Human Spinal Motor Neurons Are Particularly Vulnerable to Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients

 , and
, and 
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
2.2. CSF Affects Proliferation of NPCs
2.3. No Neuronal Loss or Neuronal Network Degeneration by ALS-CSF
2.4. No Signs of Pathological Aggregate Formation by ALS-CSF
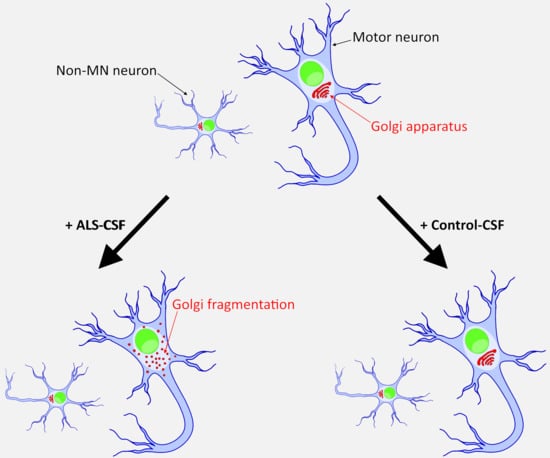
2.5. ALS-CSF Induces Golgi Fragmentation in Patient-Derived Motor Neurons
3. Discussion
4. Materials and Methods
4.1. Patient Material
4.2. Generation and Expansion of iPSCs, in Vitro Differentiation of Embryoid Bodies, AP Staining and Immunofluorescence on iPSC Colonies and Derivation of iPSC-Derived Neuroprecursor Cells
4.3. Motor Neuron Differentiation
4.4. CSF Treatment (See also Figure 1a)
4.5. Immunofluorescence of Spinal Motor Neurons
4.6. Quantification and Statistics
4.7. Analysis of Neuronal Network Degeneration
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| CSF | Cerebrospinal fluid |
| FTLD | Frontotemporal dementia |
| FUS | Fused in sarcoma |
| iPSC | human patient-derived induced pluripotent stem cell |
| MN | Motor neuron |
| NPC | Neural progenitor cells |
| PBS | Phosphate-buffered-saline |
| SOD1 | Superoxide dismutase 1 |
| TDP-43 | Transactive response DNA binding protein 43 kDA |
References
- Brauer, S.; Zimyanin, V.; Hermann, A. Prion-like properties of disease-relevant proteins in amyotrophic lateral sclerosis. J. Neural Transm. 2018, 125, 591–613. [Google Scholar] [CrossRef]
- Smith, R.; Myers, K.; Ravits, J.; Bowser, R. Amyotrophic lateral sclerosis: Is the spinal fluid pathway involved in seeding and spread? Med. Hypotheses 2015, 85, 576–583. [Google Scholar] [CrossRef]
- Couratier, P.; Hugon, J.; Sindou, P.; Vallat, J.M.; Dumas, M. Cell culture evidence for neuronal degeneration in amyotrophic lateral sclerosis being linked to glutamate AMPA/kainate receptors. Lancet 1993, 341, 265–268. [Google Scholar] [CrossRef]
- Gomez-Pinedo, U.; Galan, L.; Yanez, M.; Matias-Guiu, J.; Valencia, C.; Guerrero-Sola, A.; Lopez-Sosa, F.; Brin, J.R.; Benito-Martin, M.S.; Leon-Espinosa, G.; et al. Histological changes in the rat brain and spinal cord following prolonged intracerebroventricular infusion of cerebrospinal fluid from amyotrophic lateral sclerosis patients are similar to those caused by the disease. Neurologia 2018, 33, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinedo, U.; Yanez, M.; Matias-Guiu, J.; Galan, L.; Guerrero-Sola, A.; Benito-Martin, M.S.; Vela, A.; Arranz-Tagarro, J.A.; Garcia, A.G. Cellular changes in motor neuron cell culture produced by cytotoxic cerebrospinal fluid from patients with amyotrophic lateral sclerosis. Neurologia 2014, 29, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Vijayalakshmi, K.; Alladi, P.A.; Ghosh, S.; Prasanna, V.K.; Sagar, B.C.; Nalini, A.; Sathyaprabha, T.N.; Raju, T.R. Evidence of endoplasmic reticular stress in the spinal motor neurons exposed to CSF from sporadic amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2011, 41, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Yanez, M.; Galan, L.; Matias-Guiu, J.; Vela, A.; Guerrero, A.; Garcia, A.G. CSF from amyotrophic lateral sclerosis patients produces glutamate independent death of rat motor brain cortical neurons: Protection by resveratrol but not riluzole. Brain Res. 2011, 1423, 77–86. [Google Scholar] [CrossRef]
