Vitamin D in Triple-Negative and BRCA1-Deficient Breast Cancer—Implications for Pathogenesis and Therapy

 ,
,  , and
, and {kind=link}
{kind=link}
Abstract
1. Introduction
2. Vitamin D—an Essential Nutrient and a Potential Preventive Agent and Pharmaceutical in Breast Cancer
3. Vitamin D in Breast Cancer
4. Vitamin D3 Signaling in Triple-Negative Breast Cancer
4.1. Observational Studies
4.2. Oxidative Stress, Proliferation, Differentiation and Inflammation
4.3. Metabolism
4.4. Epithelial–Mesenchymal Transition
4.5. Epigenetics
4.6. Therapeutic Resistance
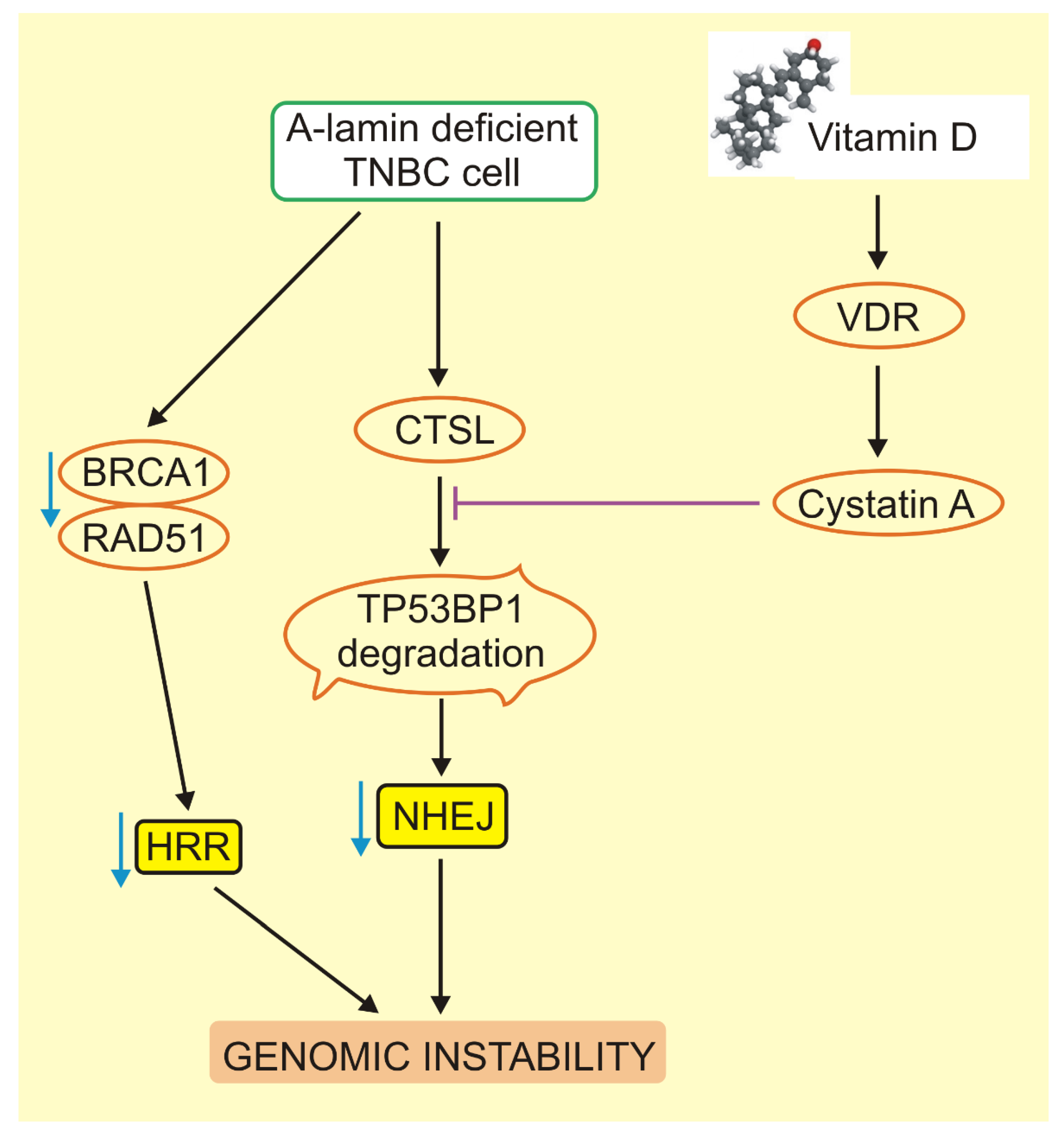
5. Critical Role of BRCA1 and TP53BP1 in VD3 Signaling in Triple-Negative Breast Cancer
6. GADD45A—a New Player in Vitamin D Signaling in Triple-Negative Breast Cancer
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B. Review of Recent Advances in Understanding the Role of Vitamin D in Reducing Cancer Risk: Breast, Colorectal, Prostate, and Overall Cancer. Anticancer Res. 2020, 40, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Ballinger, T.J.; Bouwman, B.A.M.; Mirzazadeh, R.; Garnerone, S.; Crosetto, N.; Semple, C.A. Modeling double strand break susceptibility to interrogate structural variation in cancer. Genome Biol. 2019, 20, 28. [Google Scholar] [CrossRef]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.; Pietenpol, J.A. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer 2014, 121, 8–16. [Google Scholar] [CrossRef]
- Ballinger, T.; Kremer, J.; Miller, K. Triple Negative Breast Cancer—Review of Current and Emerging Therapeutic Strategies. Oncol. Hematol. Rev. 2016, 12, 89. [Google Scholar] [CrossRef][Green Version]
- Narvaez, C.J.; Matthews, D.G.; LaPorta, E.; Simmons, K.M.; Beaudin, S.; Welsh, J. The impact of vitamin D in breast cancer: Genomics, pathways, metabolism. Front. Physiol. 2014, 5, 213. [Google Scholar] [CrossRef]
- O’Brien, K.M.; Sandler, D.P.; Xu, Z.; Kinyamu, H.K.; Taylor, J.; Weinberg, C.R. Vitamin D, DNA methylation, and breast cancer. Breast Cancer Res. 2018, 20, 70. [Google Scholar] [CrossRef]
- Welsh, J. Vitamin D and breast cancer: Past and present. J. Steroid Biochem. Mol. Biol. 2018, 177, 15–20. [Google Scholar] [CrossRef]
- Carlberg, C. Nutrigenomics of Vitamin D. Nutrients 2019, 11, 676. [Google Scholar] [CrossRef]
- Norman, A.W. Vitamin D Receptor: New Assignments for an Already Busy Receptor. Endocrinology 2006, 147, 5542–5548. [Google Scholar] [CrossRef] [PubMed]
- Richard, C.L.; Farach-Carson, M.C.; Rohe, B.; Nemere, I.; Meckling, K.A. Involvement of 1,25D3-MARRS (membrane associated, rapid response steroid-binding), a novel vitamin D receptor, in growth inhibition of breast cancer cells. Exp. Cell Res. 2010, 316, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Goeman, F.; De Nicola, F.; De Meo, P.D.; Pallocca, M.; Elmi, B.; Castrignanò, T.; Pesole, G.; Strano, S.; Blandino, G.; Fanciulli, M.; et al. VDR primary targets by genome-wide transcriptional profiling. J. Steroid Biochem. Mol. Biol. 2014, 143, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Franza, L.; Mandolini, C.; Conti, P. Immune Modulation by Vitamin D: Special Emphasis on Its Role in Prevention and Treatment of Cancer. Clin. Ther. 2017, 39, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, E.; Wysokinski, D.; Blasiak, J. Nucleotide Excision Repair and Vitamin D—Relevance for Skin Cancer Therapy. Int. J. Mol. Sci. 2016, 17, 372. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Vitamin D Genomics: From In Vitro to In Vivo. Front. Endocrinol. 2018, 9, 250. [Google Scholar] [CrossRef]
- Sequeira, V.B.; Rybchyn, M.; Tongkao-On, W.; Gordon-Thomson, C.; Malloy, P.J.; Nemere, I.; Norman, A.W.; Reeve, V.E.; Halliday, G.M.; Feldman, D.; et al. The role of the vitamin D receptor and ERp57 in photoprotection by 1α,25-dihydroxyvitamin D3. Mol. Endocrinol. 2012, 26, 574–582. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Hoffman, R.M.; Slominski, A.T. Relevance of Vitamin D in Melanoma Development, Progression and Therapy. Anticancer Res. 2020, 40, 473–489. [Google Scholar] [CrossRef]
- Abe, E.; Miyaura, C.; Sakagami, H.; Takeda, M.; Konno, K.; Yamazaki, T.; Yoshiki, S.; Suda, T. Differentiation of mouse myeloid leukemia cells induced by 1 alpha,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1981, 78, 4990–4994. [Google Scholar] [CrossRef]
- Colston, K.; Feldman, D. 1,25-dihydroxyvitamin D3 and malignant melanoma: the presence of receptors and inhibition of cell growth in culture. Endocrinology 1981, 108, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Yaghjyan, L.; Colditz, G.A. Estrogens in the breast tissue: A systematic review. Cancer Causes Control. 2011, 22, 529–540. [Google Scholar] [CrossRef] [PubMed]
