2-(3-Hydroxy-5-phosphonooxymethyl-2-methyl-4-pyridyl)-1,3-thiazolidine-4-carboxylic Acid, Novel Metabolite of Pyridoxal 5′-Phosphate and Cysteine Is Present in Human Plasma—Chromatographic Investigations



Abstract
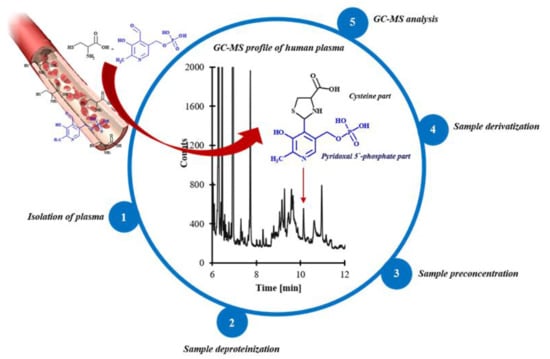
:
1. Introduction
2. Results and Discussion
2.1. Sample Preparation
2.1.1. Proteins Removal
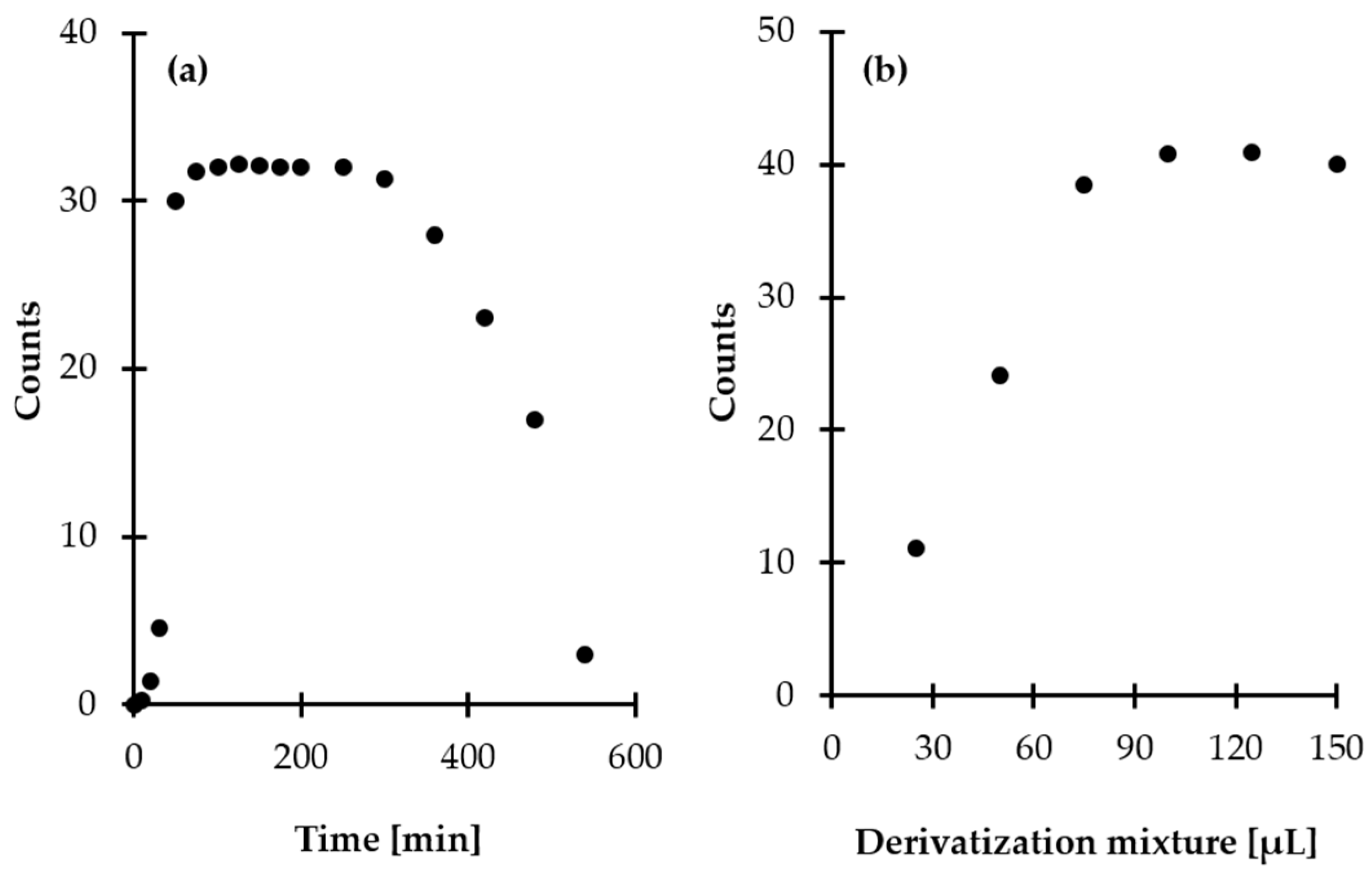
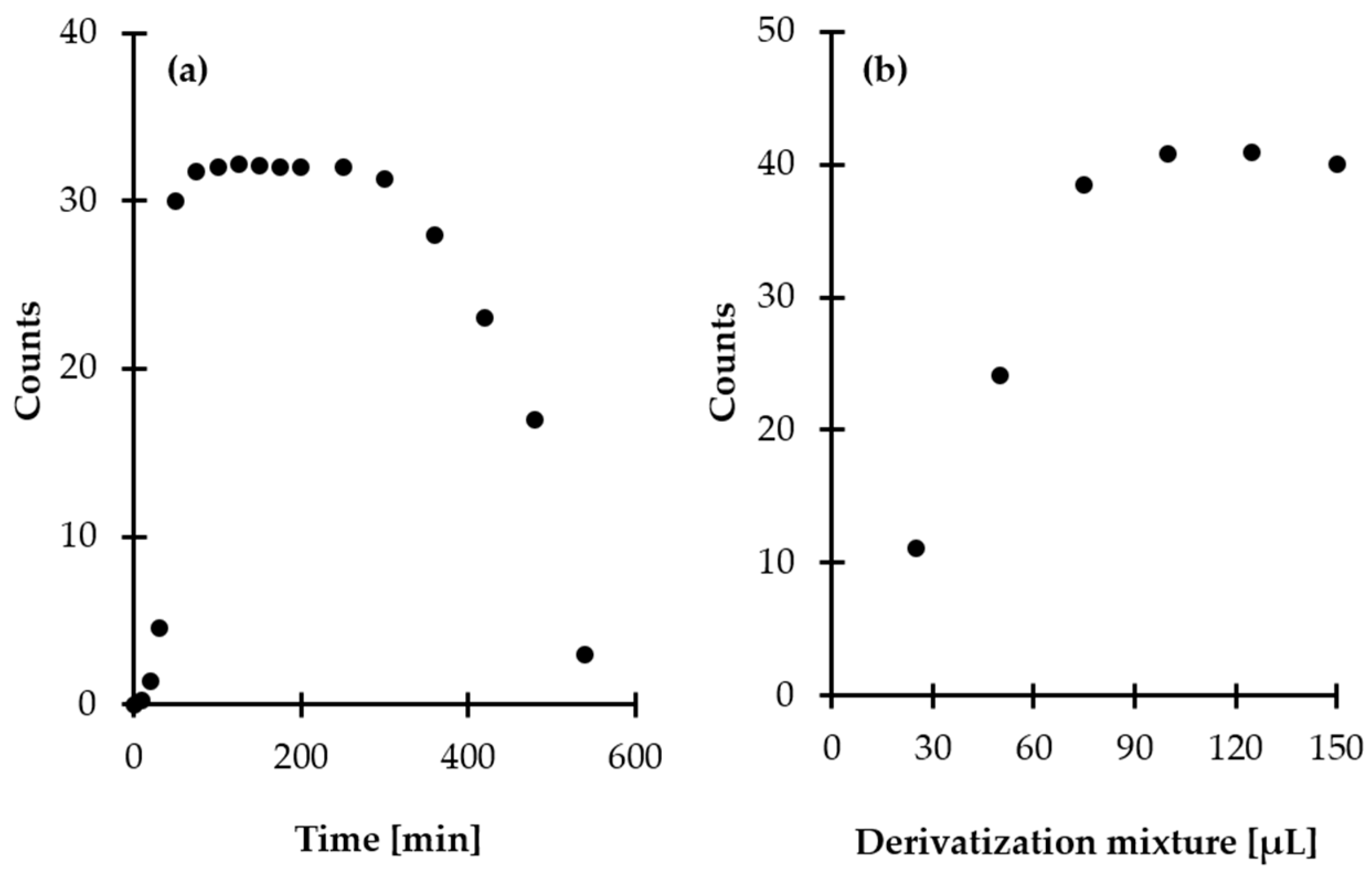
2.1.2. Preconcentration
