Abstract
Amyotrophic lateral sclerosis (ALS) is the most common and devastating motor neuron (MN) disease. Its pathophysiological cascade is still enigmatic. More than 90% of ALS patients suffer from sporadic ALS, which makes it specifically demanding to generate appropriate model systems. One interesting aspect considering the seeding, spreading and further disease development of ALS is the cerebrospinal fluid (CSF). We therefore asked whether CSF from sporadic ALS patients is capable of causing disease typical changes in human patient-derived spinal MN cultures and thus could represent a novel model system for sporadic ALS. By using induced pluripotent stem cell (iPSC)-derived MNs from healthy controls and monogenetic forms of ALS we could demonstrate a harmful effect of ALS-CSF on healthy donor-derived human MNs. Golgi fragmentation—a typical finding in lower organism models and human postmortem tissue—was induced solely by addition of ALS-CSF, but not control-CSF. No other neurodegenerative hallmarks—including pathological protein aggregation—were found, underpinning Golgi fragmentation as early event in the neurodegenerative cascade. Of note, these changes occurred predominantly in MNs, the cell type primarily affected in ALS. We thus present a novel way to model early features of sporadic ALS.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is the most common and devastating motor neuron (MN) disease. Its initial cause and the mechanisms of further progression are still enigmatic nowadays. The cerebrospinal fluid (CSF) seems to play a major role within the deathly cascade, which is particularly true concerning pathological protein aggregates and their spreading across the central nervous system [1,2].
Several studies have demonstrated diverse pathological effects of ALS-CSF in different in vivo and in vitro models [3,4,5,6,7]. These models mainly include primary murine cortical or spinal MN cultures and spinal cord neuroblastoma hybrid cell line NSC34 cultures. A multitude of phenotypes could be observed by this including aggregation of transactive response DNA binding protein 43 kDA (TDP-43) in the case of ALS-frontotemporal dementia (FTLD)-CSF [8], neurofilament abnormalities [9], gliosis [10], endoplasmic reticulum (ER)-stress [6], mitochondrial dysfunction [11] and Golgi fragmentation [6,12]. In addition, intrathecal infusion of ALS-CSF in an in vivo rat model mimicked neuropathological changes similar to the ones found in postmortem tissue of ALS patients [4]. Furthermore, interesting is the fact that not only CSF but also extracellular mutant superoxide dismutase 1 (SOD1) itself was able to induce Golgi fragmentation and the inhibition of ER-Golgi trafficking [13]. The term Golgi fragmentation describes a loss of the intactness of this cisternal structure and its dispersion across the cell [14,15]. However, nothing is known about how ALS-CSF affects human patient-derived induced pluripotent stem cell (iPSC)-derived MNs, currently believed to be the gold standard for in vitro disease modeling of ALS. Additionally, effects on typical ALS aggregating proteins are barely reported.
Golgi fragmentation in MNs themselves has been reported in different in vitro and in vivo ALS models [16,17]; reviewed by Sundaramoorthy and colleagues [15]. It is also present in postmortem sections of ALS patients [18,19,20,21,22,23,24]. Moreover, Golgi fragmentation is reported in several other neurodegenerative diseases like Alzheimer’s disease and Parkinson’s disease [25,26,27,28]. There is convincing evidence that Golgi fragmentation is an early event in the disease process [23,26,29,30] and not only a consequence of apoptosis [30] but rather inducing it [26]. Thus, similar to other neurodegenerative disease conditions [31], Golgi fragmentation in ALS could be an event that takes place before seeding of the aggregates, axon degeneration and manifestation of clinical symptoms occurs [30,32,33].
The main goal of our study was to establish a new model system for sporadic ALS by investigating the ability of CSF derived from sporadic ALS patients to induce motor neuronal degeneration in the current gold standard cell culture model of ALS, namely patient specific iPSC-derived MNs.
2. Results
2.1. Patient Characteristics
We used human iPSC-derived neural progenitor cells (NPC) from healthy subjects [34,35,36,37,38]. These cell lines were genotyped for most common monogenetic causes of ALS (namely C9ORF72, SOD1, FUS, TARDBP) excluding presymptomatic donors (female, 50 years; female, 45 years; male, 60 years; female, 48 years). Furthermore, we analyzed the effects on monogenetic ALS cell lines (SOD1-p.R115G, male, 59 years; FUS-p.R521C, female, 58 years; TDP-43p.S393L, female, 87 years). CSF was collected from 19 patients. The ALS group consisted of 11 patients (3 women and 8 men) with a mean age of 63 (SD = 12.2) years. The group of 8 control patients (3 women and 5 men) had a mean age of 47 (SD = 21.4) years (See materials and methods Table 1 and Table 2 for further information).

Table 1.
Laboratory values of the used CSF and patient characteristics; data are depicted as mean (SD).
Table 2.
Patient/proband characteristics.
2.2. CSF Affects Proliferation of NPCs
One important observation was that CSF, irrespective of whether from ALS patients or healthy controls, caused a significant proliferation of remaining NPCs in our neuronal cell culture in a clear dose dependent manner, correlating with the amount of CSF and length of exposition. As shown in Figure 1, increasing amount of CSF (10%, 20% and 50%) dramatically increased NPC (GFAP−, Vimentin+) numbers, while not affecting neuronal morphology. Prolonged incubation time additionally increased non neuronal cell numbers. This effect was still obvious, albeit to a lesser extent, when cells were allowed to differentiate for 10 days prior to being incubated with CSF. Therefore, all further experiments were performed under the latter condition (Figure 1a). To avoid that the effects of CSF are mainly due to lack dilution of standard medium/growth factors we diluted the medium of untreated MNs with PBS and further varied the concentration of the added factors (GDNF, BDNF, cAMP) in the CSF blended medium. However, we found no significant influence of this. Since we used this long-term culture protocol with significant amount of CSF needed, we had to pool the CSF samples from controls and ALS patients in all following experiments focusing on group differences.
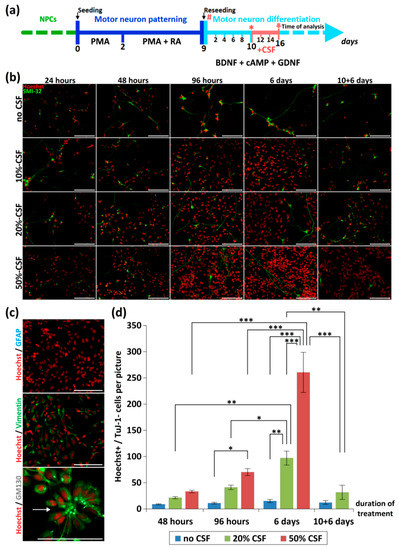
Figure 1.
