17-Aminogeldanamycin Inhibits Constitutive Nuclear Factor-Kappa B (NF-κB) Activity in Patient-Derived Melanoma Cell Lines

 ,
,  and
and
Abstract
1. Introduction
2. Results
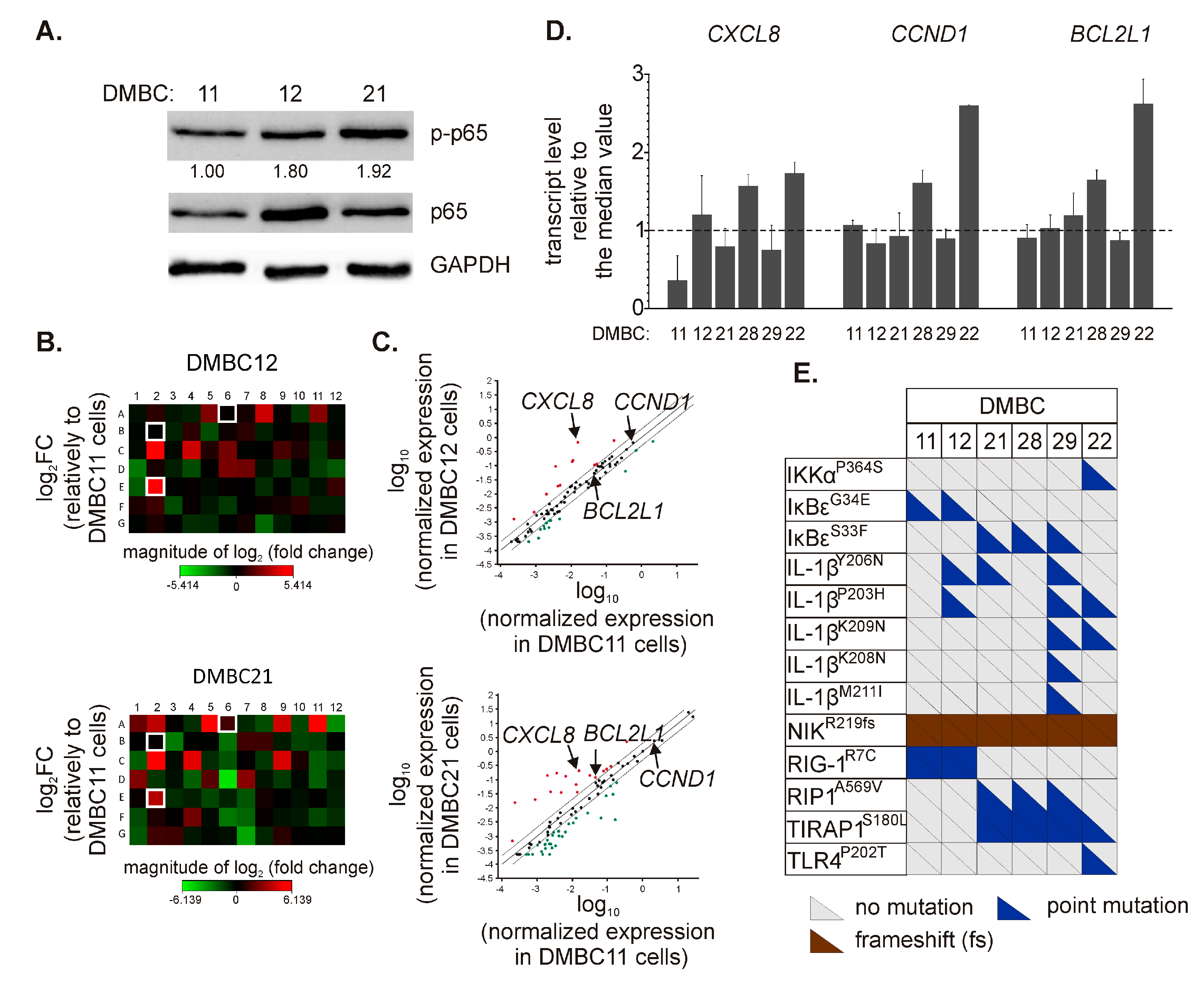
2.1. Patient-Derived Melanoma Cell Lines Differently Execute the p65/NF-κB-Dependent Program
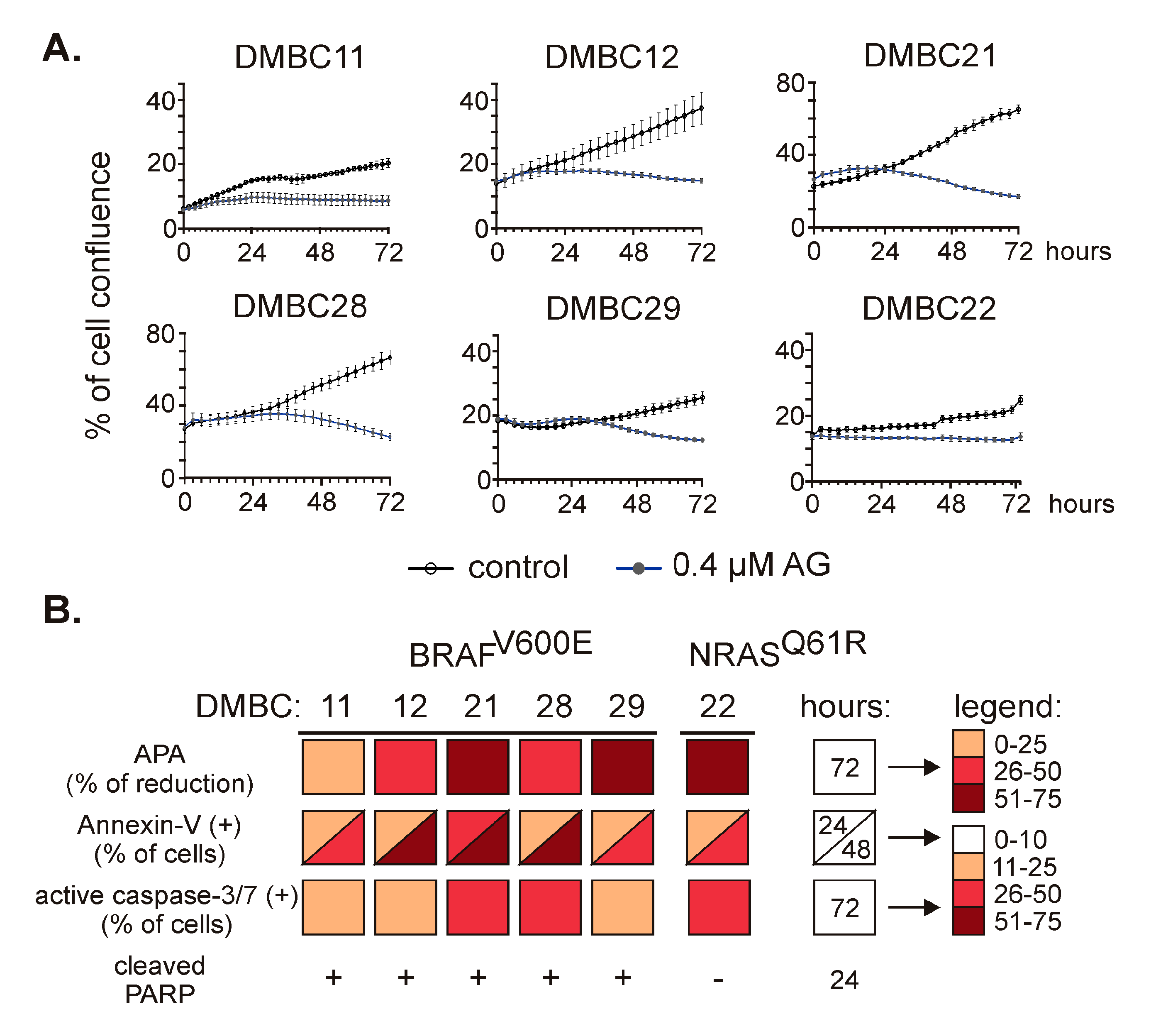
2.2. 17-Aminogeldanamycin (AG) Reduces Viable Cell Number in Melanoma Cell Lines, which is Associated with Induction of Apoptosis
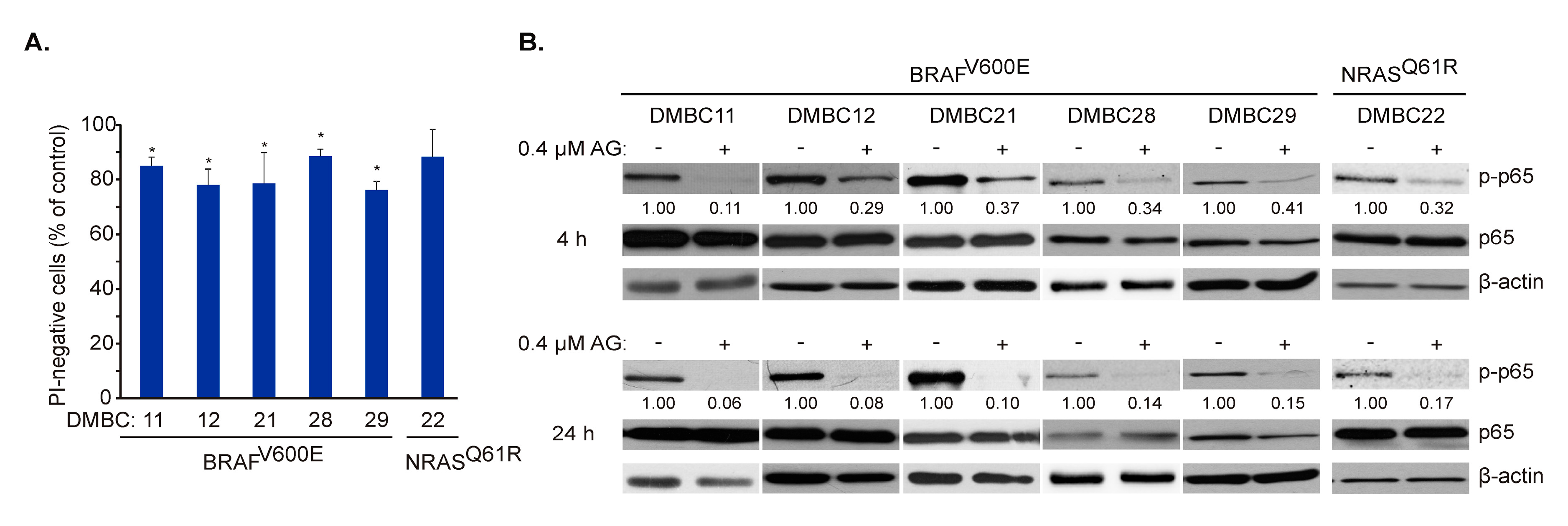
2.3. AG Inhibits p65/NF-κB Activity in Melanoma Cells of BRAFV600E and NRASQ61R Subtypes
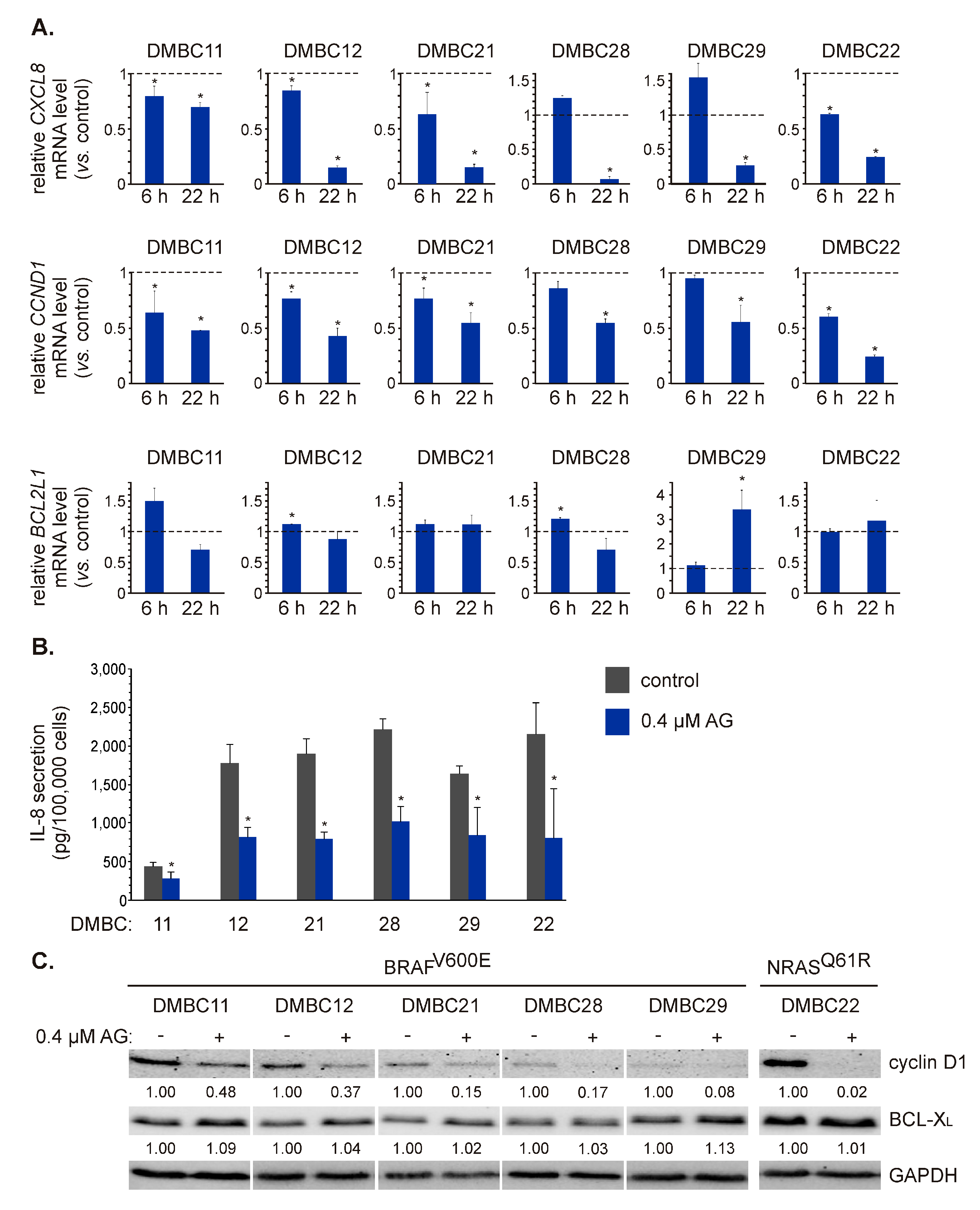
2.4. AG Diversely Affects Transcript and Proteins Levels of Different NF-κB-Dependent Genes
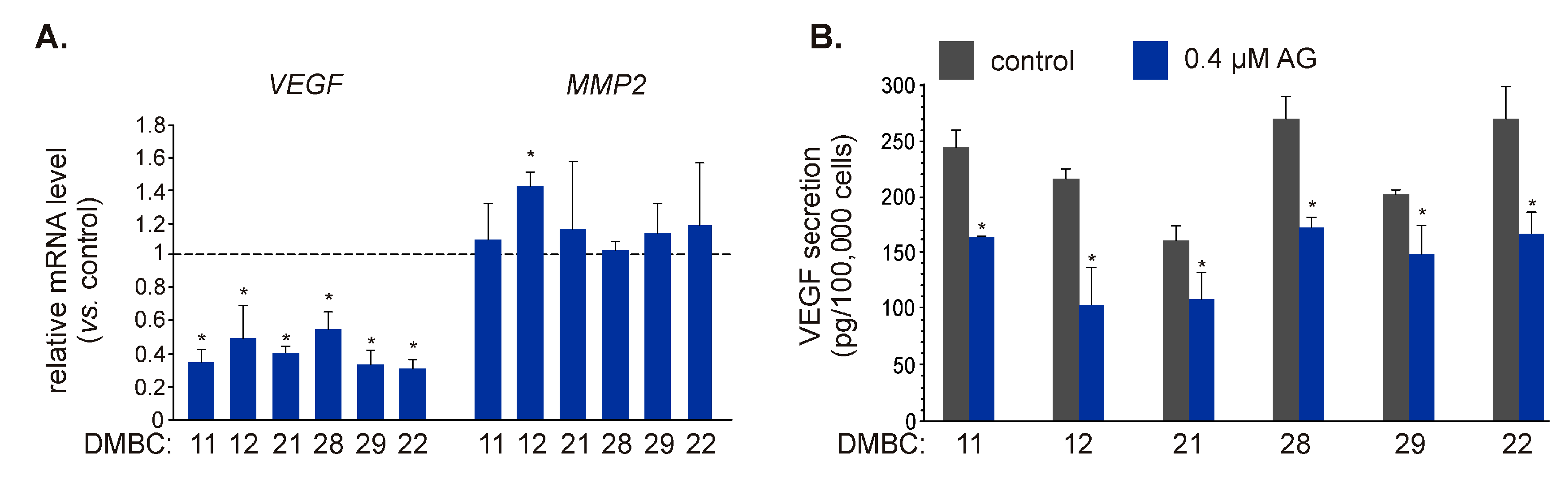
2.5. AG Affects Transcript Level and Secretion of VEGF
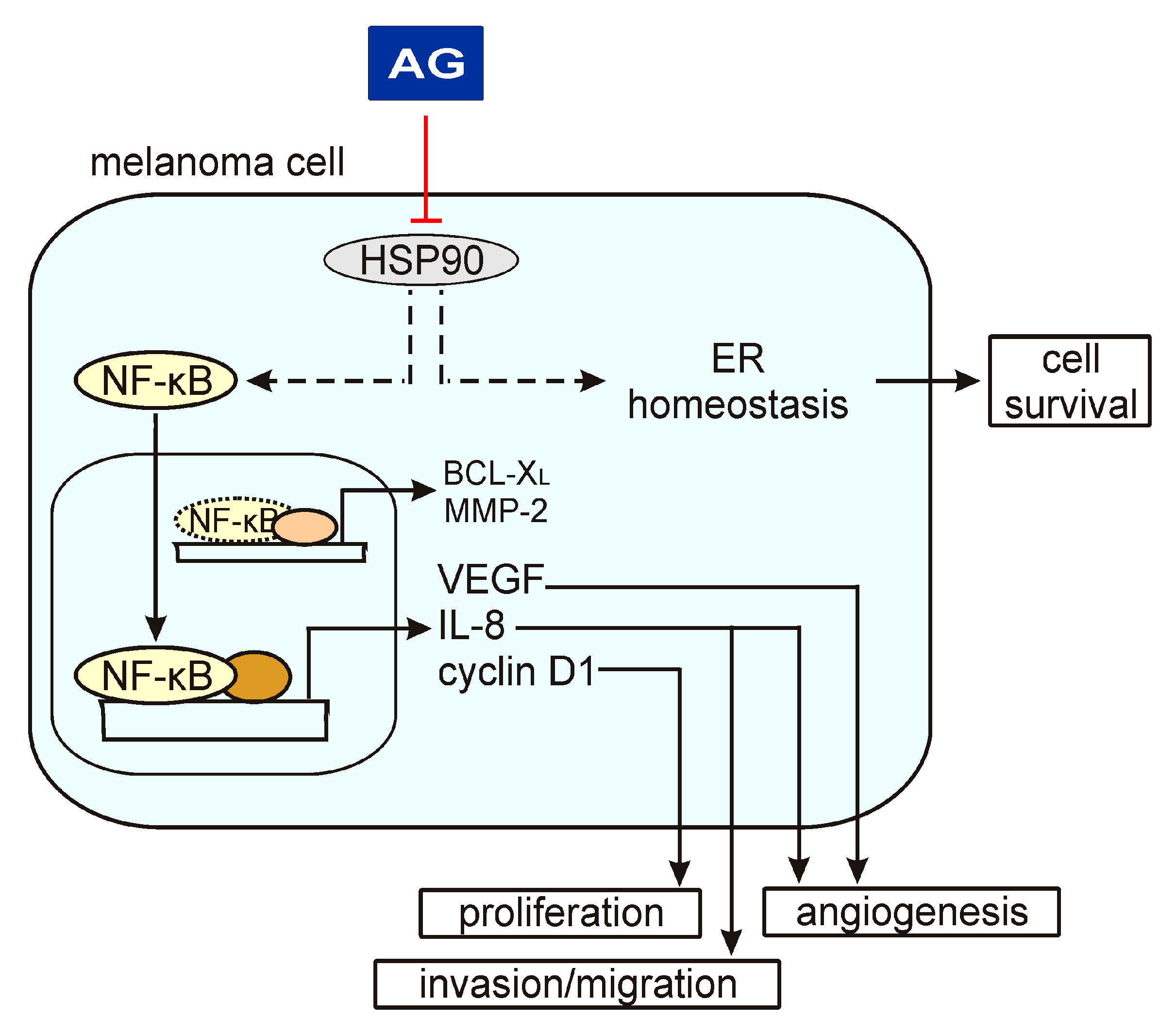
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Compounds
4.3. Time-Lapse Microscopy
4.4. Acid Phosphatase Activity Assay
4.5. Flow Cytometry
4.6. Cell Lysate Preparation and Western Blotting
4.7. RNA Isolation, Synthesis of cDNA and Quantitative Real-Time PCR (qRT-PCR)
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. RT2 Profiler PCR Array
4.10. Whole-Exome Sequencing
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCB5 AG BCL2L1 | ATP-binding cassette member B5 17-aminogeldanamycin BCL-2-like 1 |
| BRAF | B-Raf proto-oncogene serine/threonine kinase |
| CCND1 CDK6 | cyclin D1 cyclin-dependent kinase 6 |
| CXCL8 DMBC ERK1/2 EZH2 | C-X-C motif chemokine ligand 8 Department of Molecular Biology of Cancer extracellular signal-regulated kinase 1/2 enhancer of zeste homolog 2 |
| HSP90 IFN-γ IKK IL-1β IL-6 IL-8 MMP-2 | heat shock protein 90 interferon-gamma IκB kinase interleukin-1β interleukin-6 interleukin-8 matrix metalloproteinase-2 |
| NF-κB NFAT NIK NRAS PAX3 PD-L1 RSK1 STAT3 TBK1 TNFα VEGF | nuclear factor-kappa B nuclear factor of activated T-cells nuclear factor-kappa B-inducing kinase neuroblastoma RAS viral oncogene homolog paired box 3 programmed death-ligand 1 ribosomal kinase 1 signal transducer and activator of transcription 3 TANK-binding kinase 1 tumor necrosis factor alpha vascular endothelial growth factor |
References
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Jemal, A.; Miller, K.D. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Dummer, R.; Goldinger, S.M.; Turtschi, C.P.; Eggmann, N.B.; Michielin, O.; Mitchell, L.; Veronese, L.; Hilfiker, P.R.; Felderer, L.; Rinderknecht, J.D. Vemurafenib in patients with BRAF(V600) mutation-positive melanoma with symptomatic brain metastases: Final results of an open-label pilot study. Eur. J. Cancer 2014, 50, 611–621. [Google Scholar] [CrossRef]
- Rutkowski, P.; Blank, C. Dabrafenib for the treatment of BRAF V600-positive melanoma: A safety evaluation. Expert Opin. Drug Saf. 2014, 13, 1249–1258. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Flaherty, K.T. Resistance to BRAF-targeted therapy in melanoma. Eur. J. Cancer 2013, 49, 1297–1304. [Google Scholar] [CrossRef]
- Luebker, S.A.; Koepsell, S.A. Diverse mechanisms of BRAF inhibitor resistance in melanoma identified in clinical and preclinical studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Rose, A. Encorafenib and Binimetinib for the Treatment of BRAF V600E/K-Mutated Melanoma. Drugs Today (Barc.) 2019, 55, 247–264. [Google Scholar] [CrossRef]
- Welsh, S.J.; Rizos, H.; Scolyer, R.A.; Long, G.V. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur. J. Cancer 2016, 62, 76–85. [Google Scholar] [CrossRef]