- James, S.Y.; Mackay, A.G.; Binderup, L.; Colston, K.W. Effects of a new synthetic vitamin D analogue, EB1089, on the oestrogen-responsive growth of human breast cancer cells. J. Endocrinol. 1994, 141, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Swami, S.; Peng, L.; Wang, J.; Moreno, J.; Feldman, D. Tissue-selective regulation of aromatase expression by calcitriol: Implications for breast cancer therapy. Endocrinology 2010, 151, 32–42. [Google Scholar] [CrossRef]
- Simboli-Campbell, M.; Narvaez, C.J.; VanWeelden, K.; Tenniswood, M.; Welsh, J. Comparative effects of 1,25(OH)2D3 and EB1089 on cell cycle kinetics and apoptosis in MCF-7 breast cancer cells. Breast Cancer Res. Treat. 1997, 42, 31–41. [Google Scholar] [CrossRef]
- Swami, S.; Krishnan, A.V.; Feldman, D. 1alpha,25-Dihydroxyvitamin D3 down-regulates estrogen receptor abundance and suppresses estrogen actions in MCF-7 human breast cancer cells. Clin. Cancer Res. 2000, 6, 3371–3379. [Google Scholar]
- Swami, S.; Krishnan, A.V.; Peng, L.; Lundqvist, J.; Feldman, D. Transrepression of the estrogen receptor promoter by calcitriol in human breast cancer cells via two negative vitamin D response elements. Endocrine-Relat. Cancer 2013, 20, 565–577. [Google Scholar] [CrossRef]
- Swami, S.; Krishnan, A.V.; Wang, J.Y.; Jensen, K.; Peng, L.; Albertelli, M.; Feldman, D. Inhibitory Effects of Calcitriol on the Growth of MCF-7 Breast Cancer Xenografts in Nude Mice: Selective Modulation of Aromatase Expression in vivo. Horm. Cancer 2011, 2, 190–202. [Google Scholar] [CrossRef]
- LaPorta, E.; Welsh, J. Modeling vitamin D actions in triple negative/basal-like breast cancer. J. Steroid Biochem. Mol. Biol. 2013, 144, 65–73. [Google Scholar] [CrossRef]
- Lowe, L.; Hansen, C.M.; Senaratne, S.; Colston, K.W. Mechanisms implicated in the growth regulatory effects of vitamin D compounds in breast cancer cells. Methods Mol. Biol. 2003, 164, 99–110. [Google Scholar] [CrossRef]
- Johnson, A.L.; Zinser, G.M.; Waltz, S.E. Loss of vitamin D receptor signaling from the mammary epithelium or adipose tissue alters pubertal glandular development. Am. J. Physiol. Metab. 2014, 307, E674–E685. [Google Scholar] [CrossRef]
- Zinser, G.; Packman, K.; Welsh, J. Vitamin D(3) receptor ablation alters mammary gland morphogenesis. Development 2002, 129, 3067–3076. [Google Scholar] [PubMed]
- Colston, K.W.; Hansen, C.M. Mechanisms implicated in the growth regulatory effects of vitamin D in breast cancer. Endocrine-Relat. Cancer 2002, 9, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Villena-Heinsen, C.; Tilgen, W.; Schmidt, W.; Reichrat, J.; Axt-Fliedner, R. Vitamin D receptor (VDR) expression is not a prognostic factor in breast cancer. Anticancer Res. 2002, 22, 1919–1924. [Google Scholar] [PubMed]
- Al-Azhri, J.; Zhang, Y.; Bshara, W.; Zirpoli, G.R.; McCann, S.E.; Khoury, T.; Morrison, C.D.; Edge, S.B.; Ambrosone, C.B.; Yao, S. Tumor Expression of Vitamin D Receptor and Breast Cancer Histopathological Characteristics and Prognosis. Clin. Cancer Res. 2016, 23, 97–103. [Google Scholar] [CrossRef]
- Marik, R.; Fackler, M.; Gabrielson, E.; Zeiger, M.A.; Stearns, V.; Umbricht, C.B.; Sukumar, S. DNA methylation-related vitamin D receptor insensitivity in breast cancer. Cancer Biol. Ther. 2010, 10, 44–53. [Google Scholar] [CrossRef]
- Tavera-Mendoza, L.E.; Westerling, T.; Libby, E.; Marusyk, A.; Cato, L.; Cassani, R.; Cameron, L.A.; Ficarro, S.B.; Marto, J.A.; Klawitter, J.; et al. Vitamin D receptor regulates autophagy in the normal mammary gland and in luminal breast cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2186. [Google Scholar] [CrossRef]
- Bauer, S.; Hankinson, S.E.; Bertone-Johnson, E.R.; Ding, E.L. Plasma Vitamin D Levels, Menopause, and Risk of Breast Cancer. Medicine 2013, 92, 123–131. [Google Scholar] [CrossRef]
- Crew, K.D.; Gammon, M.D.; Steck, S.E.; Hershman, D.L.; Cremers, S.; Dworakowski, E.; Shane, E.; Terry, M.B.; Desai, M.; Teitelbaum, S.L.; et al. Association between plasma 25-hydroxyvitamin D and breast cancer risk. Cancer Prev. Res. 2009, 2, 598–604. [Google Scholar] [CrossRef]
- Kudela, E.; Samec, M.; Kubatka, P.; Nachajova, M.; Laucekova, Z.; Liskova, A.; Dokus, K.; Biringer, K.; Simova, D.; Gabonova, E.; et al. Breast Cancer in Young Women: Status Quo and Advanced Disease Management by a Predictive, Preventive, and Personalized Approach. Cancers 2019, 11, 1791. [Google Scholar] [CrossRef]
- Giovannucci, E. Vitamin D and Cancer Incidence in the Harvard Cohorts. Ann. Epidemiol. 2009, 19, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Albertson, D.G.; Ylstra, B.; Segraves, R.; Collins, C.; Dairkee, S.; Kowbel, D.; Kuo, W.-L.; Gray, J.W.; Pinkel, D. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat. Genet. 2000, 25, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Lope, V.; Castelló, A.; Mena-Bravo, A.; Amiano, P.; Aragonés, N.; Fernández-Villa, T.; Guevara, M.; Dierssen-Sotos, T.; Fernández-Tardon, G.; Castaño-Vinyals, G.; et al. Serum 25-hydroxyvitamin D and breast cancer risk by pathological subtype (MCC-Spain). J. Steroid Biochem. Mol. Biol. 2018, 182, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Peppone, L.J.; Rickles, A.S.; Janelsins, M.C.; Insalaco, M.R.; Skinner, K.A.; Huston, A.J.; Reid, M.E.; Rosier, R.N.; Zakharia, Y.; Trump, N.L.; et al. The association between breast cancer prognostic indicators and serum 25-OH vitamin D levels. Ann. Surg. Oncol. 2012, 19, 2590–2599. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, Y.M.; Ko, B.S.; Lee, J.W.; Yu, J.H.; Son, B.H.; Gong, G.-Y.; Kim, S.B.; Lee, J.W. Vitamin D deficiency is correlated with poor outcomes in patients with luminal-type breast cancer. Ann. Surg. Oncol. 2010, 18, 1830–1836. [Google Scholar] [CrossRef][Green Version]