Effect of CSF-application on neurons and NPCs. (a) Depicted is the differentiation and treatment scheme. During the initial dose finding experiments, the time point of CSF-application was varied (# and *). For the final experiments CSF was applied 10 days after reseeding (*) and cells were analyzed after 6 days of CSF treatment (red arrow). (b) Different concentrations of CSF (0%, 10%, 20%, 50%) were applied to human iPSC-derived MN-cell cultures directly after seeding for 24 h, 48 h, 96 h and 6 days. Additionally, we included a scenario in which we initiated treatment after 10 days of differentiation, followed by 6 days CSF treatment (10 + 6 days). Depending on the duration and concentration of CSF-incubation, a proliferation of NPCs could be seen. (c) This effect was independent from the CSF-origin (data not shown) and led to a dramatic overgrowth of GFAP−/Vimentin+ NPCs, which partially formed neural rosettes (arrow). (d) Quantification of non-neuronal cells (Hoechst+/Tuj-1−) during CSF treatment. */**/*** represents p < 0.05/0.01/0.001 as calculated by two-way ANOVA with Bonferroni post-hoc test). Scale bar = 100 µm.
2.3. No Neuronal Loss or Neuronal Network Degeneration by ALS-CSF
We found a significant influence of CSF treatment itself on the number of neurons relative to total cells (TuJ-1/Hoechst) in control cultures, but no ALS-CSF specific effect. In detail, the amount of neuronal cells treated with either ALS- or control-CSF was significantly decreased compared to non-treated cells (44.95% vs. 69.18%, p < 0.05; 45.33% vs. 69.18%, p < 0.05; one-way ANOVA with Tukey post-hoc test; Figure 2a). However, no significant difference was found in the comparison of cells treated with ALS-CSF and those exposed to CSF from controls (44.95% vs. 45.33%, p = 0.9990 by one-way ANOVA with Tukey post-hoc test). Furthermore, the control cell line showed neither significant difference in the number of MNs relative to total cells (MN/Hoechst, Figure 2b) nor in the number of MNs relative to total neurons (MN/TuJ-1, Figure 2c), when exposed to either control-CSF or ALS-CSF.
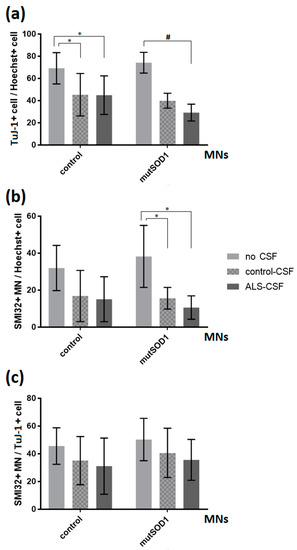
Figure 2.
Effects of ALS-CSF on the relations of neuronal cells in control- and mutant SOD1-MNs. (a) Left: A significant decrease in the relations of TuJ-1+cell/Hoechst when treated with either control- (* p < 0.05 by one-way ANOVA with Tukey post-hoc test) or ALS-CSF (* p < 0.05 by one-way ANOVA with Tukey post-hoc test), but no significant effect comparing respective CSF conditions. Right: The mutant SOD1-cell line exhibited a significant decrease in the relations of TuJ-1+cell/Hoechst when exposed to ALS-CSF compared to the no CSF condition (# = Kruskal-Wallis test H(2) = 7.636, p < 0.05, z = 2.763, p < 0.05 with Dunn-Bonferroni post-hoc test), but again no significant effect comparing control- with ALS-CSF (29.21% vs. 39.91%, z = 1.279, p = 0.602 with Dunn-Bonferroni post-hoc test). (b) Left: The application of CSF had no significant effect in either cell line analyzing the relation of SMI32+MN/Hoechst in the control-cell line. Right: In SOD1 mutant cells, we detected a significant decrease in the amount of total MNs in relation to all cells (MN/Hoechst) comparing either ALS- or control-CSF-treatment condition and non-treated cells (* p < 0.05 by one-way ANOVA with Tukey post-test). (c) The application of CSF had no significant effect in either cell line analyzing the relation SMI32+MN/TuJ-1+cell. Data are depicted as mean ± SEM.
When analyzing a monogenetic ALS cell line, namely the SOD-1-mutant cell line (p.R115G), we saw a significant decrease in the number of MNs relative to total cells (MN/Hoechst) when comparing ALS- or control-CSF-treatment to non-treated cells (10.59% vs. 38.24%, p < 0.05; 15.59% vs. 38.24%, p < 0.05; one-way ANOVA with Tukey post-hoc test) (Figure 2b). Of note, there was no difference between ALS-CSF treatment and control-CSF exposition in this cell line. Furthermore, the treatment with CSF showed a significant influence on the relation of total neurons (TuJ1/Hoechst) (Kruskal-Wallis test H(2) = 7.636, p < 0.05; Figure 2a). We could detect a significant decrease comparing ALS- and no CSF treatment condition (29.21% vs. 74.33%, z = 2.763, p < 0.05 with Dunn-Bonferroni post-hoc test), but no difference between ALS- and control-CSF exposition (29.21% vs. 39.91%, z = 1.279, p = 0.602 with Dunn-Bonferroni post-hoc test).
There was no overt visible difference in the neuronal network comparing cells treated with control-CSF or ALS-CSF (Figure 3). We therefore analyzed the total size of the SMI-32 and TuJ-1 network (Figure 3b) and calculated the network degeneration (Figure 3c). There was no significant difference between either CSF treated condition and, furthermore, no difference when comparing them with the no CSF treatment, neither in the control- or in the mutant-SOD1 (p.R115G) cell line. We could not detect a significant distinction between the control- and mutant-SOD1 (p.R115G) cell line, either.
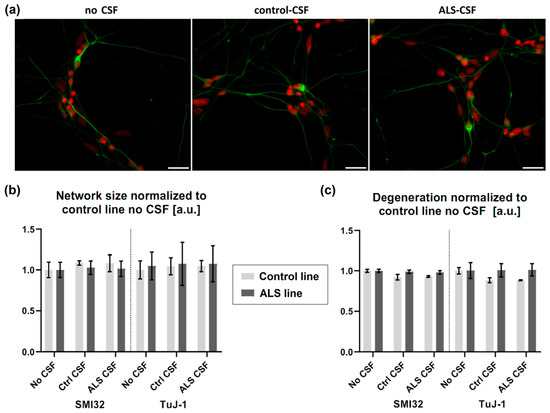
Figure 3.