- Lorusso, P.M.; Schalper, K.; Sosman, J. Targeted therapy and immunotherapy: Emerging biomarkers in metastatic melanoma. Pigment. Cell Melanoma Res. 2019. epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.; Pick, E.; Kluger, Y.; Gould-Rothberg, B.; Lazova, R.; Camp, R.; Rimm, D.; Kluger, H. HSP90 as a marker of progression in melanoma. Ann. Oncol. 2008, 19, 590–594. [Google Scholar] [CrossRef]
- Dias, S.D.R.; Friedlos, F.; Light, Y.; Springer, C.; Workman, P.; Marais, R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005, 65, 10686–10691. [Google Scholar] [CrossRef] [PubMed]
- Grbovic, O.M.; Basso, A.D.; Sawai, A.; Ye, Q.; Friedlander, P.; Solit, D.; Rosen, N. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Mielczarek-Lewandowska, A.; Hartman, M.L.; Czyz, M. Inhibitors of HSP90 in melanoma. Apoptosis 2020, 25, 12–28. [Google Scholar] [CrossRef]
- Shevtsov, M.; Multhoff, G.; Mikhaylova, E.; Shibata, A.; Guzhova, I.; Margulis, B. Combination of anti-cancer drugs with molecular chaperone inhibitors. Int. J. Mol. Sci. 2019, 20, 5284. [Google Scholar] [CrossRef]
- Sztiller-Sikorska, M.; Koprowska, K.; Majchrzak, K.; Hartman, M.; Czyz, M. Natural compounds’ activity against cancer stem-like or fast-cycling melanoma cells. PLoS ONE 2014, 9, e90783. [Google Scholar] [CrossRef]
- Mielczarek-Lewandowska, A.; Sztiller-Sikorska, M.; Osrodek, M.; Czyz, M.; Hartman, M.L. 17-Aminogeldanamycin selectively diminishes IRE1α-XBP1s pathway activity and cooperatively induces apoptosis with MEK1/2 and BRAFV600E inhibitors in melanoma cells of different genetic subtypes. Apoptosis 2019, 24, 596–611. [Google Scholar] [CrossRef]
- Hartman, M.L.; Talar, B.; Sztiller-Sikorska, M.; Nejc, D.; Czyz, M. Parthenolide induces MITF-M downregulation and senescence in patient-derived MITF-MHigh melanoma cell populations. Oncotarget 2016, 7, 9026–9040. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Rozanski, M.; Osrodek, M.; Zalesna, I.; Czyz, M. Vemurafenib and trametinib reduce expression of CTGF and IL-8 in V600EBRAF melanoma cells. Lab. Investig. 2017, 97, 217–227. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M. Whole-exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol. Carcinog. 2019, 58, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Osrodek, M.; Hartman, M.L. Plasticity of drug-naïve and vemurafenib- or trametinib-resistant melanoma cells in execution of differentiation/pigmentation program. J. Oncol. 2019, 2019, 1697913. [Google Scholar] [CrossRef] [PubMed]
- Osrodek, M.; Hartman, M.L.; Czyz, M. Physiologically relevant oxygen concentration (6% O2) as an important component of the microenvironment impacting melanoma phenotype and melanoma response to targeted therapeutics in vitro. Int. J. Mol. Sci. 2019, 20, 4203. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Czyz, M. Dissecting mechanisms of melanoma resistance to BRAF and MEK inhibitors revealed genetic and non-genetic patient- and drug-specific alterations and remarkable phenotypic plasticity. Cells 2020, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Yokoyama, S.; Hawryluk, E.B.; Jönsson, G.B.; Frederick, D.T.; McHenry, K.; Porter, D.; Tran, T.N.; Love, K.T.; Langer, R.; et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc. Natl. Acad. Sci. USA 2013, 110, 4321–4326. [Google Scholar] [CrossRef]
- Hartman, M.L.; Talar, B.; Noman, M.Z.; Gajos-Michniewicz, A.; Chouaib, S.; Czyz, M. Gene expression profiling identifies microphthalmia-associated transcription factor (MITF) and Dickkopf-1 (DKK1) as regulators of microenvironment-driven alterations in melanoma phenotype. PLoS ONE 2014, 9, e95157. [Google Scholar] [CrossRef]