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.H.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 2015, 6, 24178–24191. [Google Scholar] [CrossRef]
- Rao, M.S.; Devi, M.G.; Nalini, A.; Shahani, N.; Raju, T.R. Neurofilament phosphorylation is increased in ventral horn neurons of neonatal rat spinal cord exposed to cerebrospinal fluid from patients with amyotrophic lateral sclerosis. Neurodegeneration 1995, 4, 397–401. [Google Scholar] [CrossRef]
- Shobha, K.; Alladi, P.A.; Nalini, A.; Sathyaprabha, T.N.; Raju, T.R. Exposure to CSF from sporadic amyotrophic lateral sclerosis patients induces morphological transformation of astroglia and enhances GFAP and S100beta expression. Neurosci. Lett. 2010, 473, 56–61. [Google Scholar] [CrossRef]
- Sharma, A.; Varghese, A.M.; Vijaylakshmi, K.; Sumitha, R.; Prasanna, V.K.; Shruthi, S.; Chandrasekhar Sagar, B.K.; Datta, K.K.; Gowda, H.; Nalini, A.; et al. Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces mitochondrial and lysosomal dysfunction. Neurochem. Res. 2016, 41, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Ramamohan, P.Y.; Gourie-Devi, M.; Nalini, A.; Shobha, K.; Ramamohan, Y.; Joshi, P.; Raju, T.R. Cerebrospinal fluid from amyotrophic lateral sclerosis patients causes fragmentation of the Golgi apparatus in the neonatal rat spinal cord. Amyotroph. Lateral Scler. 2007, 8, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, V.; Walker, A.K.; Yerbury, J.; Soo, K.Y.; Farg, M.A.; Hoang, V.; Zeineddine, R.; Spencer, D.; Atkin, J.D. Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell. Mol. Life Sci. 2013, 70, 4181–4195. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, S.; Barsoum, M.J.; Bossy-Wetzel, E.; Sutterlin, C.; Malhotra, V.; Lipton, S.A. A Golgi fragmentation pathway in neurodegeneration. Neurobiol. Dis. 2008, 29, 221–231. [Google Scholar] [CrossRef]
- Sundaramoorthy, V.; Sultana, J.M.; Atkin, J.D. Golgi fragmentation in amyotrophic lateral sclerosis, an overview of possible triggers and consequences. Front. Neurosci. 2015, 9, 400. [Google Scholar] [CrossRef]
- Stieber, A.; Gonatas, J.O.; Collard, J.; Meier, J.; Julien, J.; Schweitzer, P.; Gonatas, N.K. The neuronal Golgi apparatus is fragmented in transgenic mice expressing a mutant human SOD1, but not in mice expressing the human NF-H gene. J. Neurol. Sci. 2000, 173, 63–72. [Google Scholar] [CrossRef]
- Stieber, A.; Gonatas, J.O.; Moore, J.S.; Bantly, A.; Yim, H.S.; Yim, M.B.; Gonatas, N.K. Disruption of the structure of the Golgi apparatus and the function of the secretory pathway by mutants G93A and G85R of Cu, Zn superoxide dismutase (SOD1) of familial amyotrophic lateral sclerosis. J. Neurol. Sci. 2004, 219, 45–53. [Google Scholar] [CrossRef]
- Fujita, Y.; Mizuno, Y.; Takatama, M.; Okamoto, K. Anterior horn cells with abnormal TDP-43 immunoreactivities show fragmentation of the Golgi apparatus in ALS. J. Neurol. Sci. 2008, 269, 30–34. [Google Scholar] [CrossRef]