- Rainville, C.; Khan, Y.; Tisman, G. Triple negative breast cancer patients presenting with low serum vitamin D levels: A case series. Cases J. 2009, 2, 8390. [Google Scholar] [CrossRef]
- Tommie, J.L.; Pinney, S.M.; Nommsen-Rivers, L.A. Serum Vitamin D Status and Breast Cancer Risk by Receptor Status: A Systematic Review. Nutr. Cancer 2018, 70, 804–820. [Google Scholar] [CrossRef]
- Viala, M.; Chiba, A.; Thezenas, S.; Delmond, L.; Lamy, P.-J.; Mott, S.L.; Schroeder, M.C.; Thomas, A.; Jacot, W. Impact of vitamin D on pathological complete response and survival following neoadjuvant chemotherapy for breast cancer: A retrospective study. BMC Cancer 2018, 18, 770. [Google Scholar] [CrossRef]
- Yao, S.; Sucheston, L.E.; Millen, A.E.; Johnson, C.S.; Trump, N.L.; Nesline, M.K.; Davis, W.; Hong, C.-C.; McCann, S.E.; Hwang, H.; et al. Pretreatment Serum Concentrations of 25-Hydroxyvitamin D and Breast Cancer Prognostic Characteristics: A Case-Control and a Case-Series Study. PLoS ONE 2011, 6, e17251. [Google Scholar] [CrossRef]
- Mutlu, H.; Buyukcelik, A.; Çolak, T.; Ozdogan, M.; Erden, A.; Aslan, T.; Akca, Z. Is Sunlight a Predisposing Factor for Triple Negative Breast Cancer in Turkey? Asian Pac. J. Cancer Prev. 2013, 14, 801–803. [Google Scholar] [CrossRef]
- Qin, B.; Xu, B.; Ji, N.; Yao, S.; Pawlish, K.; Llanos, A.A.M.; Lin, Y.; Demissie, K.; Ambrosone, C.B.; Hong, C.-C.; et al. Intake of vitamin D and calcium, sun exposure, and risk of breast cancer subtypes among black women. Am. J. Clin. Nutr. 2020, 111, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Amend, K.; Hicks, D.; Ambrosone, C.B. Breast cancer in African-American women: differences in tumor biology from European-American women. Cancer Res. 2006, 66, 8327–8330. [Google Scholar] [CrossRef] [PubMed]
- Ping, J.; Guo, X.; Ye, F.; Long, J.; Lipworth, L.; Cai, Q.; Blot, W.; Shu, X.-O.; Zheng, W. Differences in gene-expression profiles in breast cancer between African and European-ancestry women. Carcinogenesis 2020. [Google Scholar] [CrossRef] [PubMed]
- Purrington, K.S.; Gorski, D.; Simon, M.S.; Hastert, T.A.; Kim, S.; Rosati, R.; Schwartz, A.G.; Ratnam, M. Racial differences in estrogen receptor staining levels and implications for treatment and survival among estrogen receptor positive, HER2-negative invasive breast cancers. Breast Cancer Res. Treat. 2020, 181, 145–154. [Google Scholar] [CrossRef]
- Nesby-O’Dell, S.; Scanlon, K.S.; Cogswell, M.E.; Gillespie, C.; Hollis, B.W.; Looker, A.C.; Allen, C.; Doughertly, C.; Gunter, E.W.; Bowman, B.A. Hypovitaminosis D prevalence and determinants among African American and white women of reproductive age: Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Clin. Nutr. 2002, 76, 187–192. [Google Scholar] [CrossRef]
- Wejse, C.; Olesen, R.; Rabna, P.; Kaestel, P.; Gustafson, P.; Aaby, P.; Andersen, P.L.; Glerup, H.; Sodemann, M. Serum 25-hydroxyvitamin D in a West African population of tuberculosis patients and unmatched healthy controls. Am. J. Clin. Nutr. 2007, 86, 1376–1383. [Google Scholar] [CrossRef]
- Yao, S.; Ambrosone, C.B. Associations between vitamin D deficiency and risk of aggressive breast cancer in African-American women. J. Steroid Biochem. Mol. Biol. 2013, 136, 337–341. [Google Scholar] [CrossRef]
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018, 29, 895–902. [Google Scholar] [CrossRef]
- Werner, H.; Sarfstein, R.; Bruchim, I. Investigational IGF1R inhibitors in early stage clinical trials for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 1101–1112. [Google Scholar] [CrossRef]
- Soljic, M.; Mrklic, I.; Tomic, S.; Omrcen, T.; Sutalo, N.; Bevanda, M.; Vrdoljak, E. Prognostic value of vitamin D receptor and insulin-like growth factor receptor 1 expression in triple-negative breast cancer. J. Clin. Pathol. 2017, 71, 34–39. [Google Scholar] [CrossRef]
- Bohl, L.P.; Liaudat, A.C.; Picotto, G.; Marchionatti, A.M.; Narvaez, C.J.; Welsh, J.; Rodriguez, V.A.; De Talamoni, N.G.T. Buthionine Sulfoximine and 1,25-Dihydroxyvitamin D Induce Apoptosis in Breast Cancer Cells via Induction of Reactive Oxygen Species. Cancer Investig. 2012, 30, 560–570. [Google Scholar] [CrossRef]