- Hartman, M.L.; Talar, B.; Gajos-Michniewicz, A.; Czyz, M. MCL-1, BCL-XL and MITF are diversely employed in adaptive response of melanoma cells to changes in microenvironment. PLoS ONE 2015, 10, e0128796. [Google Scholar] [CrossRef]
- Sztiller-Sikorska, M.; Hartman, M.L.; Talar, B.; Jakubowska, J.; Zalesna, I.; Czyz, M. Phenotypic diversity of patient-derived melanoma populations in stem cell medium. Lab. Investig. 2015, 95, 672–683. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. TYRP1 mRNA level is stable and MITF-M-independent in drug-naïve, vemurafenib- and trametinib-resistant BRAFV600E melanoma cells. Arch. Dermatol. Res. 2019. epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Bellail, A.C.; Van Meir, E.G. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro. Oncol. 2005, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Villarete, L.H.; Remick, D.G. Transcriptional and post-transcriptional regulation of interleukin-8. Am. J. Pathol. 1996, 149, 1685–1693. [Google Scholar] [PubMed]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Role of VEGFs/VEGFR-1 Signaling and its inhibition in modulating tumor invasion: Experimental evidence in different metastatic cancer models. Int. J. Mol. Sci. 2020, 21, 1388. [Google Scholar] [CrossRef]
- Jászai, J.; Schmidt, M.H.H. Trends and challenges in tumor anti-Angiogenic therapies. Cells 2019, 8, 1102. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Chen, Z.; Sternlicht, M.D.; Hidalgo, M.; Steffensen, B. Matrix metalloproteinase-2 contributes to cancer cell migration on collagen. Cancer Res. 2005, 65, 130–136. [Google Scholar]
- Xie, T.X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 2010, 23, 725–732. [Google Scholar]
- Philip, S.; Kundu, G.C. Osteopontin induces nuclear factor kappa B-mediated promatrix metalloproteinase-2 activation through I kappa B alpha/IKK signaling pathways, and curcumin (diferulolylmethane) down-regulates these pathways. J. Biol. Chem. 2003, 278, 14487–14497. [Google Scholar] [CrossRef]
- Philip, S.; Bulbule, A.; Kundu, G.C. Osteopontin stimulates tumor growth and activation of promatrix metalloproteinase-2 through nuclear factor-kappa B-mediated induction of membrane type 1 matrix metalloproteinase in murine melanoma cells. J. Biol. Chem. 2001, 276, 44926–44935. [Google Scholar] [CrossRef]
- Madonna, G.; Ullman, C.; Gentilcore, G.; Palmieri, G.; Ascierto, P. NF-kB as potential target in the treatment of melanoma. J. Transl. Med. 2012, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M.E. BCL-w: Apoptotic and non-apoptotic role in health and disease. Cell Death Dis. 2020, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L. Non-apoptotic cell death signaling pathways in melanoma. Int. J. Mol. Sci. 2020, 21, 2980. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M. HGF/c-MET signaling in melanocytes and melanoma. Int. J. Mol. Sci. 2018, 19, 3844. [Google Scholar] [CrossRef]
- Wang, S.; Tang, L.; Lin, J.; Shen, Z.; Yao, Y.; Wang, W.; Tao, S.; Gu, C.; Ma, J.; Xie, Y.; et al. ABCB5 promotes melanoma metastasis through enhancing NF-ΚB p65 protein stability. Biochem. Biophys. Res. Commun. 2017, 492, 18–26. [Google Scholar] [CrossRef]
- Smith, M.P.; Sanchez-Laorden, B.; Obrien, K.; Brunton, H.; Ferguson, J.; Young, H.; Dhomen, N.; Flaherty, K.T.; Frederick, D.T.; Cooper, Z.A.; et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNF. Cancer Discov. 2014, 4, 1214–1229. [Google Scholar] [CrossRef]