- Fujita, Y.; Okamoto, K.; Sakurai, A.; Amari, M.; Nakazato, Y.; Gonatas, N.K. Fragmentation of the Golgi apparatus of Betz cells in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1999, 163, 81–85. [Google Scholar] [CrossRef]
- Fujita, Y.; Okamoto, K.; Sakurai, A.; Gonatas, N.K.; Hirano, A. Fragmentation of the Golgi apparatus of the anterior horn cells in patients with familial amyotrophic lateral sclerosis with SOD1 mutations and posterior column involvement. J. Neurol. Sci. 2000, 174, 137–140. [Google Scholar] [CrossRef]
- Gonatas, N.K.; Stieber, A.; Mourelatos, Z.; Chen, Y.; Gonatas, J.O.; Appel, S.H.; Hays, A.P.; Hickey, W.F.; Hauw, J.J. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Am. J. Pathol. 1992, 140, 731–737. [Google Scholar] [PubMed]
- Mourelatos, Z.; Adler, H.; Hirano, A.; Donnenfeld, H.; Gonatas, J.O.; Gonatas, N.K. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis revealed by organelle-specific antibodies. Proc. Natl. Acad. Sci. USA 1990, 87, 4393–4395. [Google Scholar] [CrossRef] [PubMed]
- Mourelatos, Z.; Hirano, A.; Rosenquist, A.C.; Gonatas, N.K. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis (ALS). Clinical studies in ALS of Guam and experimental studies in deafferented neurons and in beta, beta’-iminodipropionitrile axonopathy. Am. J. Pathol. 1994, 144, 1288–1300. [Google Scholar] [PubMed]
- Mourelatos, Z.; Yachnis, A.; Rorke, L.; Mikol, J.; Gonatas, N.K. The Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 1993, 33, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Ohama, E.; Takatama, M.; Al-Sarraj, S.; Okamoto, K. Fragmentation of Golgi apparatus of nigral neurons with alpha-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathol. 2006, 112, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Gonatas, N.K.; Stieber, A.; Gonatas, J.O. Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J. Neurol. Sci. 2006, 246, 21–30. [Google Scholar] [CrossRef]
- Maekawa, T.; Mori, S.; Sasaki, Y.; Miyajima, T.; Azuma, S.; Ohta, E.; Obata, F. The I2020T Leucine-rich repeat kinase 2 transgenic mouse exhibits impaired locomotive ability accompanied by dopaminergic neuron abnormalities. Mol. Neurodegener. 2012, 7, 15. [Google Scholar] [CrossRef]
- Stieber, A.; Mourelatos, Z.; Gonatas, N.K. In Alzheimer’s disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am. J. Pathol. 1996, 148, 415–426. [Google Scholar]
- Stieber, A.; Chen, Y.; Wei, S.; Mourelatos, Z.; Gonatas, J.; Okamoto, K.; Gonatas, N.K. The fragmented neuronal Golgi apparatus in amyotrophic lateral sclerosis includes the trans-Golgi-network: Functional implications. Acta Neuropathol. 1998, 95, 245–253. [Google Scholar] [CrossRef]
- van Dis, V.; Kuijpers, M.; Haasdijk, E.D.; Teuling, E.; Oakes, S.A.; Hoogenraad, C.C.; Jaarsma, D. Golgi fragmentation precedes neuromuscular denervation and is associated with endosome abnormalities in SOD1-ALS mouse motor neurons. Acta Neuropathol. Commun. 2014, 2, 38. [Google Scholar] [CrossRef]