No signs of structural degeneration. (a) There was no differing degeneration of the neuronal network (green = SMI32, red = Hoechst) detectable in either cell line treated with CSF. (b) There was no significant effect of ALS-CSF on neither the neuronal network size, (c) nor network degeneration. Scale bar = 50 µm.
2.4. No Signs of Pathological Aggregate Formation by ALS-CSF
Intrathecal infusion of ALS-CSF in an in vivo rat model caused pathological abnormalities similar to those found in the postmortem sections of ALS patients, namely accumulation of cytoplasmic TDP-43, indicating a spread of the disease via CSF [4]. Further investigations showed the induction of TDP-43 aggregation in the case of ALS-FTD-CSF using a glioblastoma cell model [8]. We therefore asked whether ALS-CSF was able to induce aggregate formation in healthy donor-derived MNs. Following our treatment protocol, there were no signs of significant aggregates containing disease relevant proteins such as TDP-43, fused in sarcoma (FUS), or SOD-1. Additionally, no relevant cytoplasmic mislocalization of TDP-43 and/or FUS was detected in iPSC-derived neurons (Figure 4a–c). We next asked whether ALS-CSF is able to seed aggregation in patient-derived MNs expressing mutations in ALS aggregating proteins, namely TDP43, SOD1 and FUS mutant. Interestingly, by using SOD1 p.R115G (Figure 4d–f), TDP-43 p.S393L (data not shown) and FUS p.R521C cell lines (Figure 4g–i), we could detect neither aggregation of any disease relevant protein nor cytoplasmic mislocalization of FUS and/or TDP-43 (Figure 4).
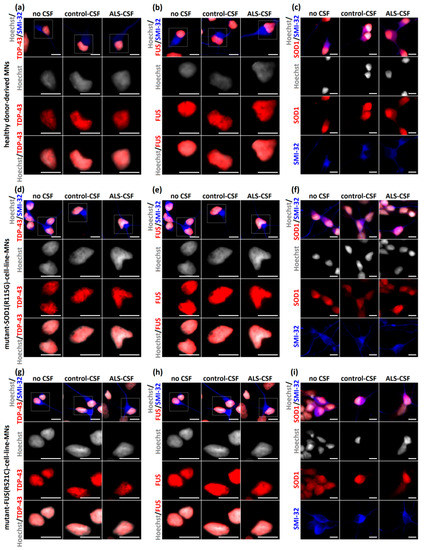
Figure 4.
No aggregation of disease-relevant proteins after exposition to ALS-CSF. (a–c) ALS-CSF caused no detectable aggregates of TDP-43 (a), FUS (b), or SOD1 (c) when applied to healthy donor-derived MNs. Furthermore, there were no differences of cytoplasmic or nuclear localization of any disease relevant protein and—importantly—no cytoplasmic mislocalization of TDP-43 and FUS, respectively. (d–f) Similar results were detected after applying CSF to the mutant-SOD1-(p.R115G)- and (g–i) mutant-FUS-(p.R521C)-cell-line MNs. Scale bar = 10 µm.
2.5. ALS-CSF Induces Golgi Fragmentation in Patient-Derived Motor Neurons
Golgi fragmentation has been reported in different ALS-models and postmortem sections of ALS patients [18,19,22,23] reviewed by Sundaramoorthy and colleagues [15]. In addition, Golgi fragmentation has been extensively described as an early event in the neurodegenerative cascade [16,17,25,26,27,28]. We therefore investigated whether the addition of patient-CSF is sufficient to cause this early hallmark in healthy donor-derived iPSC-derived motor neurons. After CSF exposition, we classified the Golgi apparatus depending on its structure in four different categories (Figure 5a). As further shown in Figure 5, the application of ALS-CSF to healthy donor-derived MNs caused a significant higher rate of Golgi fragmentation in MNs when compared to cells treated with control-CSF (22.99% vs. 2.35%, p < 0.001 by one-way ANOVA with Tukey post-hoc test) or without CSF (22.99% vs. 2.21%, p < 0.001 by one-way ANOVA with Tukey post-hoc test). Control-CSF had no effect when compared to the non-treated condition (2.35% vs. 2.21%, p = 1.00 by one-way ANOVA with Tukey post-hoc test).
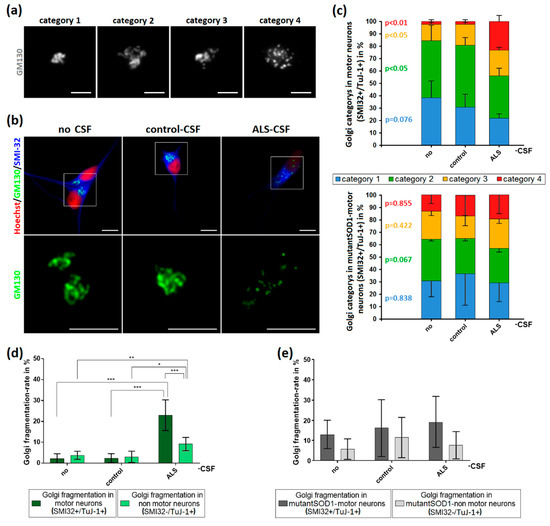
Figure 5.
Induction of Golgi fragmentation predominantly in MNs by the application of ALS-CSF. (a) For analysis, Golgi complexes were categorized depending on their structure. Category 1 was defined as small, compact Golgi apparatus. Single cisterns were difficult to differentiate. In contrast, category 2 included bigger “loosened” Golgi complexes. Cisterns were easier to differentiate. Unlike in the previous ones, category 3 Golgi complexes were not closed compartments anymore. They still formed a cisternal structure, but it was possible to differentiate small cisterns/vesicles not being in contact with the main part of the Golgi. The Golgi apparatus in category 4 was fragmented and only this category accounted for the fragmented ones in our statistics in (d,e). It lost its cohesion and cisternal structure thus became vesicular and the small compartments were dispersed across the cell. (b) The structural Golgi changes in control-human iPSC-induced MNs after ALS-CSF application comprised the loss of cisternal configuration and overall Golgi integrity. (c) The diagrams depict the distribution of the respective Golgi categories in control- (upper diagram) and mutant-SOD1-MNs (lower diagram) ± CSF treatment. (d) The exposition to ALS-CSF caused a significantly higher rate of Golgi fragmentation in WT-human iPSC-induced MNs compared to MNs treated with control-CSF or without CSF. Similar results could be observed comparing SMI32−/TuJ-1+-non MNs treated with ALS-CSF to control-CSF or no CSF-. Of note, SMI32+-MNs exposed to ALS-CSF had a significantly higher Golgi fragmentation rate compared to SMI32−/TuJ-1+-non MNs. (e) The application of ALS-CSF to mutant-SOD1 cells caused no significant rate of Golgi fragmentation neither in SMI32+-MNs nor SMI32-/TuJ-1+-non MNs. Data are depicted as mean ± SEM. */**/** p < 0.05/0.01/0.001 by one-way ANOVA with Tukey post-hoc test. Scale bar = 5 µm.