- Lehraiki, A.; Cerezo, M.; Rouaud, F.; Abbe, P.; Allegra, M.; Kluza, J.; Marchetti, P.; Imbert, V.; Cheli, Y.; Bertolotto, C.; et al. Increased CD271 expression by the NF-KB pathway promotes melanoma cell Survival and drives acquired resistance to BRAF inhibitor vemurafenib. Cell Discov. 2015, 1, 15030. [Google Scholar] [CrossRef]
- Shao, Y.; Le, K.; Cheng, H.; Aplin, A.E. NF-ΚB regulation of c-FLIP promotes TNFα-mediated RAF inhibitor resistance in melanoma. J. Investig. Dermatol. 2015, 135, 1839–1848. [Google Scholar] [CrossRef]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-Diaz, A.; Moreno, B.H.; Kim, J.; Ng, R.H.; Lee, J.W.; et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-ΚB. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef]
- García-Aranda, M.; Redondo, M. Targeting protein kinases to enhance the response to anti-PD-1/PD-L1 immunotherapy. Int. J. Mol. Sci. 2019, 20, 2296. [Google Scholar] [CrossRef] [PubMed]
- Christian, F.; Smith, E.; Carmody, R. The regulation of NF-ΚB subunits by phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Buss, H.; Dörrie, A.; Schmitz, M.L.; Hoffmann, E.; Resch, K.; Kracht, M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-ΚB at serine 536 is mediated by multiple protein kinases including IκB Kinase (IKK)-α, IKKβ, IKKϵ, TRAF Family Member-Associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-Binding Protein-Associated Factor II31-mediated interleukin-8 transcription. J. Biol. Chem. 2004, 279, 55633–55643. [Google Scholar]
- Sakurai, H.; Suzuki, S.; Kawasaki, N.; Nakano, H.; Okazaki, T.; Chino, A.; Doi, T.; Saiki, I. Tumor Necrosis Factor-α-induced IKK phosphorylation of NF-ΚB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J. Biol. Chem. 2003, 278, 36916–36923. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Ciechanover, A.; Marzese, D.M.; Hata, K.; Bustos, M.; Ono, S.; Wang, J.; Salomon, M.P.; Tran, K.; Lam, S.; et al. Epigenetic regulation of KPC1 ubiquitin ligase affects the NF-ΚB pathway in melanoma. Clin. Cancer Res. 2017, 23, 4831–4842. [Google Scholar] [CrossRef]
- Zalesna, I.; Osrodek, M.; Hartman, M.L.; Rozanski, M.; Sztiller-Sikorska, M.; Niewinna, K.; Nejc, D.; Czyz, M. Exogenous growth factors bFGF, EGF and HGF do not influence viability and phenotype of V600EBRAF melanoma cells and their response to vemurafenib and trametinib in vitro. PLoS ONE 2017, 12, e0183498. [Google Scholar] [CrossRef]
- Shoshan, E.; Braeuer, R.R.; Kamiya, T.; Mobley, A.K.; Huang, L.; Vasquez, M.E.; Velazquez-Torres, G.; Chakravarti, N.; Ivan, C.; Prieto, V.; et al. NFAT1 directly regulates IL8 and MMP3 to promote melanoma tumor growth and metastasis. Cancer Res. 2016, 76, 3145–3155. [Google Scholar] [CrossRef]
- Oka, M.; Sakaguchi, M.; Okada, T.; Nagai, H.; Ozaki, M.; Yoshioka, T.; Inoue, H.; Mukaida, N.; Kikkawa, U.; Nishigori, C. Signal Transducer and Activator of Transcription 3 upregulates interleukin-8 expression at the level of transcription in human melanoma cells. Exp. Dermatol. 2009, 19, 50–55. [Google Scholar] [CrossRef]
- Wilson, B.J.; Saab, K.R.; Ma, J.; Schatton, T.; Putz, P.; Zhan, Q.; Murphy, G.F.; Gasser, M.; Waaga-Gasser, A.M.; Frank, N.Y.; et al. ABCB5 maintains melanoma-initiating cells through a proinflammatory cytokine signaling circuit. Cancer Res. 2014, 74, 4196–4207. [Google Scholar] [CrossRef]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1β. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef]