- Marchionatti, A.M.; Picotto, G.; Narvaez, C.J.; Welsh, J.; De Talamoni, N.T. Antiproliferative action of menadione and 1,25(OH)2D3 on breast cancer cells. J. Steroid Biochem. Mol. Biol. 2009, 113, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Bohl, L.; Guizzardi, S.; Rodríguez, V.; Hinrichsen, L.; Rozados, V.; Cremonezzi, D.; De Talamoni, N.T.; Picotto, G. Combined calcitriol and menadione reduces experimental murine triple negative breast tumor. Biomed. Pharmacother. 2017, 94, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Körner, C.; Ben-Baruch, A. Tumor-Stroma-Inflammation Networks Promote Pro-metastatic Chemokines and Aggressiveness Characteristics in Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Malaguarnera, M.; Nicoletti, F.; Malaguarnera, M. Vitamin D3: A helpful immuno-modulator. Immunology 2011, 134, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reza, I.; Díaz, L.; Barrera, D.; Segovia-Mendoza, M.; Pedraza-Sánchez, S.; Soca-Chafre, G.; Larrea, F.; García-Becerra, R. Calcitriol Inhibits the Proliferation of Triple-Negative Breast Cancer Cells through a Mechanism Involving the Proinflammatory Cytokines IL-1βand TNF-α. J. Immunol. Res. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Almendro, V.; Merino, V.F.; Wu, Z.; Maruyama, R.; Su, Y.; Martins, F.C.; Fackler, M.J.; Bessarabova, M.; Kowalczyk, A.; et al. Molecular profiling of human mammary gland links breast cancer risk to a p27(+) cell population with progenitor characteristics. Cell Stem Cell 2013, 13, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Wahler, J.; So, J.Y.; Cheng, L.; Maehr, H.; Uskokovic, M.; Suh, N. Vitamin D compounds reduce mammosphere formation and decrease expression of putative stem cell markers in breast cancer. J. Steroid Biochem. Mol. Biol. 2014, 148, 148–155. [Google Scholar] [CrossRef]
- Shan, N.L.; Wahler, J.; Lee, H.J.; Bak, M.J.; Das Gupta, S.; Maehr, H.; Suh, N. Vitamin D compounds inhibit cancer stem-like cells and induce differentiation in triple negative breast cancer. J. Steroid Biochem. Mol. Biol. 2016, 173, 122–129. [Google Scholar] [CrossRef]
- Zheng, W.; Duan, B.; Zhang, Q.; Ouyang, L.; Peng, W.; Qian, F.; Wang, Y.; Huang, S. Vitamin D-induced vitamin D receptor expression induces tamoxifen sensitivity in MCF-7 stem cells via suppression of Wnt/β-catenin signaling. Biosci. Rep. 2018, 38, BSR20180595. [Google Scholar] [CrossRef]
- Abu El Maaty, M.; Dabiri, Y.; Almouhanna, F.; Blagojevic, B.; Theobald, J.; Büttner, M.; Wölfl, S. Activation of pro-survival metabolic networks by 1,25(OH)2D3 does not hamper the sensitivity of breast cancer cells to chemotherapeutics. Cancer Metab. 2018, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 1994, 1219, 26–32. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, Q.; Chng, W.J. TXNIP (VDUP-1, TBP-2): A major redox regulator commonly suppressed in cancer by epigenetic mechanisms. Int. J. Biochem. Cell Biol. 2011, 43, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.E.; Weierstahl, K.A.; Kelts, J.L. Vitamin D effect on growth and vitamin D metabolizing enzymes in triple-negative breast cancer. Anticancer Res. 2015, 35, 805–810. [Google Scholar]
- Gwin, K.; Buell-Gutbrod, R.; Tretiakova, M.; Montag, A. Epithelial-to-Mesenchymal Transition in Metaplastic Breast Carcinomas with Chondroid Differentiation. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 526–531. [Google Scholar] [CrossRef]
- Sikandar, S.S.; Kuo, A.H.; Kalisky, T.; Cai, S.; Zabala, M.; Hsieh, R.W.; Lobo, N.A.; Scheeren, F.A.; Sim, S.; Qian, D.; et al. Role of epithelial to mesenchymal transition associated genes in mammary gland regeneration and breast tumorigenesis. Nat. Commun. 2017, 8, 1669. [Google Scholar] [CrossRef]
- Larriba, M.J.; De Herreros, A.G.; Muñoz, A. Vitamin D and the Epithelial to Mesenchymal Transition. Stem Cells Int. 2016, 2016, 6213872. [Google Scholar] [CrossRef]
- Simeone, P.; Trerotola, M.; Franck, J.; Cardon, T.; Marchisio, M.; Fournier, I.; Salzet, M.; Maffia, M.; Vergara, D.; Tristan, C. The multiverse nature of epithelial to mesenchymal transition. Semin. Cancer Biol. 2019, 58, 1–10. [Google Scholar] [CrossRef]
- Shen, T.; Zhang, K.; Siegal, G.P.; Wei, S. Prognostic Value of E-Cadherin and β-Catenin in Triple-Negative Breast Cancer. Am. J. Clin. Pathol. 2016, 146, 603–610. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Milanezi, F.; Paredes, J.; Silva, P.; Pereira, E.M.; Maeda, S.A.; De Carvalho, L.V.; Schmitt, D.E. Novel and Classic Myoepithelial/Stem Cell Markers in Metaplastic Carcinomas of the Breast. Appl. Immunohistochem. Mol. Morphol. 2003, 11, 1–8. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Milanezi, F.; Steele, D.; Savage, K.; Simpson, P.; Nesland, J.M.; Pereira, E.M.; Lakhani, S.; Schmitt, F.C. Metaplastic breast carcinomas are basal-like tumours. Histopathology 2006, 49, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Xie, G.; Yao, Q.; Liu, Y.; Du, S.; Liu, A.; Guo, Z.; Sun, A.; Ruan, J.; Chen, L.; et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int. J. Oncol. 2011, 40, 1171–1179. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; Hollander, P.D.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, M.A.; Deif, S.M.A.; Abou-Shamaa, L.A. IL-6 Impairs the Activity of Vitamin D3 in the Regulation of Epithelial-Mesenchymal Transition in Triple Negative Breast Cancer. Asian Pac. J. Cancer Prev. 2019, 20, 2267–2273. [Google Scholar] [CrossRef] [PubMed]