- Rendon, W.O.; Martinez-Alonso, E.; Tomas, M.; Martinez-Martinez, N.; Martinez-Menarguez, J.A. Golgi fragmentation is Rab and SNARE dependent in cellular models of Parkinson’s disease. Histochem. Cell Biol. 2013, 139, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Okamoto, K. Golgi apparatus of the motor neurons in patients with amyotrophic lateral sclerosis and in mice models of amyotrophic lateral sclerosis. Neuropathology 2005, 25, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Mourelatos, Z.; Gonatas, N.K.; Stieber, A.; Gurney, M.E.; Dal Canto, M.C. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc. Natl. Acad. Sci. USA 1996, 93, 5472–5477. [Google Scholar] [CrossRef] [PubMed]
- Japtok, J.; Lojewski, X.; Naumann, M.; Klingenstein, M.; Reinhardt, P.; Sterneckert, J.; Putz, S.; Demestre, M.; Boeckers, T.M.; Ludolph, A.C.; et al. Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol. Dis. 2015, 82, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Gunther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef]
- Abo-Rady, M.; Kalmbach, N.; Pal, A.; Schludi, C.; Janosch, A.; Richter, T.; Freitag, P.; Bickle, M.; Kahlert, A.K.; Petri, S.; et al. Knocking out C9ORF72 Exacerbates Axonal Trafficking Defects Associated with Hexanucleotide Repeat Expansion and Reduces Levels of Heat Shock Proteins. Stem Cell Rep. 2020. [Google Scholar] [CrossRef]
- Kreiter, N.; Pal, A.; Lojewski, X.; Corcia, P.; Naujock, M.; Reinhardt, P.; Sterneckert, J.; Petri, S.; Wegner, F.; Storch, A.; et al. Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 2018, 115, 167–181. [Google Scholar] [CrossRef]
- Pal, A.; Glass, H.; Naumann, M.; Kreiter, N.; Japtok, J.; Sczech, R.; Hermann, A. High content organelle trafficking enables disease state profiling as powerful tool for disease modelling. Sci. Data 2018, 5, 180241. [Google Scholar] [CrossRef]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Watanabe, S.; Kaneko, K.; Yamanaka, K.; Nukina, N.; Furukawa, Y. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 2014, 289, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Prudlo, J.; Konig, J.; Schuster, C.; Kasper, E.; Buttner, A.; Teipel, S.; Neumann, M. TDP-43 pathology and cognition in ALS: A prospective clinicopathologic correlation study. Neurology 2016, 87, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Budini, M.; Romano, V.; Avendano-Vazquez, S.E.; Bembich, S.; Buratti, E.; Baralle, F.E. Role of selected mutations in the Q/N rich region of TDP-43 in EGFP-12xQ/N-induced aggregate formation. Brain Res. 2012, 1462, 139–150. [Google Scholar] [CrossRef]
- Guo, W.; Chen, Y.; Zhou, X.; Kar, A.; Ray, P.; Chen, X.; Rao, E.J.; Yang, M.; Ye, H.; Zhu, L.; et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 2011, 18, 822–830. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Galan, L.; Matias-Guiu, J.; Matias-Guiu, J.A.; Yanez, M.; Pytel, V.; Guerrero-Sola, A.; Vela-Souto, A.; Arranz-Tagarro, J.A.; Gomez-Pinedo, U.; Garcia, A.G. Cerebrospinal fluid cytotoxicity does not affect survival in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2017, 136, 212–216. [Google Scholar] [CrossRef]