- Strozyk, E.A.; Desch, A.; Poeppelmann, B.; Magnolo, N.; Wegener, J.; Huck, V.; Schneider, S.W. Melanoma-derived IL-1 converts vascular endothelium to a proinflammatory and procoagulatory phenotype via NFκB activation. Exp. Dermatol. 2014, 23, 670–676. [Google Scholar] [CrossRef]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.A.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef]
- Dhawan, P.; Richmond, A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J. Biol. Chem. 2002, 277, 7920–7928. [Google Scholar] [CrossRef]
- Thu, Y.M.; Su, Y.; Yang, J.; Splittgerber, R.; Na, S.; Boyd, A.; Mosse, C.; Simons, C.; Richmond, A. NF-κB inducing kinase (NIK) modulates melanoma tumorigenesis by regulating expression of pro-survival factors through the β-catenin pathway. Oncogene 2012, 31, 2580–2592. [Google Scholar] [CrossRef] [PubMed]
- Wells, K.; Hintzsche, J.; Amato, C.M.; Tobin, R.; Vorwald, V.; Mccarter, M.; Shellman, Y.; Tan, A.C.; Robinson, W. Abstract 5002: Investigating the role of NF-ΚB signaling and immune checkpoint blockade therapy in melanoma. In Proceedings of the American Association for Cancer Research Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019; AACR: Philadelphia, PA, USA, 2019. [Google Scholar]
- Broemer, M.; Krappmann, D.; Scheidereit, C. Requirement of Hsp90 activity for IκB Kinase (IKK) biosynthesis and for constitutive and inducible IKK and NF-ΚB activation. Oncogene 2004, 23, 5378–5386. [Google Scholar] [CrossRef]
- Malhotra, V.; Shanley, T.P.; Pittet, J.F.; Welch, W.J.; Wong, H.R. Geldanamycin inhibits NF-κB activation and interleukin-8 gene expression in cultured human respiratory epithelium. Am. J. Respir. Cell Mol. Biol. 2001, 25, 92–97. [Google Scholar] [CrossRef]
- Yeramian, A.; Vea, A.; Benítez, S.; Ribera, J.; Domingo, M.; Santacana, M.; Martinez, M.; Maiques, O.; Valls, J.; Dolcet, X.; et al. 2-Phenylethynesulphonamide (PFT-μ) enhances the anticancer effect of the novel hsp90 inhibitor NVP-AUY922 in melanoma, by reducing GSH levels. Pigment. Cell Melanoma Res. 2016, 29, 352–371. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.Y.; Barberi, T.J.; Ghosh, P.; Longo, D.L. Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-ΚB pathway. J. Biol. Chem. 2005, 280, 34538–34547. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-kB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Kwon, H.J.; Choi, G.E.; Ryu, S.; Kwon, S.J.; Kim, S.C.; Booth, C.; Nichols, K.E.; Kim, H.S. Stepwise phosphorylation of p65 promotes NF-κB activation and NK cell responses during target cell recognition. Nat. Commun. 2016, 7, 11686. [Google Scholar] [CrossRef] [PubMed]
- Handschick, K.; Beuerlein, K.; Jurida, L.; Bartkuhn, M.; Müller, H.; Soelch, J.; Weber, A.; Dittrich-Breiholz, O.; Schneider, H.; Scharfe, M.; et al. Cyclin-dependent kinase 6 is a chromatin-bound cofactor for NF-κB-dependent gene expression. Mol. Cell 2014, 53, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Buss, H.; Handschick, K.; Jurrmann, N.; Pekkonen, P.; Beuerlein, K.; Müller, H.; Wait, R.; Saklatvala, J.; Ojala, P.M.; Schmitz, M.L.; et al. Cyclin-dependent kinase 6 phosphorylates NF-κB p65 at serine 536 and contributes to the regulation of inflammatory gene expression. PLoS ONE 2012, 7, e51847. [Google Scholar] [CrossRef]
- Sarnico, I.; Lanzillotta, A.; Boroni, F.; Benarese, M.; Alghisi, M.; Schwaninger, M.; Inta, I.; Battistin, L.; Spano, P.; Pizzi, M. NF-ΚB p50/RelA and c-Rel-containing dimers: Opposite regulators of neuron vulnerability to ischaemia. J. Neurochem. 2009, 108, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Margue, C.M.; Bernasconi, M.; Barr, F.G.; Schäfer, B.W. Transcriptional modulation of the anti-apoptotic protein BCL-XL by the paired box transcription factors PAX3 and PAX3/FKHR. Oncogene 2000, 19, 2921–2929. [Google Scholar] [CrossRef][Green Version]