- Hummel, D.M.; Fetahu, I.S.; Gröschel, C.; Manhardt, T.; Kallay, E. Role of proinflammatory cytokines on expression of vitamin D metabolism and target genes in colon cancer cells. J. Steroid Biochem. Mol. Biol. 2013, 144, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Ricca, C.; Aillon, A.; Viano, M.; Bergandi, L.; Aldieri, E.; Silvagno, F. Vitamin D inhibits the epithelial-mesenchymal transition by a negative feedback regulation of TGF-β activity. J. Steroid Biochem. Mol. Biol. 2019, 187, 97–105. [Google Scholar] [CrossRef]
- Fischer, K.D.; Agrawal, D. Vitamin D regulating TGF-β induced epithelial-mesenchymal transition. Respir. Res. 2014, 15, 146. [Google Scholar] [CrossRef]
- Lopes, N.; Carvalho, J.; Durães, C.; Sousa, B.; Gomes, M.; Costa, J.L.; Oliveira, C.; Paredes, J.; Schmitt, F. 1Alpha,25-dihydroxyvitamin D3 induces de novo E-cadherin expression in triple-negative breast cancer cells by CDH1-promoter demethylation. Anticancer Res. 2012, 32, 249–257. [Google Scholar]
- Saramäki, A.; Diermeier, S.; Kellner, R.; Laitinen, H.; Vaïsänen, S.; Carlberg, C. Cyclical Chromatin Looping and Transcription Factor Association on the Regulatory Regions of the p21 (CDKN1A) Gene in Response to 1α,25-Dihydroxyvitamin D3. J. Biol. Chem. 2009, 284, 8073–8082. [Google Scholar] [CrossRef]
- Bijian, K.; Kaldre, D.; Wang, T.-T.; Su, J.; Bouttier, M.; Boucher, A.; Alaoui-Jamali, M.; White, J.H.; Gleason, J.L. Efficacy of hybrid vitamin D receptor agonist/histone deacetylase inhibitors in vitamin D-resistant triple-negative 4T1 breast cancer. J. Steroid Biochem. Mol. Biol. 2018, 177, 135–139. [Google Scholar] [CrossRef]
- Alimirah, F.; Peng, X.; Gupta, A.; Yuan, L.; Welsh, J.; Cleary, M.; Mehta, R.G. Crosstalk between the vitamin D receptor (VDR) and miR-214 in regulating SuFu, a hedgehog pathway inhibitor in breast cancer cells. Exp. Cell Res. 2016, 349, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Mohri, T.; Nakajima, M.; Takagi, S.; Komagata, S.; Yokoi, T. MicroRNA regulates human vitamin D receptor. Int. J. Cancer 2009, 125, 1328–1333. [Google Scholar] [CrossRef]
- Klopotowska, D.; Matuszyk, J.; Wietrzyk, J. Steroid hormone calcitriol and its analog tacalcitol inhibit miR-125b expression in a human breast cancer MCF-7 cell line. Steroids 2019, 141, 70–75. [Google Scholar] [CrossRef]
- Dastmalchi, N.; Safaralizadeh, R.; Baradaran, B.; Hosseinpourfeizi, M.; Baghbanzadeh, A. An update review of deregulated tumor suppressive microRNAs and their contribution in various molecular subtypes of breast cancer. Gene 2020, 729, 144301. [Google Scholar] [CrossRef] [PubMed]
- Kalecky, K.; Modisette, R.; Pena, S.; Cho, Y.-R.; Taube, J.H. Integrative analysis of breast cancer profiles in TCGA by TNBC subgrouping reveals novel microRNA-specific clusters, including miR-17-92a, distinguishing basal-like 1 and basal-like 2 TNBC subtypes. BMC Cancer 2020, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-Y.; Jiang, Z.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. Molecular stratification within triple-negative breast cancer subtypes. Sci. Rep. 2019, 9, 19107–19110. [Google Scholar] [CrossRef]
- Anfossi, S.; Calin, G.A. When non-coding is not enough. J. Exp. Med. 2020, 217, 20192009. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, S.; Zhu, X.; Zhang, L.; Deng, J.; Li, F.; Guo, B.; Zhang, S.; Wu, R.; Zhang, Z.; et al. LncRNA-encoded polypeptide ASRPS inhibits triple-negative breast cancer angiogenesis. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Fan, H.; Yuan, J.; Li, X.; Ma, Y.; Wang, X.; Xu, B.; Li, X. LncRNA LINC00173 enhances triple-negative breast cancer progression by suppressing miR-490-3p expression. Biomed. Pharmacother. 2020, 125, 109987. [Google Scholar] [CrossRef]
- Han, C.; Fu, Y.; Zeng, N.; Yin, J.; Li, Q. LncRNA FAM83H-AS1 promotes triple-negative breast cancer progression by regulating the miR-136-5p/metadherin axis. Aging 2020, 12, 3594–3616. [Google Scholar] [CrossRef]
- Escrich, E.; Moral, R.; García, G.; Costa, I.; Sánchez-Espigares, J.A.; Solanas, M.; Garcia, M.S. Identification of novel differentially expressed genes by the effect of a high-fat n-6 diet in experimental breast cancer. Mol. Carcinog. 2004, 40, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Oskooei, V.K.; Geranpayeh, L.; Omrnai, M.D.; Ghafouri-Fard, S. Assessment of functional variants and expression of long noncoding RNAs in vitamin D receptor signaling in breast cancer. Cancer Manag. Res. 2018, 10, 3451–3462. [Google Scholar] [CrossRef] [PubMed]
