. 2000, 275, 14336–14345. [Google Scholar] [CrossRef]
- Yabuki, Y.; Wu, L.; Fukunaga, K. Cognitive enhancer ST101 improves schizophrenia-like behaviors in neonatal ventral hippocampus-lesioned rats in association with improved CaMKII/PKC pathway. J. Pharmacol. Sci. 2019, 140, 263–272. [Google Scholar] [CrossRef]
- Fukunaga, K.; Goto, S.; Miyamoto, E. Immunohistochemical localization of Ca2+/calmodulin-dependent protein kinase II in rat brain and various tissues. J. Neurochem. 1988, 51, 1070–1078. [Google Scholar] [CrossRef]
- Shioda, N.; Beppu, H.; Fukuda, T.; Li, E.; Kitajima, I.; Fukunaga, K. Aberrant calcium/calmodulin-dependent protein kinase II (CaMKII) activity is associated with abnormal dendritic spine morphology in the ATRX mutant mouse brain. J. Neurosci. 2011, 31, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Mucha, M.; Skrzypiec, A.E.; Schiavon, E.; Attwood, B.K.; Kucerova, E.; Pawlak, R. Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. Proc. Natl. Acad. Sci. USA 2011, 108, 18436–18441. [Google Scholar] [CrossRef] [PubMed]
- Risher, W.C.; Ustunkaya, T.; Singh Alvarado, J.; Eroglu, C. Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS ONE 2014, 9, e107591. [Google Scholar] [CrossRef] [PubMed]
- Horner, A.E.; Heath, C.J.; Hvoslef-Eide, M.; Kent, B.A.; Kim, C.H.; Nilsson, S.R.; Alsio, J.; Oomen, C.A.; Holmes, A.; Saksida, L.M.; et al. The touchscreen operant platform for testing learning and memory in rats and mice. Nat. Protoc. 2013, 8, 1961–1984. [Google Scholar] [CrossRef]
- Oomen, C.A.; Hvoslef-Eide, M.; Heath, C.J.; Mar, A.C.; Horner, A.E.; Bussey, T.J.; Saksida, L.M. The touchscreen operant platform for testing working memory and pattern separation in rats and mice. Nat. Protoc. 2013, 8, 2006–2021. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izumi, H.; Kawahata, I.; Shinoda, Y.; Helmstetter, F.J.; Fukunaga, K. SAK3 Administration Improves Spine Abnormalities and Cognitive Deficits in AppNL-G-F/NL-G-F Knock-in Mice by Increasing Proteasome Activity through CaMKII/Rpt6 Signaling. Int. J. Mol. Sci. 2020, 21, 3833. https://doi.org/10.3390/ijms21113833
Izumi H, Kawahata I, Shinoda Y, Helmstetter FJ, Fukunaga K. SAK3 Administration Improves Spine Abnormalities and Cognitive Deficits in AppNL-G-F/NL-G-F Knock-in Mice by Increasing Proteasome Activity through CaMKII/Rpt6 Signaling. International Journal of Molecular Sciences. 2020; 21(11):3833. https://doi.org/10.3390/ijms21113833
Chicago/Turabian StyleIzumi, Hisanao, Ichiro Kawahata, Yasuharu Shinoda, Fred J. Helmstetter, and Kohji Fukunaga. 2020. "SAK3 Administration Improves Spine Abnormalities and Cognitive Deficits in AppNL-G-F/NL-G-F Knock-in Mice by Increasing Proteasome Activity through CaMKII/Rpt6 Signaling" International Journal of Molecular Sciences 21, no. 11: 3833. https://doi.org/10.3390/ijms21113833
APA StyleIzumi, H., Kawahata, I., Shinoda, Y., Helmstetter, F. J., & Fukunaga, K. (2020). SAK3 Administration Improves Spine Abnormalities and Cognitive Deficits in AppNL-G-F/NL-G-F Knock-in Mice by Increasing Proteasome Activity through CaMKII/Rpt6 Signaling. International Journal of Molecular Sciences, 21(11), 3833. https://doi.org/10.3390/ijms21113833