- Medic, S.; Rizos, H.; Ziman, M. Differential PAX3 functions in normal skin melanocytes and melanoma cells. Biochem. Biophys. Res. Commun. 2011, 411, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.; Boyle, G.M.; Ziman, M.; Medic, S. Mechanisms contributing to differential regulation of PAX3 downstream target genes in normal human epidermal melanocytes versus melanoma cells. PLoS ONE 2015, 10, e0124154. [Google Scholar] [CrossRef]
- Zhuang, L.; Lee, C.S.; Scolyer, R.A.; Mccarthy, S.W.; Zhang, X.D.; Thompson, J.F.; Hersey, P. Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Mod. Pathol. 2007, 20, 416–426. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M.; et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef]
- Tiffen, J.; Gallagher, S.J.; Hersey, P. EZH2: An emerging role in melanoma biology and strategies for targeted therapy. Pigment. Cell Melanoma Res. 2015, 28, 21–30. [Google Scholar] [CrossRef]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.M.; Liou, Y.C.; Yu, Q. Context-specific regulation of NF-ΚB target gene expression by EZH2 in breast cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; He, S.; Tian, Y.; Gu, Y.; Chen, P.; Li, C.; Huang, J.; Liu, Y.; Yu, H.; Jin, M.; et al. Hsp90 inhibition destabilizes EZH2 protein in alloreactive T cells and reduces graft-versus-host disease in mice. Blood 2017, 129, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Chang, W.W.; Chen, Y.Y.; Tsai, Y.H.; Chou, Y.H.; Tseng, H.C.; Chen, H.L.; Wu, C.C.; Chang-Chien, J.; Lee, H.T.; et al. Hsp90α mediates BMI1 expression in breast cancer stem/progenitor cells through facilitating nuclear translocation of c-Myc and EZH2. Int. J. Mol. Sci. 2017, 18, 1986. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Buckley, K.; Rao, R.; Mandawat, A.; Yang, Y.; Joshi, R.; Wang, Y.; Balusu, R.; Chen, J.; Koul, S.; et al. Panobinostat treatment depletes EZH2 and DNMT1 levels and enhances decitabine mediated de-repression of JunB and loss of survival of human acute leukemia cells. Cancer Biol. Ther. 2009, 8, 939–950. [Google Scholar] [CrossRef]
- Rooswinkel, R.W.; van de Kooij, B.; de Vries, E.; Paauwe, M.; Braster, R.; Verheij, M.; Borst, J. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood 2014, 123, 2806–2815. [Google Scholar] [CrossRef]
- Miyakawa, Y.; Matsushime, H. Rapid downregulation of cyclin D1 mRNA and protein levels by ultraviolet irradiation in murine macrophage cells. Biochem. Biophys. Res. Commun. 2001, 284, 71–76. [Google Scholar] [CrossRef]
- Gabellini, C.; Gómez-Abenza, E.; Ibáñez-Molero, S.; Tupone, M.G.; Pérez-Oliva, A.B.; Oliveira, S.D.; Bufalo, D.D.; Mulero, V. Interleukin 8 mediates Bcl-XL-induced enhancement of human melanoma cell dissemination and angiogenesis in a zebrafish xenograft model. Int. J. Cancer 2017, 142, 584–596. [Google Scholar] [CrossRef]
- Chelouche-Lev, D.; Miller, C.P.; Tellez, C.; Ruiz, M.; Bar-Eli, M.; Price, J.E. Different signalling pathways regulate VEGF and IL-8 expression in breast cancer: Implications for therapy. Eur. J. Cancer 2004, 40, 2509–2518. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2019. epub ahead of print. [Google Scholar] [CrossRef]
- Martin, D.; Galisteo, R.; Gutkind, J. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J. Biol. Chem. 2009, 284, 6038–6042. [Google Scholar] [CrossRef]