- Oskooei, V.K.; Ghafouri-Fard, S.; Mir, D.O. A Combined Bioinformatics and Literature Based Approach for Identification of Long Non-coding RNAs That Modulate Vitamin D Receptor Signaling in Breast Cancer. Klin. Onkol. 2018, 31, 264–269. [Google Scholar] [CrossRef]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.Z.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef]
- Schneider, B.P.; Winer, E.P.; Foulkes, W.D.; Garber, J.E.; Perou, C.M.; Richardson, A.; Sledge, G.W.; Carey, L.A. Triple-Negative Breast Cancer: Risk Factors to Potential Targets. Clin. Cancer Res. 2008, 14, 8010–8018. [Google Scholar] [CrossRef]
- Muley, H.; Fadó, R.; Rodríguez-Rodríguez, R.; Casals, N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem. Pharmacol. 2020, 177, 113959. [Google Scholar] [CrossRef]
- Jeon, S.-M.; Shin, E.-A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 20. [Google Scholar] [CrossRef]
- Guo, L.-S.; Li, H.-X.; Li, C.-Y.; Zhang, S.-Y.; Chen, J.; Wang, Q.-L.; Gao, J.-M.; Liang, J.-Q.; Gao, M.-T.; Wu, Y.-J. Synergistic antitumor activity of vitamin D3 combined with metformin in human breast carcinoma MDA-MB-231 cells involves m-TOR related signaling pathways. Die Pharm. 2015, 70, 117–122. [Google Scholar]
- Effects of Combined Treatment with Vitamin D and COX2 Inhibitors on Breast Cancer Cell Lines. Anticancer Res. 2018, 38, 1201–1207. [CrossRef]
- Thill, M.; Reichert, K.; Woeste, A.; Polack, S.; Fischer, D.; Hoellen, F.; Rody, A.; Friedrich, M.; Köster, F. Combined treatment of breast cancer cell lines with vitamin D and COX-2 inhibitors. Anticancer Res. 2015, 35, 1189–1195. [Google Scholar] [PubMed]
- Guo, W.; Deng, L.; Yu, J.; Chen, Z.; Woo, Y.; Liu, H.; Li, T.; Lin, T.; Chen, H.; Zhao, M.; et al. Sericin nanomicelles with enhanced cellular uptake and pH-triggered release of doxorubicin reverse cancer drug resistance. Drug Deliv. 2018, 25, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Kutlehria, S.; Behl, G.; Patel, K.; Doddapaneni, R.; Vhora, I.; Chowdhury, N.; Bagde, A.; Singh, M. Cholecalciferol-PEG Conjugate Based Nanomicelles of Doxorubicin for Treatment of Triple-Negative Breast Cancer. AAPS PharmSciTech 2017, 19, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Thakkar, A.; Ergonul, A.; Wang, B.; Woo, T.; Hu, R.; Harrell, J.C.; McNamara, G.; Schwede, M.; Culhane, A.C.; et al. Taxonomy of breast cancer based on normal cell phenotype predicts outcome. J. Clin. Investig. 2014, 124, 859–870. [Google Scholar] [CrossRef]
- Thakkar, A.; Wang, B.; López-Ruiz, E.; Buchwald, P.; Ince, T.A. Vitamin D and androgen receptor-targeted therapy for triple-negative breast cancer. Breast Cancer Res. Treat. 2016, 157, 77–90. [Google Scholar] [CrossRef]
- Abu Samaan, T.M.; Samec, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef]
- Wilhelm, C.A.; Clor, Z.J.; Kelts, J.L. Effect of Vitamin D on Paclitaxel Efficacy in Triple-negative Breast Cancer Cell Lines. Anticancer Res. 2018, 38, 5043–5048. [Google Scholar] [CrossRef]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science (N.Y.) 1994, 266, 66–71. [Google Scholar] [CrossRef]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.E.; Hurvitz, S.A. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context 2018, 7, 212540. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, C.A.; Mountzios, G.; Papadimitriou, C.A. The role of PARP inhibition in triple-negative breast cancer: Unraveling the wide spectrum of synthetic lethality. Cancer Treat. Rev. 2018, 67, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Gombart, A.F.; Kwok, S.H.; Park, S.; Koeffler, H.P. The anti-proliferative effects of 1α,25(OH)2D3 on breast and prostate cancer cells are associated with induction of BRCA1 gene expression. Oncogene 2000, 19, 5091–5097. [Google Scholar] [CrossRef]
- Graziano, S.; Johnston, R.; Deng, O.; Zhang, J.; Gonzalo, S. Vitamin D/vitamin D receptor axis regulates DNA repair during oncogene-induced senescence. Oncogene 2016, 35, 5362–5376. [Google Scholar] [CrossRef]
- Luan, H.; Mohapatra, B.; Bielecki, T.A.; Mushtaq, I.; Mirza, S.; Jennings, T.A.; Clubb, R.J.; An, W.; Ahmed, D.; El Ansari, R.; et al. Loss of the Nuclear Pool of Ubiquitin Ligase CHIP/STUB1 in Breast Cancer Unleashes the MZF1-Cathepsin Pro-oncogenic Program. Cancer Res. 2018, 78, 2524–2535. [Google Scholar] [CrossRef]
- Sudhan, D.; Rabaglino, M.B.; Wood, C.E.; Siemann, D.W. Cathepsin L in tumor angiogenesis and its therapeutic intervention by the small molecule inhibitor KGP. Clin. Exp. Metastasis 2016, 33, 461–473. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R.; Cai, K.; He, H.; Liu, Y.; Yen, J.; Wang, Z.; Xu, M.; Sun, Y.; Zhou, X.; et al. Selective in vivo metabolic cell-labeling-mediated cancer targeting. Nat. Methods 2017, 13, 415–424. [Google Scholar] [CrossRef]