Similar results could be seen in the Golgi fragmentation rates of non-MN-neurons (SMI32−/ TuJ-1+), albeit to a much smaller extent compared to MNs (22.99% vs. 9.15%, p < 0.001 by two-way ANOVA with Bonferroni post-hoc test), indicating a higher vulnerability of MNs to ALS-CSF.
In contrast, there was no significant increase of the Golgi fragmentation rates when ALS-CSF was applied to the mutant SOD1 cell line. Of note, the fragmentation rate of not treated mutant SOD-1 neurons was remarkably higher compared to the control cell line.
3. Discussion
There is evidence that CSF from ALS patients can directly harm neurons and induce changes associated with ALS pathophysiology. However, data does not exist about such effects on human patient-derived MNs, nor have the effects on modern molecular markers of ALS been well investigated. By using iPSC-derived MNs from healthy controls and monogenetic forms of ALS we demonstrate a harmful effect of CSF from patients suffering from sporadic ALS on healthy donor-derived human MNs. Golgi fragmentation—a typical finding in lower organism ALS models and human post mortem tissue of ALS patients—was induced solely by the addition of ALS- but not control-CSF. Strikingly, these changes occurred predominantly in MNs, the cell type primarily affected in ALS [39,40]. Thus, the presented system might serve as valuable human MN model system for sporadic ALS (Figure 6). Interestingly, this was not obvious in SOD1 p.R115G mutant MNs most likely due to already slightly increased fragmentation rates in the SOD1 mutant MNs. It might also point towards genotype specific effects, which, however, needs to be proven in future systematic studies across a variety of ALS genes and different mutations.
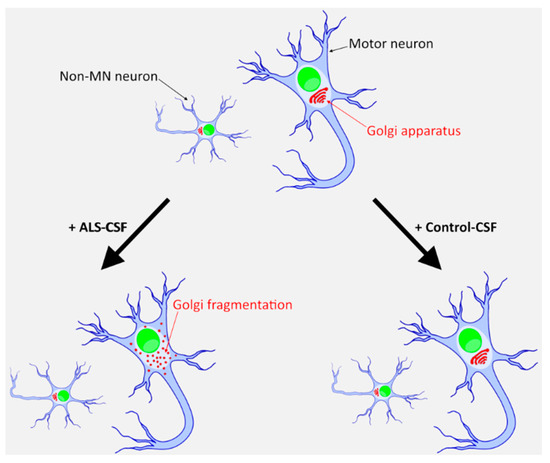
Figure 6.
An iPSC-derived neuronal system to model sporadic ALS. Only ALS-CSF, and not healthy donor CSF, induces a degenerative phenotype in healthy donor derived iPSCs. This effect is mainly seen in MNs and only to a much lesser extent in other neuronal subtypes.
In our study, we did not observe significant neuronal network degeneration, MN-loss or protein aggregates containing SOD1, FUS or TDP-43 (Figure 3 and Figure 4. Indeed, there is a large debate as to whether protein aggregations can be detected in iPSC-derived motor neurons [37], but there is at least clear evidence that it is found in FUS-ALS models [34,35,41]. Thus, even though it might not be surprising that we could not induce any kind of cytoplasmic aggregation of TDP-43 and/or SOD1, it was also not visible in the FUS-ALS model. Another explanation might be the protocol used in the study, which is known to induce structural neurodegeneration only at later stages [35]. Since Golgi fragmentation is expected to be an early event in the pathophysiological cascade [23,26,29,30], it is plausible that it occurs prior to further changes, which could develop either later or under additional stress conditions [34,35,36,37]. Furthermore, ALS-CSF might not be “toxic enough” to induce the complete spectrum of pathological changes seen in postmortem tissue, which is particularly true for inducing pathological aggregates via CSF [8]. Concentration of disease relevant proteins and/or aggregates in patient-derived CSF might be too low to act as agent to induce further aggregation (seeding). This would fit with a recent study by Ding and colleagues using CSF from ALS and additionally ALS-FTLD patients in which only ALS-FTLD-CSF was able to induce TDP-43 aggregates in target cells. It is well known that ALS-FTLD patients have a much more widespread TDP-43 pathology compared to ALS patients without co-occurrence of FTLD [42]. The main differences from our study were the use of a glioblastoma cell line (U251) with obvious differences in general cell metabolism and a longer incubation time with CSF (21 days vs. 6 days). Of note, we additionally did not observe pathological protein mislocalization or aggregate formation when treating monogenetic ALS-patient-derived MNs with ALS-CSF (Figure 4), which are known to have a higher propensity to serve as seeding acceptors [43]. Furthermore, the seeding ability of the mutant protein also differs significantly between different mutations [1,41,44,45,46], which might explain why we did not find seeding events in patient-derived MNs. Since we modelled spinal MNs, we cannot exclude that this might differ in cortical neuronal subtypes. Finally, the employed methods might not have been able to visualize smaller aggregates or oligomers of misfolded proteins. However, we intended to analyze the samples as similar as possible to human postmortem sections in which cytoplasmic protein localization and aggregation is the hallmark pathology.
We could not observe a significant reduction of MNs or neurons in general by exposition to ALS-CSF compared to control-CSF, when healthy donor-derived MNs were treated. Several reasons could explain this difference from results from murine primary cortical/spinal motor neuron cultures [3,47], including human vs. murine cell culture system, medium composition or preanalytic CSF preparation. Another major concern regards the cellular age. Because iPSC-derived neurons do resemble fetal neurons and are not as old as the ones normally affected in ALS patients. Even though all primary neuron cell culture models are derived from fetal brains as well—and thus this cannot be the sole reason for the differences—human development takes much longer, and hence neuronal maturation affects phenotype appearance more severely especially in stem cell derived models [34,35]. The reduction of neurons and MNs by exposition to control CSF with no differences to ALS-CSF (Figure 2a,b) indicated a disease-independent effect on human iPSC-derived neurons. This finding can likely be caused by a reduction of specific medium components in case of CSF-concentrations above 10%. Interestingly, this seems to be similar in adult human neural precursor cells in which CSF promotes stemness [48]. Our data point more towards a decreased relative amount of MNs and neurons compared to total cells due to a proliferative effect on NPCs rather than neuronal cell death.