- Björndahl, M.; Cao, R.; Eriksson, A.; Cao, Y. Blockage of VEGF-induced angiogenesis by preventing VEGF secretion. Circ. Res. 2004, 94, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, I.; Kowalska, M.; Fuksiewicz, M.; Kotowicz, B.; Mierzejewska, E.; Koseła-Paterczyk, H.; Szamotulska, K.; Rutkowski, P. Serum markers in early-stage and locally advanced melanoma. Tumour Biol. 2015, 36, 8277–8285. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Floris, C.; Mangieri, D.; Piras, F.; Ennas, M.G.; Vacca, A.; Sirigu, P. Microvascular density, vascular endothelial growth factor immunoreactivity in tumor cells, vessel diameter and intussusceptive microvascular growth in primary melanoma. Oncol. Rep. 2005, 14, 81–84. [Google Scholar] [PubMed]
- Xu, X.; Zong, Y.; Gao, Y.; Sun, X.; Zhao, H.; Luo, W.; Jia, S. VEGF induce vasculogenic mimicry of choroidal melanoma through the PI3k signal pathway. Biomed. Res. Int. 2019, 2019, 3909102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Zhou, H.; Fan, G.; Li, Q. Molecular mechanisms and anticancer therapeutic strategies in vasculogenic mimicry. J. Cancer. 2019, 10, 6327–6340. [Google Scholar] [CrossRef]
- Zhang, Z.; Imani, S.; Shasaltaneh, M.D.; Hosseinifard, H.; Zou, L.; Fan, Y.; Wen, Q. The role of vascular mimicry as a biomarker in malignant melanoma: A systematic review and meta-analysis. BMC Cancer 2019, 19, 1134. [Google Scholar] [CrossRef]
- Liu, Z.J.; Zhou, Y.J.; Ding, R.L.; Xie, F.; Fu, S.Z.; Wu, J.B.; Yang, L.L.; Wen, Q.L. In vitro and in vivo apatinib inhibits vasculogenic mimicry in melanoma MUM-2B cells. PLoS ONE 2018, 13, e0200845. [Google Scholar] [CrossRef]
- Bosserhoff, A.K.; Schneider, N.; Ellmann, L.; Heinzerling, L.; Kuphal, S. The neurotrophin Neuritin1 (cpg15) is involved in melanoma migration, attachment independent growth, and vascular mimicry. Oncotarget 2017, 8, 1117–1131. [Google Scholar] [CrossRef][Green Version]
- Vartanian, A.; Stepanova, E.; Grigorieva, I.; Solomko, E.; Baryshnikov, A.; Lichinitser, M. VEGFR1 and PKCα signaling control melanoma vasculogenic mimicry in a VEGFR2 kinase-independent manner. Melanoma Res. 2011, 21, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, A.; Baryshnikova, M.; Burova, O.; Afanasyeva, D.; Misyurin, V.; Belyavsky, A.; Shprakh, Z. Inhibitor of vasculogenic mimicry restores sensitivity of resistant melanoma cells to DNA-damaging agents. Melanoma Res. 2017, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Schnegg, C.I.; Yang, M.H.; Ghosh, S.K.; Hsu, M.Y. Induction of vasculogenic mimicry overrides VEGF-A silencing and enriches stem-like cancer cells in melanoma. Cancer Res. 2015, 75, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.G.; Ceci, C.; Ruffini, F.; Trapani, M.; Barbaccia, M.L.; Tentori, L.; D’Atri, S.; Lacal, P.M.; Graziani, G. Role of VEGFR-1 in melanoma acquired resistance to the BRAF inhibitor vemurafenib. J. Cell. Mol. Med. 2020, 24, 465–475. [Google Scholar] [CrossRef]
- Caporali, S.; Amaro, A.; Levati, L.; Alvino, E.; Lacal, P.M.; Mastroeni, S.; Ruffini, F.; Bonmassar, L.; Antonini Cappellini, G.C.; Felli, N.; et al. miR-126-3p down-regulation contributes to dabrafenib acquired resistance in melanoma by up-regulating ADAM9 and VEGF-A. J. Exp. Clin. Cancer Res. 2019, 38, 272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, M.; Gu, Y.; Liu, Z.; Xu, S.; Cui, Y.; Sun, B. Thalidomide influences growth and vasculogenic mimicry channel formation in melanoma. J. Exp. Clin. Cancer Res. 2008, 27, 60. [Google Scholar] [CrossRef]