- Goulet, B.; Baruch, A.; Moon, N.-S.; Poirier, M.; Sansregret, L.L.; Erickson, A.; Bogyo, M.; Nepveu, A. A Cathepsin L Isoform that Is Devoid of a Signal Peptide Localizes to the Nucleus in S Phase and Processes the CDP/Cux Transcription Factor. Mol. Cell 2004, 14, 207–219. [Google Scholar] [CrossRef]
- Sullivan, S.; Tosetto, M.; Kevans, D.; Coss, A.; Wang, L.; O’Donoghue, D.; Hyland, J.; Sheahan, K.; Mulcahy, H.; O’Sullivan, J. Localization of nuclear cathepsin L and its association with disease progression and poor outcome in colorectal cancer. Int. J. Cancer 2009, 125, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Burton, L.J.; Hawsawi, O.; Sweeney, J.; Bowen, N.; Hudson, T.; Odero-Marah, V. CCAAT-displacement protein/cut homeobox transcription factor (CUX1) represses estrogen receptor-alpha (ER-α) in triple-negative breast cancer cells and can be antagonized by muscadine grape skin extract (MSKE). PLoS ONE 2019, 14, e0214844. [Google Scholar] [CrossRef] [PubMed]
- Zámborszky, J.; Szikriszt, B.; Gervai, J.Z.; Pipek, O.; Póti, Á.; Krzystanek, M.; Ribli, D.; Szalai-Gindl, J.M.; Csabai, I.; Szallasi, Z.; et al. Erratum: Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 2017, 36, 5085–5086. [Google Scholar] [CrossRef]
- Zámborszky, J.; Szikriszt, B.; Gervai, J.Z.; Pipek, O.; Poti, A.; Krzystanek, M.; Ribli, D.; Szalai-Gindl, J.M.; Csabai, I.; Szallasi, Z.; et al. Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 2016, 36, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.; Ganesan, S. BRCA1, PARP, and 53BP1: Conditional synthetic lethality and synthetic viability. J. Mol. Cell Biol. 2011, 3, 66–74. [Google Scholar] [CrossRef]
- Schwarz, B.; Friedl, A.A.; Girst, S.; Dollinger, G.; Reindl, J. Nanoscopic analysis of 53BP1, BRCA1 and Rad51 reveals new insights in temporal progression of DNA-repair and pathway choice. Mutat. Res. Mol. Mech. Mutagen. 2019, 111675. [Google Scholar] [CrossRef]
- Squatrito, M.; Vanoli, F.; Schultz, N.; Jasin, M.; Holland, E.C. 53BP1 is a haploinsufficient tumor suppressor and protects cells from radiation response in glioma. Cancer Res. 2012, 72, 5250–5260. [Google Scholar] [CrossRef]
- Gonzalez-Suarez, I.; Redwood, A.B.; Grotsky, D.A.; Neumann, M.A.; Cheng, E.H.-Y.; Stewart, C.L.; Dusso, A.; Gonzalo, S. A new pathway that regulates 53BP1 stability implicates Cathepsin L and vitamin D in DNA repair. EMBO J. 2011, 30, 3383–3396. [Google Scholar] [CrossRef]
- Alvarez-Diaz, S.; Valle, N.; García, J.M.; Pena, C.; Freije, J.M.P.; Quesada, V.; Astudillo, A.; Bonilla, F.; Lopez-Otin, C.; Muñoz, A. Cystatin D is a candidate tumor suppressor gene induced by vitamin D in human colon cancer cells. J. Clin. Investig. 2009, 119, 2343–2358. [Google Scholar] [CrossRef]
- Redwood, A.B.; Perkins, S.M.; VanderWaal, R.P.; Feng, Z.; Biehl, K.J.; Gonzalez-Suarez, I.; Morgado-Palacin, L.; Shi, W.; Sage, J.; Roti-Roti, J.L.; et al. A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle 2011, 10, 2549–2560. [Google Scholar] [CrossRef]
- Gonzalo, S. Novel roles of 1α,25(OH)2D3 on DNA repair provide new strategies for breast cancer treatment. J. Steroid Biochem. Mol. Biol. 2013, 144, 59–64. [Google Scholar] [CrossRef]
- Grotsky, D.A.; Gonzalez-Suarez, I.; Novell, A.; Neumann, M.A.; Yaddanapudi, S.C.; Croke, M.; Martinez-Alonso, M.; Redwood, A.B.; Ortega-Martinez, S.; Feng, Z.; et al. BRCA1 loss activates cathepsin L–mediated degradation of 53BP1 in breast cancer cells. J. Cell Biol. 2013, 200, 187–202. [Google Scholar] [CrossRef]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Physiol. 2005, 289, F8–F28. [Google Scholar] [CrossRef]
- Redwood, A.B.; Gonzalez-Suarez, I.; Gonzalo, S. Regulating the levels of key factors in cell cycle and DNA repair. Cell Cycle 2011, 10, 3652–3657. [Google Scholar] [CrossRef]
- Heublein, S.; Mayr, D.; Meindl, A.; Kircher, A.; Jeschke, U.; Ditsch, N. Vitamin D receptor, Retinoid X receptor and peroxisome proliferator-activated receptor γ are overexpressed in BRCA1 mutated breast cancer and predict prognosis. J. Exp. Clin. Cancer Res. 2017, 36, 57. [Google Scholar] [CrossRef][Green Version]
- Wingert, S.; Rieger, M.A. Terminal differentiation induction as DNA damage response in hematopoietic stem cells by GADD45A. Exp. Hematol. 2016, 44, 561–566. [Google Scholar] [CrossRef]
- Tront, J.S.; Hoffman, B.; Liebermann, D.A. Gadd45a Suppresses Ras-Driven Mammary Tumorigenesis by Activation of c-Jun NH2-Terminal Kinase and p38 Stress Signaling Resulting in Apoptosis and Senescence. Cancer Res. 2006, 66, 8448–8454. [Google Scholar] [CrossRef]
- Tront, J.S.; Willis, A.I.; Huang, Y.; Hoffman, B.; Liebermann, D.A. Gadd45a levels in human breast cancer are hormone receptor dependent. J. Transl. Med. 2013, 11, 131. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Long, F.; Yan, F.; Wang, N.; Wang, Y. The expression and clinical significance of GADD45A in breast cancer patients. PeerJ 2018, 6, e5344. [Google Scholar] [CrossRef]