Our study has some limitations. First, the amount, duration and time point of CSF treatment was limited due to the significant proliferation of NPCs induced by CSF and thus an overgrowth after longer incubation times. This effect was independent of the CSF-type. One additional limitation of our study is the use of pooled CSF. However, this was necessary due to the long time culture conditions and the protocol for human iPSC-derived MNs with the need for medium change every other day (Figure 1). Thus, we cannot answer the question as to whether single ALS patient-derived CSF might vary depending on clinical parameters such as ALS subtype, disease stage or even family background [47]. However, the study rationale was to investigate whether CSF from sporadic ALS patients could induce an MN-like degeneration in the dish, and thus we intended to analyze group differences rather than ALS subtype or individual patient’s specific differences. Furthermore, the model system presented represents one aspect of ALS (namely a model for spinal cord degeneration), and thus there might be differences when analyzing other neuronal cell populations affected, e.g., cortical neurons. The control group were not healthy controls but consisted of patients suffering from non-inflammatory, non-neurodegenerative neurological disorders (e.g., headache), who underwent CSF puncture during clinical routine conditions. However, CSF samples depicting signs of inflammation and/or classical neuropathological markers were excluded (see Table 1). Additionally, we used the marker SMI-32 for the staining of MNs. However, as described in the methods section, following this particular protocol for MN differentiation, all SMI-32 stained MNs were additionally positive for specific motor neuronal markers including Hb9 (Homeobox gene 9), Cholinacetyltransferase (ChAt) and ISLET1 (ISL LH4 homeobox 1) [34,35]. We clearly show, for the first time, that sporadic ALS-patient-derived CSF is sufficient to induce typical signs of neurodegeneration (Golgi fragmentation) and that human iPSC-derived motor neurons, the predominantly affected cell type in ALS, seem to be specifically vulnerable.
4. Materials and Methods
4.1. Patient Material
We included patient material for (i) CSF derivation and (ii) cell derivation.
Add (i): CSF was taken from 11 ALS patients and 8 control subjects (who suffered from non-inflammatory, non-neurodegenerative neurological disorders) under clinical routine conditions and independent of this study. The use of CSF was approved by the local Ethics committee (EK393122012, EK 49022016). CSF was immediately centrifuged (2100 rpm at room temperature for 10 min) and supernatant was stored at −80 °C until use. We included patients with definite, probable or possible amyotrophic lateral sclerosis, according to the revised El Escorial criteria [49]. Patients suffering from genetically proven spinal muscular atrophy, spinal bulbar muscular atrophy, genetic ALS and FTD overlap syndromes were excluded. Details on patients and CSF characteristics are shown in Table 1.
Add (ii): We included cell lines carrying FUS (p.R521C), TDP-43 (p.S393L), SOD1 (p.R115G) mutations and systematically compared them to four control iPSC lines from healthy volunteers (Table 2). All procedures were in accordance with the Helsinki convention and approved by the local Ethical Committee of the Technische Universität Dresden, Germany (EK45022009, approved 21 April 2009; EK393122012 approved 11 March 2013). Patients as well as controls gave their written informed consent prior to skin biopsy.
4.2. Generation and Expansion of iPSCs, in Vitro Differentiation of Embryoid Bodies, AP Staining and Immunofluorescence on iPSC Colonies and Derivation of iPSC-Derived Neuroprecursor Cells
These procedures were performed as previously described by Nauman et al., Japtok et al., and Kreiter et al. [34,35,37].
4.3. Motor Neuron Differentiation
The basal medium (N2B27) for the final MN differentiation consisted of 48.75% DMEM/F12 (Invitrogen, Carlsbad, CA, USA), 48.75% Neurobasal medium (Invitrogen, Carlsbad, CA, USA), 1% penicillin/streptomycin/glutamine (Thermo Fisher Scientific, Waltham, MA, USA), 1% B27 supplement without vitamin A (Invitrogen, Carlsbad, CA, USA) and 0.5% N2 supplement (Invitrogen, Carlsbad, CA, USA). NPCs were taken from the liquid-nitrogen-storage (−196 °C), then seeded and expanded using N2B27 medium containing additional 150 µM ascorbic acid (Sigma Aldrich, St. Louis, MO, USA), 3 µM CHIR99021 (Cayman chemical company, Ann Arbor, MI, USA) and 0.5 µM purmorphamin (Cayman chemical company, Ann Arbor, MI, USA). After at least two more passages during NPC expansion, patterning was started after reseeding, using N2B27 medium with 1 µM purmorphamin and 1 µM retinoic acid (Sigma Aldrich, St. Louis, MO, USA). After 9 days cells were re-seeded with a density of 50,000 cells per well (26.316 cells/cm²) on PLO/laminin-coated coverslips and differentiation was started using N2B27 medium additionally containing 10 ng/mL rhBDNF (Promega, Madison, WI, USA), 500 µM dbcAMP (Sigma Aldrich, St. Louis, MO, USA) and 10 ng/ mL rhGDNF (Sigma Aldrich, St. Louis, MO, USA). Following this protocol all SMI-32+ cells correspond to MNs and were positively co-stained for Hb9 (Homeobox gene 9), Cholinacetyltransferase (ChAt) and ISLET1 (ISL LH4 homeobox 1) [35,37]. All further analyses were performed according to this protocol.
4.4. CSF Treatment (See also Figure 1a)
Since CSF caused a significant proliferation of NPCs in our cell cultures (Figure 1), we had to find a suitable protocol for our experiments. We therefore analyzed the optimal time point of CSF application. We tested different options (immediately after seeding, 24 h, 48 h and 10 days later) using different concentrations (10%, 20%, 50%) of test-CSF. We had the best results initiating the treatment at day 10 after seeding for differentiation, using CSF concentrations of 10% and 20%. We then evaluated the duration of CSF-treatment (24 h, 48 h, 72 h, 96 h, 6 d) and the optimal concentration (10%, 20%, 50%). For this part of our experiment we started using ALS- and control-CSF. This evaluation led to the decision of treating with 20% CSF for 6 days, the highest possible concentration with no extensive NPC proliferation. For the final experiments, CSF from 11 ALS patients and 8 control patients was mixed in equal parts. CSF treatment was started 10 days after the reseeding, blending the differentiation-medium with CSF in the proportion 1:5. Cells were either treated with 20% ALS-, 20% control-, or no CSF. Since the number of seeded cells was constant throughout all experiments, the ratio of CSF to the initial amount of cells was the same in all experiments. Medium was changed every second day throughout the whole protocol. After 6 days of treatment, cells were fixated for immunofluorescence.