- Gong, F.; Chen, M.F.; Zhang, Y.Y.; Li, C.Y.; Zhou, C.X.; Hong, P.Z.; Sun, S.L.; Qian, Z.J. A novel peptide from abalone (Haliotis discus hannai) to suppress metastasis and vasculogenic mimicry of tumor cells and enhance anti-tumor effect in vitro. Mar. Drugs 2019, 17, 244. [Google Scholar] [CrossRef]
- Sztiller-Sikorska, M.; Koprowska, K.; Jakubowska, J.; Zalesna, I.; Stasiak, M.; Duechler, M.; Czyz, M.E. Sphere formation and self-renewal capacity of melanoma cells is affected by the microenvironment. Melanoma Res. 2012, 22, 215–224. [Google Scholar] [CrossRef]
- Koprowska, K.; Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M.E. Parthenolide enhances dacarbazine activity against melanoma cells. Anticancer Drugs 2013, 24, 835–845. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes Overexpressed in DMBC12 vs. DMBC11 | Genes Underexpressed in DMBC12 vs. DMBC11 | ||
|---|---|---|---|
| Gene Symbol | Fold Regulation | Gene Symbol | Fold Regulation |
| CXCL8 | 42.64 | ICAM1 | −5.60 |
| CXCL1 | 25.41 | TNFSF10 | −4.50 |
| CXCL2 | 13.27 | IL6 | −3.74 |
| BIRC3 | 12.79 | IL4 | −3.68 |
| CCL2 | 5.28 | IL12B | −3.02 |
| BCL2A1 | 4.88 | NQO1 | −3.00 |
| IL1A | 4.65 | CD69 | −3.11 |
| EGFR | 4.38 | IL2RA | −2.52 |
| IL1B | 4.09 | CCL11 | −2.38 |
| F8 | 2.53 | PLAU | −2.38 |
| NFKBIA | 2.23 | FASLG | −2.27 |
| NFKB1 | 2.10 | IL1RN | −2.17 |
| STAT3 | −2.05 | ||
| Genes Overexpressed in DMBC21 vs. DMBC11 | Genes Underexpressed in DMBC21 vs. DMBC11 | ||
| Gene Symbol | Fold Regulation | Gene Symbol | Fold Regulation |
| BCL2A1 | 70.48 | IL1A | −43.81 |
| CCL2 | 64.05 | TNFRSF1B | −15.27 |
| CXCL1 | 46.70 | CCL22 | −10.14 |
| CXCL2 | 26.45 | CCR5 | −7.35 |
| F8 | 22.12 | PLAU | −6.88 |
| C3 | 19.83 | CD80 | −6.39 |
| AGT | 15.05 | PTGS2 | −5.34 |
| CXCL8 | 13.33 | IL12B | −4.77 |
| IL1B | 7.03 | IL2RA | −4.72 |
| ICAM1 | 6.19 | FASLG | −4.69 |
| NR4A2 | 5.18 | CSF3 | −4.60 |
| ADM | 4.43 | MAP2K6 | −4.25 |
| IL15 | 3.39 | SNAP25 | −4.24 |
| BCL2L1 | 2.61 | EGFR | −4.12 |
| SOD2 | 2.57 | BIRC3 | −3.68 |
| CD83 | 2.56 | NFKB2 | −3.48 |
| CDKN1A | 2.44 | CCL11 | −3.34 |
| STAT1 | 2.35 | SELE | −3.32 |
| LTA | −3.27 | ||
| CSF1 | −3.09 | ||
| GADD45B | −3.02 | ||
| CSF2 | −2.78 | ||
| IL1RN | −2.73 | ||
| F3 | −2.54 | ||
| IFNB1 | −2.53 | ||
| ALDH3A2 | −2.33 | ||
| SELP | −2.21 | ||
| INS | −2.17 | ||
| CXCL9 | −2.16 | ||
| BIRC2 | −2.11 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartman, M.L.; Rogut, M.; Mielczarek-Lewandowska, A.; Wozniak, M.; Czyz, M. 17-Aminogeldanamycin Inhibits Constitutive Nuclear Factor-Kappa B (NF-κB) Activity in Patient-Derived Melanoma Cell Lines. Int. J. Mol. Sci. 2020, 21, 3749. https://doi.org/10.3390/ijms21113749
Hartman ML, Rogut M, Mielczarek-Lewandowska A, Wozniak M, Czyz M. 17-Aminogeldanamycin Inhibits Constitutive Nuclear Factor-Kappa B (NF-κB) Activity in Patient-Derived Melanoma Cell Lines. International Journal of Molecular Sciences. 2020; 21(11):3749. https://doi.org/10.3390/ijms21113749
Chicago/Turabian StyleHartman, Mariusz L., Magdalena Rogut, Aleksandra Mielczarek-Lewandowska, Michal Wozniak, and Malgorzata Czyz. 2020. "17-Aminogeldanamycin Inhibits Constitutive Nuclear Factor-Kappa B (NF-κB) Activity in Patient-Derived Melanoma Cell Lines" International Journal of Molecular Sciences 21, no. 11: 3749. https://doi.org/10.3390/ijms21113749
APA StyleHartman, M. L., Rogut, M., Mielczarek-Lewandowska, A., Wozniak, M., & Czyz, M. (2020). 17-Aminogeldanamycin Inhibits Constitutive Nuclear Factor-Kappa B (NF-κB) Activity in Patient-Derived Melanoma Cell Lines. International Journal of Molecular Sciences, 21(11), 3749. https://doi.org/10.3390/ijms21113749