- Buddensiek, J.; Dressel, A.; Kowalski, M.; Runge, U.; Schroeder, H.; Hermann, A.; Kirsch, M.; Storch, A.; Sabolek, M. Cerebrospinal fluid promotes survival and astroglial differentiation of adult human neural progenitor cells but inhibits proliferation and neuronal differentiation. BMC Neurosci. 2010, 11, 48. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Glass, H.; Neumann, P.; Pal, A.; Reinhardt, P.; Storch, A.; Sterneckert, J.; Hermann, A. Combined dendritic and axonal deterioration are responsible for motoneuronopathy in patient-derived neuronal cell models of chorea-acanthocytosis. Int. J. Mol. Sci. 2020, 21, 1797. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSF/Patient-Parameter | Control-CSF | ALS-CSF | p-Value |
|---|---|---|---|
| Number | 8 | 11 | |
| Gender m:f | 5:3 | 7:4 | 0.96 1 |
| Age at lumbal puncture, years | 47 (21.4) | 63 (12.2) | 0.08 2 |
| ALS disease onset, spinal:bulbar | n.a. | 5:6 | |
| ALS-FSR-R at date of lumbal puncture | n.a. | 40.36 (3.67) | |
| ALS genetic, sporadic:familiar | n.a. | 11:0 | |
| Total cell count, MPt/L | 1.63 (0.74) | 1.27 (0.65) | 0.21 2 |
| Total protein, mg/L | 412.88 (311.13) | 469.55 (223.75) | 0.23 2 |
| Albumin, mg/L | 308.13 (249.13) | 312.82 (148.51) | 0.41 2 |
| Glucose, mmol/L | 3.59 (0.41) | 4.11 (0.92) | 0.11 2 |
| Lactate, mmol/L | 1.52 (0.22) | 1.77 (0.31) | 0.10 2 |
| Intrathecal IgG production, yes:no | 0:8 | 0:11 | n.a. |
| Oligoclonal bands, yes:no | 1:7 | 0:11 | n.a. |
| Blood-CSF-barrier dysfunction, yes:no | 1:7 | 3:8 | 0.44 1 |
| Genotyp | Cell Culture Model | Sex | Age at Biopsy (Years) | Mutation | Family History | Age at Disease Onset | Clinical Phenotype | Disease Duration (Months) |
|---|---|---|---|---|---|---|---|---|
| controls | hiPSC | |||||||
| female | 48 | - | - | - | - | |||
| male | 60 | - | - | - | - | |||
| female | 45 | - | - | - | - | |||
| female | 50 | - | - | - | - | |||
| FUS-ALS | hiPSC | |||||||
| female | 58 | p.R521C | Pos. for ALS | 57 | spinal | 7 | ||
| SOD1-ALS | hiPSC | male | 59 | p.R115G | Pos. for ALS | n.a. | spinal | n.a. |
| TDP-43-ALS | hiPSC | female | 87 | p.S393L | Pos. for ALS | n.a. | bulbar | n.a. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bräuer, S.; Günther, R.; Sterneckert, J.; Glaß, H.; Hermann, A. Human Spinal Motor Neurons Are Particularly Vulnerable to Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients. Int. J. Mol. Sci. 2020, 21, 3564. https://doi.org/10.3390/ijms21103564
Bräuer S, Günther R, Sterneckert J, Glaß H, Hermann A. Human Spinal Motor Neurons Are Particularly Vulnerable to Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients. International Journal of Molecular Sciences. 2020; 21(10):3564. https://doi.org/10.3390/ijms21103564
Chicago/Turabian StyleBräuer, Stefan, René Günther, Jared Sterneckert, Hannes Glaß, and Andreas Hermann. 2020. "Human Spinal Motor Neurons Are Particularly Vulnerable to Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients" International Journal of Molecular Sciences 21, no. 10: 3564. https://doi.org/10.3390/ijms21103564
APA StyleBräuer, S., Günther, R., Sterneckert, J., Glaß, H., & Hermann, A. (2020). Human Spinal Motor Neurons Are Particularly Vulnerable to Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients. International Journal of Molecular Sciences, 21(10), 3564. https://doi.org/10.3390/ijms21103564