4.5. Immunofluorescence of Spinal Motor Neurons
For immunofluorescence staining, cells were washed twice with phosphate-buffered-saline (PBS) without Ca2+/Mg2+ (LifeTechnologies, Carlsbad, CA, USA) and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Paraformaldehyde was aspirated and cells were washed three times with PBS. Fixed cells were first permeabilized for 10 min in 0.2% Triton X solution and subsequently incubated for 1 h at room temperature in blocking solution (1% bovine serum albumin, 5% donkey serum, 0.3M glycine (Carl Roth GmBH & Co. KG, Germany) and 0.02% Triton X-100 (Thermo Fisher Scientific, Waltham, MA, USA) in PBS). Following blocking, primary antibodies were diluted in blocking solution and cells were incubated with primary antibody solution for 12 h at 4 °C. The following primary antibodies were used: chicken anti-SMI-32 (1:10.000, Covance, Princeton, NJ, USA), mouse anti-FUS (1:5000, Sigma Aldrich, St. Louis, MO, USA), rabbit anti-beta-III-Tubulin (1:3000, Covance, Princeton, NJ, USA), mouse anti-GM-130 (1:200, BD Pharmingen, Franklin Lakes, NJ, USA), mouse anti-SOD1 (1:100, Cell Applications Inc., San Diego, CA, USA), rabbit anti-TDP-43 (1:400, Abcam, Cambridge, UK), rabbit anti-GFAP (1:1000 Chemicon, Temecula, CA, USA), mouse anti-vimentin (1:100, Sigma Aldrich, St. Louis, MO, USA). The nuclei were counterstained using Hoechst 33342 (LifeTechnologies, Carlsbad, CA, USA).
4.6. Quantification and Statistics
After immunofluorescence staining, cells were counted, and relative amounts were calculated. We took at least 15 pictures of each condition at random fields of view across the respective well. This resulted in from 350 to up to 3000 cells counted in each condition for one individual experiment, which we always performed at least three times. Furthermore, the morphology of the Golgi apparatuses was analyzed using a Zeiss Axio Observer Z1 microscope (Carl Zeiss AG, Oberkochen, Germany) and classified in different categories (Figure 5). One category was defined as Golgi fragmentation (loss of the intactness of the cisternal structure and dispersion of the vesicular compartments across the cell; see Figure 5) and relations were calculated using this number. We analyzed the Golgi apparatus of at least 140 neurons per condition of each individual experiment, which we again always performed at least three times. For the analysis of aggregates containing disease relevant protein such as FUS, TDP-43 and SOD1, we examined at least 200 cells per condition of each experiment, which we performed at least four times. Statistical analyses were done using either one-way ANOVA with Tukey post-hoc test, independent samples t-test, or a two-way ANOVA with Bonferroni post-hoc test using GraphPad prism 7 (Version 7.03, GraphPad Software, San Dieago, CA, USA). In case of non-normal distributed data, analyses were performed using Kruskal-Wallis-test with Dunn-Bonferroni post-hoc test. Analysis of normal distribution was performed using Shapiro-Wilk test. Data are depicted as mean ± SEM from at least three independent experiments each (independent differentiation batches).
4.7. Analysis of Neuronal Network Degeneration
We performed the analysis as previously described by Glaß et al. [50]. After imaging a neuron or neuronal network, we converted the picture into a 2-bit image using the “threshold” function from Fiji (ImageJ-win64, open source). Following this, we transformed the obtained mask into a skeletonized network using the skeletonize plugin. A full intact axon without any disruption will yield a ratio of perimeter/section-length of 2. A disrupted axon will have an increased perimeter/section-length of up to 4, when the axon is solely represented by alternating pixel/blank spaces. We then rescaled the value range from 2–4 to 0–1. We normalized the data within each experiment to control-no-CSF condition. For the analysis we took at least 15 pictures at random spots across the respective well of each condition of one individual experiment, which we always performed at least three times.
Author Contributions
Conceptualization, A.H. and S.B.; methodology, A.H. and S.B.; validation, A.H. and S.B.; formal analysis, S.B.; investigation, S.B. and H.G.; resources, A.H., J.S., R.G., H.G.; data curation, S.B.; writing—original draft preparation, S.B.; writing—review and editing, A.H., R.G., J.S., H.G. and S.B.; visualization, S.B.; supervision, A.H.; project administration, A.H.; funding acquisition, A.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported, in part, by the “Deutsche Gesellschaft für Muskelerkrankungen (He2/2)” to A.H., the NOMIS foundation to A.H., the Helmholtz Virtual Institute “RNA dysmetabolism in ALS and FTD (VH-VI-510)” to A.H., an unrestricted grant by a family of a deceased ALS patient to A.H, and the Stiftung zur Förderung der Hochschulmedizin in Dresden. A.H. is supported by the Hermann und Lilly Schilling-Stiftung für medizinische Forschung im Stifterverband.