- Flores, O.; Burnstein, K.L. GADD45gamma: A new vitamin D-regulated gene that is antiproliferative in prostate cancer cells. Endocrinology 2010, 151, 4654–4664. [Google Scholar] [CrossRef] [PubMed]
- Bremmer, F.; Thelen, P.; Pottek, T.; Behnes, C.L.; Radzun, H.J.; Schweyer, S. Expression and function of the vitamin D receptor in malignant germ cell tumour of the testis. Anticancer Res. 2012, 32, 341–349. [Google Scholar]
- Galbiati, F.; Polastri, L.; Thorens, B.; Dupraz, P.; Fiorina, P.; Cavallaro, U.; Christofori, G.; Davalli, A.M. Molecular Pathways Involved in the Antineoplastic Effects of Calcitriol on Insulinoma Cells. Endocrinology 2003, 144, 1832–1841. [Google Scholar] [CrossRef]
- Jiang, F.; Li, P.; Fornace, A.J.; Nicosia, S.V.; Bai, W. G2/M Arrest by 1,25-Dihydroxyvitamin D3in Ovarian Cancer Cells Mediated through the Induction ofGADD45via an Exonic Enhancer. J. Biol. Chem. 2003, 278, 48030–48040. [Google Scholar] [CrossRef]
- Prudencio, J.; Akutsu, N.; Benlimame, N.; Wang, T.; Bastien, Y.; Lin, R.; Black, M.J.; Alaoui-Jamali, M.A.; White, J.H. Action of Low Calcemic 1,25-Dihydroxyvitamin D3 Analogue EB1089 in Head and Neck Squamous Cell Carcinoma. J. Natl. Cancer Inst. 2001, 93, 745–753. [Google Scholar] [CrossRef]
- Sidhu, P.S.; Teske, K.A.; Feleke, B.; Yuan, N.Y.; Guthrie, M.L.; Fernstrum, G.B.; Vyas, N.D.; Han, L.; Preston, J.; Bogart, J.W.; et al. Anticancer activity of VDR-coregulator inhibitor PS. Cancer Chemother. Pharmacol. 2014, 74, 787–798. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, F.; Li, P.; Li, C.; Ma, Q.; Nicosia, S.V.; Bai, W. Growth suppression of ovarian cancer xenografts in nude mice by vitamin D analogue EB1089. Clin. Cancer Res. 2005, 11, 323–328. [Google Scholar]
- Pietrasik, S.; Zajac, G.; Morawiec, J.; Soszynski, M.; Fila, M.; Blasiak, J. Interplay between BRCA1 and GADD45A and Its Potential for Nucleotide Excision Repair in Breast Cancer Pathogenesis. Int. J. Mol. Sci. 2020, 21, 870. [Google Scholar] [CrossRef]
- Razzaque, M.S. Can adverse effects of excessive vitamin D supplementation occur without developing hypervitaminosis D? J. Steroid Biochem. Mol. Biol. 2018, 180, 81–86. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Fernández, E. Vitamin D receptor polymorphisms and diseases. Clin. Chim. Acta 2006, 371, 1–12. [Google Scholar] [CrossRef]
- Li, J.; Li, B.; Jiang, Q.; Zhang, Y.-S.; Liu, A.; Wang, H.; Zhang, J.; Qin, Q.; Hong, Z. Do genetic polymorphisms of the vitamin D receptor contribute to breast/ovarian cancer? A systematic review and network meta-analysis. Gene 2018, 677, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Jing, L.; Zhang, S. Vitamin D Receptor Polymorphism and Breast Cancer Risk. Medicine 2016, 95, e3535. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Aspeslagh, S.; Garaud, S.; Dupont, F.; Solinas, C.; Kok, M.; Routy, B.; Sotiriou, C.; Stagg, J.; Buisseret, L. Immuno-oncology-101: Overview of major concepts and translational perspectives. Semin. Cancer Biol. 2018, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chirumbolo, S.; Bjørklund, G.; Sboarina, A.; Vella, A. The Role of Vitamin D in the Immune System as a Pro-survival Molecule. Clin. Ther. 2017, 39, 894–916. [Google Scholar] [CrossRef]
- Altieri, B.; Muscogiuri, G.; Barrea, L.; Mathieu, C.; Vallone, C.V.; Mascitelli, L.; Bizzaro, G.; Altieri, V.M.; Tirabassi, G.; Balercia, G.; et al. Does vitamin D play a role in autoimmune endocrine disorders? A proof of concept. Rev. Endocr. Metab. Disord. 2017, 18, 335–346. [Google Scholar] [CrossRef]
- Eppensteiner, J.; Davis, R.P.; Barbas, A.S.; Kwun, J.; Lee, J. Immunothrombotic Activity of Damage-Associated Molecular Patterns and Extracellular Vesicles in Secondary Organ Failure Induced by Trauma and Sterile Insults. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blasiak, J.; Pawlowska, E.; Chojnacki, J.; Szczepanska, J.; Fila, M.; Chojnacki, C. Vitamin D in Triple-Negative and BRCA1-Deficient Breast Cancer—Implications for Pathogenesis and Therapy. Int. J. Mol. Sci. 2020, 21, 3670. https://doi.org/10.3390/ijms21103670
Blasiak J, Pawlowska E, Chojnacki J, Szczepanska J, Fila M, Chojnacki C. Vitamin D in Triple-Negative and BRCA1-Deficient Breast Cancer—Implications for Pathogenesis and Therapy. International Journal of Molecular Sciences. 2020; 21(10):3670. https://doi.org/10.3390/ijms21103670
Chicago/Turabian StyleBlasiak, Janusz, Elzbieta Pawlowska, Jan Chojnacki, Joanna Szczepanska, Michal Fila, and Cezary Chojnacki. 2020. "Vitamin D in Triple-Negative and BRCA1-Deficient Breast Cancer—Implications for Pathogenesis and Therapy" International Journal of Molecular Sciences 21, no. 10: 3670. https://doi.org/10.3390/ijms21103670
APA StyleBlasiak, J., Pawlowska, E., Chojnacki, J., Szczepanska, J., Fila, M., & Chojnacki, C. (2020). Vitamin D in Triple-Negative and BRCA1-Deficient Breast Cancer—Implications for Pathogenesis and Therapy. International Journal of Molecular Sciences, 21(10), 3670. https://doi.org/10.3390/ijms21103670