Acknowledgments
We acknowledge the great help in cell culture by Anett Böhme, Sylvia Kanzler and Katja Zoschke.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| CSF | Cerebrospinal fluid |
| FTLD | Frontotemporal dementia |
| FUS | Fused in sarcoma |
| iPSC | human patient-derived induced pluripotent stem cell |
| MN | Motor neuron |
| NPC | Neural progenitor cells |
| PBS | Phosphate-buffered-saline |
| SOD1 | Superoxide dismutase 1 |
| TDP-43 | Transactive response DNA binding protein 43 kDA |
References
- Brauer, S.; Zimyanin, V.; Hermann, A. Prion-like properties of disease-relevant proteins in amyotrophic lateral sclerosis. J. Neural Transm. 2018, 125, 591–613. [Google Scholar] [CrossRef]
- Smith, R.; Myers, K.; Ravits, J.; Bowser, R. Amyotrophic lateral sclerosis: Is the spinal fluid pathway involved in seeding and spread? Med. Hypotheses 2015, 85, 576–583. [Google Scholar] [CrossRef]
- Couratier, P.; Hugon, J.; Sindou, P.; Vallat, J.M.; Dumas, M. Cell culture evidence for neuronal degeneration in amyotrophic lateral sclerosis being linked to glutamate AMPA/kainate receptors. Lancet 1993, 341, 265–268. [Google Scholar] [CrossRef]
- Gomez-Pinedo, U.; Galan, L.; Yanez, M.; Matias-Guiu, J.; Valencia, C.; Guerrero-Sola, A.; Lopez-Sosa, F.; Brin, J.R.; Benito-Martin, M.S.; Leon-Espinosa, G.; et al. Histological changes in the rat brain and spinal cord following prolonged intracerebroventricular infusion of cerebrospinal fluid from amyotrophic lateral sclerosis patients are similar to those caused by the disease. Neurologia 2018, 33, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinedo, U.; Yanez, M.; Matias-Guiu, J.; Galan, L.; Guerrero-Sola, A.; Benito-Martin, M.S.; Vela, A.; Arranz-Tagarro, J.A.; Garcia, A.G. Cellular changes in motor neuron cell culture produced by cytotoxic cerebrospinal fluid from patients with amyotrophic lateral sclerosis. Neurologia 2014, 29, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Vijayalakshmi, K.; Alladi, P.A.; Ghosh, S.; Prasanna, V.K.; Sagar, B.C.; Nalini, A.; Sathyaprabha, T.N.; Raju, T.R. Evidence of endoplasmic reticular stress in the spinal motor neurons exposed to CSF from sporadic amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2011, 41, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Yanez, M.; Galan, L.; Matias-Guiu, J.; Vela, A.; Guerrero, A.; Garcia, A.G. CSF from amyotrophic lateral sclerosis patients produces glutamate independent death of rat motor brain cortical neurons: Protection by resveratrol but not riluzole. Brain Res. 2011, 1423, 77–86. [Google Scholar] [CrossRef]
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.H.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 2015, 6, 24178–24191. [Google Scholar] [CrossRef]
- Rao, M.S.; Devi, M.G.; Nalini, A.; Shahani, N.; Raju, T.R. Neurofilament phosphorylation is increased in ventral horn neurons of neonatal rat spinal cord exposed to cerebrospinal fluid from patients with amyotrophic lateral sclerosis. Neurodegeneration 1995, 4, 397–401. [Google Scholar] [CrossRef]
- Shobha, K.; Alladi, P.A.; Nalini, A.; Sathyaprabha, T.N.; Raju, T.R. Exposure to CSF from sporadic amyotrophic lateral sclerosis patients induces morphological transformation of astroglia and enhances GFAP and S100beta expression. Neurosci. Lett. 2010, 473, 56–61. [Google Scholar] [CrossRef]
- Sharma, A.; Varghese, A.M.; Vijaylakshmi, K.; Sumitha, R.; Prasanna, V.K.; Shruthi, S.; Chandrasekhar Sagar, B.K.; Datta, K.K.; Gowda, H.; Nalini, A.; et al. Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces mitochondrial and lysosomal dysfunction. Neurochem. Res. 2016, 41, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Ramamohan, P.Y.; Gourie-Devi, M.; Nalini, A.; Shobha, K.; Ramamohan, Y.; Joshi, P.; Raju, T.R. Cerebrospinal fluid from amyotrophic lateral sclerosis patients causes fragmentation of the Golgi apparatus in the neonatal rat spinal cord. Amyotroph. Lateral Scler. 2007, 8, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, V.; Walker, A.K.; Yerbury, J.; Soo, K.Y.; Farg, M.A.; Hoang, V.; Zeineddine, R.; Spencer, D.; Atkin, J.D. Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell. Mol. Life Sci. 2013, 70, 4181–4195. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, S.; Barsoum, M.J.; Bossy-Wetzel, E.; Sutterlin, C.; Malhotra, V.; Lipton, S.A. A Golgi fragmentation pathway in neurodegeneration. Neurobiol. Dis. 2008, 29, 221–231. [Google Scholar] [CrossRef]
- Sundaramoorthy, V.; Sultana, J.M.; Atkin, J.D. Golgi fragmentation in amyotrophic lateral sclerosis, an overview of possible triggers and consequences. Front. Neurosci. 2015, 9, 400. [Google Scholar] [CrossRef]
- Stieber, A.; Gonatas, J.O.; Collard, J.; Meier, J.; Julien, J.; Schweitzer, P.; Gonatas, N.K. The neuronal Golgi apparatus is fragmented in transgenic mice expressing a mutant human SOD1, but not in mice expressing the human NF-H gene. J. Neurol. Sci. 2000, 173, 63–72. [Google Scholar] [CrossRef]
- Stieber, A.; Gonatas, J.O.; Moore, J.S.; Bantly, A.; Yim, H.S.; Yim, M.B.; Gonatas, N.K. Disruption of the structure of the Golgi apparatus and the function of the secretory pathway by mutants G93A and G85R of Cu, Zn superoxide dismutase (SOD1) of familial amyotrophic lateral sclerosis. J. Neurol. Sci. 2004, 219, 45–53. [Google Scholar] [CrossRef]
- Fujita, Y.; Mizuno, Y.; Takatama, M.; Okamoto, K. Anterior horn cells with abnormal TDP-43 immunoreactivities show fragmentation of the Golgi apparatus in ALS. J. Neurol. Sci. 2008, 269, 30–34. [Google Scholar] [CrossRef]
- Fujita, Y.; Okamoto, K.; Sakurai, A.; Amari, M.; Nakazato, Y.; Gonatas, N.K. Fragmentation of the Golgi apparatus of Betz cells in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1999, 163, 81–85. [Google Scholar] [CrossRef]
- Fujita, Y.; Okamoto, K.; Sakurai, A.; Gonatas, N.K.; Hirano, A. Fragmentation of the Golgi apparatus of the anterior horn cells in patients with familial amyotrophic lateral sclerosis with SOD1 mutations and posterior column involvement. J. Neurol. Sci. 2000, 174, 137–140. [Google Scholar] [CrossRef]
- Gonatas, N.K.; Stieber, A.; Mourelatos, Z.; Chen, Y.; Gonatas, J.O.; Appel, S.H.; Hays, A.P.; Hickey, W.F.; Hauw, J.J. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Am. J. Pathol. 1992, 140, 731–737. [Google Scholar] [PubMed]
- Mourelatos, Z.; Adler, H.; Hirano, A.; Donnenfeld, H.; Gonatas, J.O.; Gonatas, N.K. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis revealed by organelle-specific antibodies. Proc. Natl. Acad. Sci. USA 1990, 87, 4393–4395. [Google Scholar] [CrossRef] [PubMed]
- Mourelatos, Z.; Hirano, A.; Rosenquist, A.C.; Gonatas, N.K. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis (ALS). Clinical studies in ALS of Guam and experimental studies in deafferented neurons and in beta, beta’-iminodipropionitrile axonopathy. Am. J. Pathol. 1994, 144, 1288–1300. [Google Scholar] [PubMed]
- Mourelatos, Z.; Yachnis, A.; Rorke, L.; Mikol, J.; Gonatas, N.K. The Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 1993, 33, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Ohama, E.; Takatama, M.; Al-Sarraj, S.; Okamoto, K. Fragmentation of Golgi apparatus of nigral neurons with alpha-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathol. 2006, 112, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Gonatas, N.K.; Stieber, A.; Gonatas, J.O. Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J. Neurol. Sci. 2006, 246, 21–30. [Google Scholar] [CrossRef]
- Maekawa, T.; Mori, S.; Sasaki, Y.; Miyajima, T.; Azuma, S.; Ohta, E.; Obata, F. The I2020T Leucine-rich repeat kinase 2 transgenic mouse exhibits impaired locomotive ability accompanied by dopaminergic neuron abnormalities. Mol. Neurodegener. 2012, 7, 15. [Google Scholar] [CrossRef]
- Stieber, A.; Mourelatos, Z.; Gonatas, N.K. In Alzheimer’s disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am. J. Pathol. 1996, 148, 415–426. [Google Scholar]
- Stieber, A.; Chen, Y.; Wei, S.; Mourelatos, Z.; Gonatas, J.; Okamoto, K.; Gonatas, N.K. The fragmented neuronal Golgi apparatus in amyotrophic lateral sclerosis includes the trans-Golgi-network: Functional implications. Acta Neuropathol. 1998, 95, 245–253. [Google Scholar] [CrossRef]
- van Dis, V.; Kuijpers, M.; Haasdijk, E.D.; Teuling, E.; Oakes, S.A.; Hoogenraad, C.C.; Jaarsma, D. Golgi fragmentation precedes neuromuscular denervation and is associated with endosome abnormalities in SOD1-ALS mouse motor neurons. Acta Neuropathol. Commun. 2014, 2, 38. [Google Scholar] [CrossRef]
- Rendon, W.O.; Martinez-Alonso, E.; Tomas, M.; Martinez-Martinez, N.; Martinez-Menarguez, J.A. Golgi fragmentation is Rab and SNARE dependent in cellular models of Parkinson’s disease. Histochem. Cell Biol. 2013, 139, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Okamoto, K. Golgi apparatus of the motor neurons in patients with amyotrophic lateral sclerosis and in mice models of amyotrophic lateral sclerosis. Neuropathology 2005, 25, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Mourelatos, Z.; Gonatas, N.K.; Stieber, A.; Gurney, M.E.; Dal Canto, M.C. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc. Natl. Acad. Sci. USA 1996, 93, 5472–5477. [Google Scholar] [CrossRef] [PubMed]
- Japtok, J.; Lojewski, X.; Naumann, M.; Klingenstein, M.; Reinhardt, P.; Sterneckert, J.; Putz, S.; Demestre, M.; Boeckers, T.M.; Ludolph, A.C.; et al. Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol. Dis. 2015, 82, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Gunther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef]
- Abo-Rady, M.; Kalmbach, N.; Pal, A.; Schludi, C.; Janosch, A.; Richter, T.; Freitag, P.; Bickle, M.; Kahlert, A.K.; Petri, S.; et al. Knocking out C9ORF72 Exacerbates Axonal Trafficking Defects Associated with Hexanucleotide Repeat Expansion and Reduces Levels of Heat Shock Proteins. Stem Cell Rep. 2020. [Google Scholar] [CrossRef]
- Kreiter, N.; Pal, A.; Lojewski, X.; Corcia, P.; Naujock, M.; Reinhardt, P.; Sterneckert, J.; Petri, S.; Wegner, F.; Storch, A.; et al. Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 2018, 115, 167–181. [Google Scholar] [CrossRef]
- Pal, A.; Glass, H.; Naumann, M.; Kreiter, N.; Japtok, J.; Sczech, R.; Hermann, A. High content organelle trafficking enables disease state profiling as powerful tool for disease modelling. Sci. Data 2018, 5, 180241. [Google Scholar] [CrossRef]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Watanabe, S.; Kaneko, K.; Yamanaka, K.; Nukina, N.; Furukawa, Y. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 2014, 289, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Prudlo, J.; Konig, J.; Schuster, C.; Kasper, E.; Buttner, A.; Teipel, S.; Neumann, M. TDP-43 pathology and cognition in ALS: A prospective clinicopathologic correlation study. Neurology 2016, 87, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Budini, M.; Romano, V.; Avendano-Vazquez, S.E.; Bembich, S.; Buratti, E.; Baralle, F.E. Role of selected mutations in the Q/N rich region of TDP-43 in EGFP-12xQ/N-induced aggregate formation. Brain Res. 2012, 1462, 139–150. [Google Scholar] [CrossRef]
- Guo, W.; Chen, Y.; Zhou, X.; Kar, A.; Ray, P.; Chen, X.; Rao, E.J.; Yang, M.; Ye, H.; Zhu, L.; et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 2011, 18, 822–830. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Galan, L.; Matias-Guiu, J.; Matias-Guiu, J.A.; Yanez, M.; Pytel, V.; Guerrero-Sola, A.; Vela-Souto, A.; Arranz-Tagarro, J.A.; Gomez-Pinedo, U.; Garcia, A.G. Cerebrospinal fluid cytotoxicity does not affect survival in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2017, 136, 212–216. [Google Scholar] [CrossRef]
- Buddensiek, J.; Dressel, A.; Kowalski, M.; Runge, U.; Schroeder, H.; Hermann, A.; Kirsch, M.; Storch, A.; Sabolek, M. Cerebrospinal fluid promotes survival and astroglial differentiation of adult human neural progenitor cells but inhibits proliferation and neuronal differentiation. BMC Neurosci. 2010, 11, 48. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Glass, H.; Neumann, P.; Pal, A.; Reinhardt, P.; Storch, A.; Sterneckert, J.; Hermann, A. Combined dendritic and axonal deterioration are responsible for motoneuronopathy in patient-derived neuronal cell models of chorea-acanthocytosis. Int. J. Mol. Sci. 2020, 21, 1797. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).