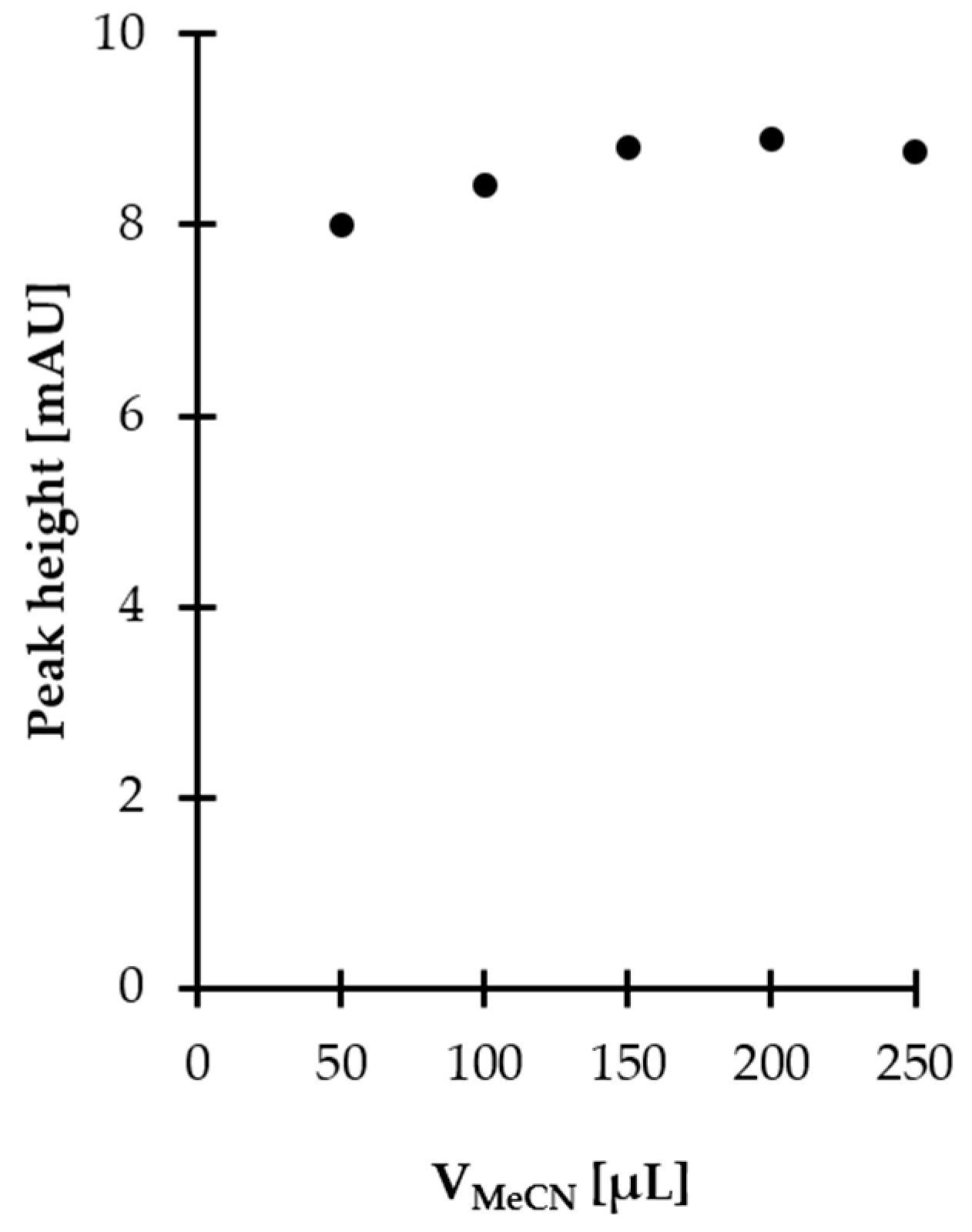
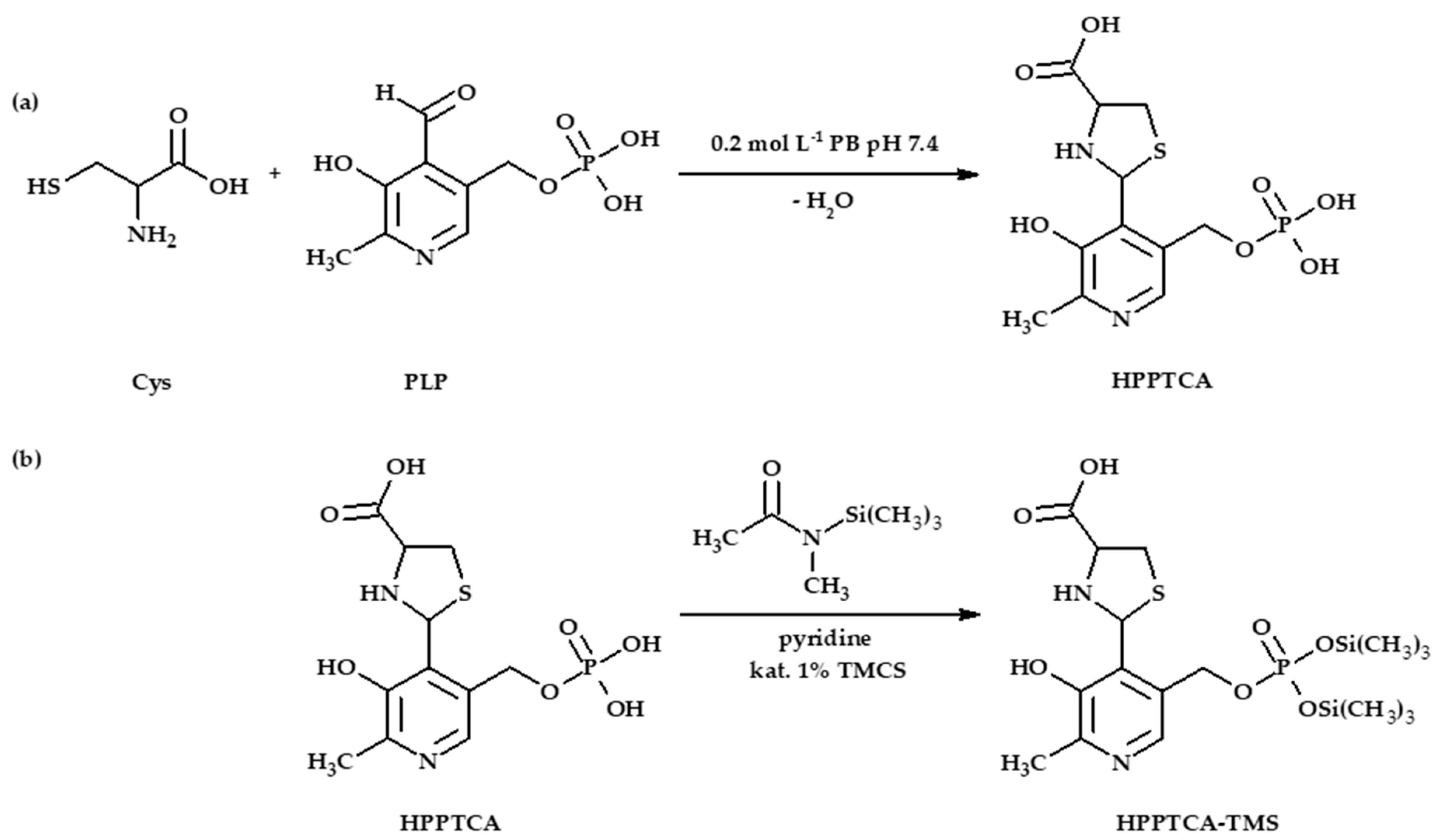
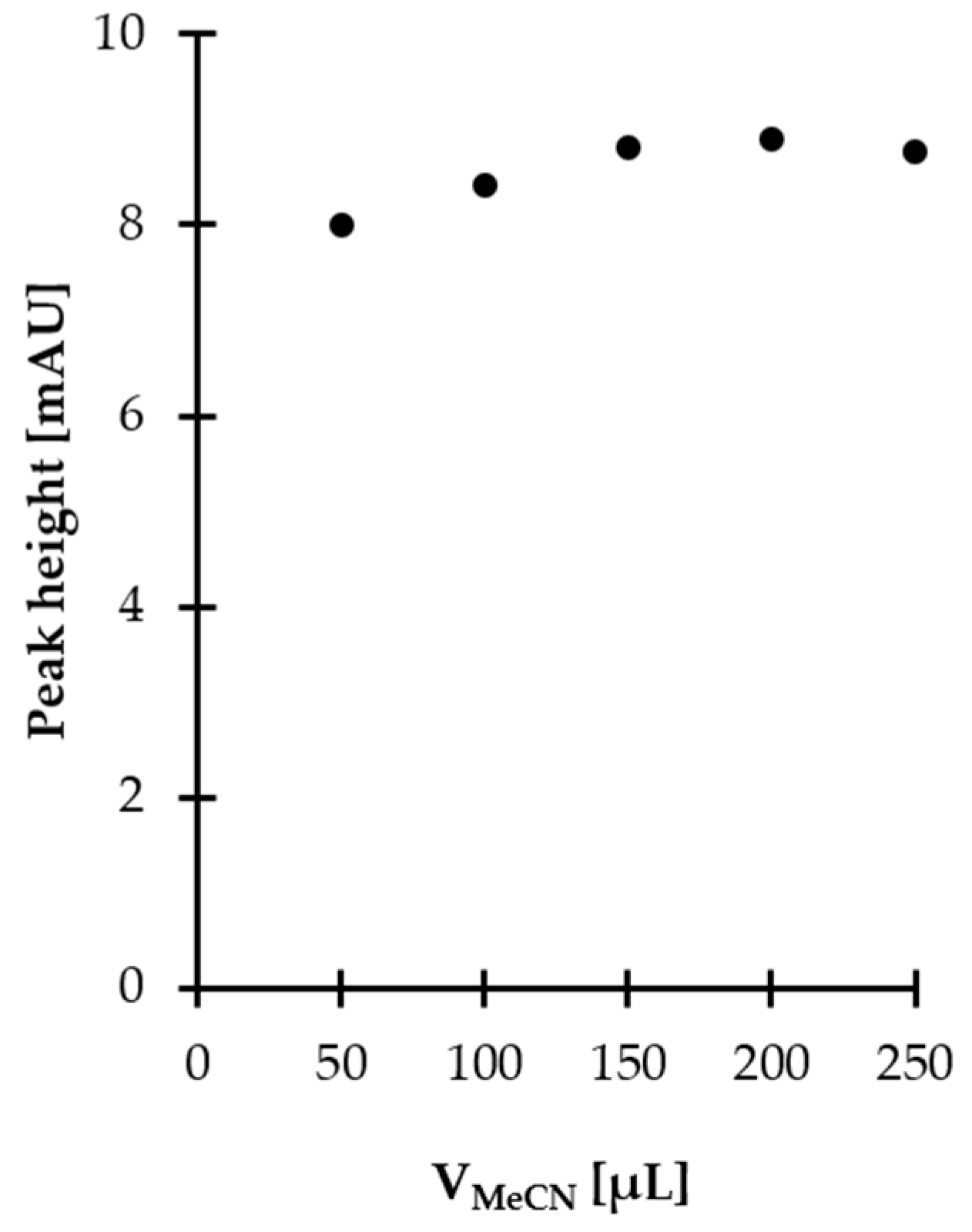
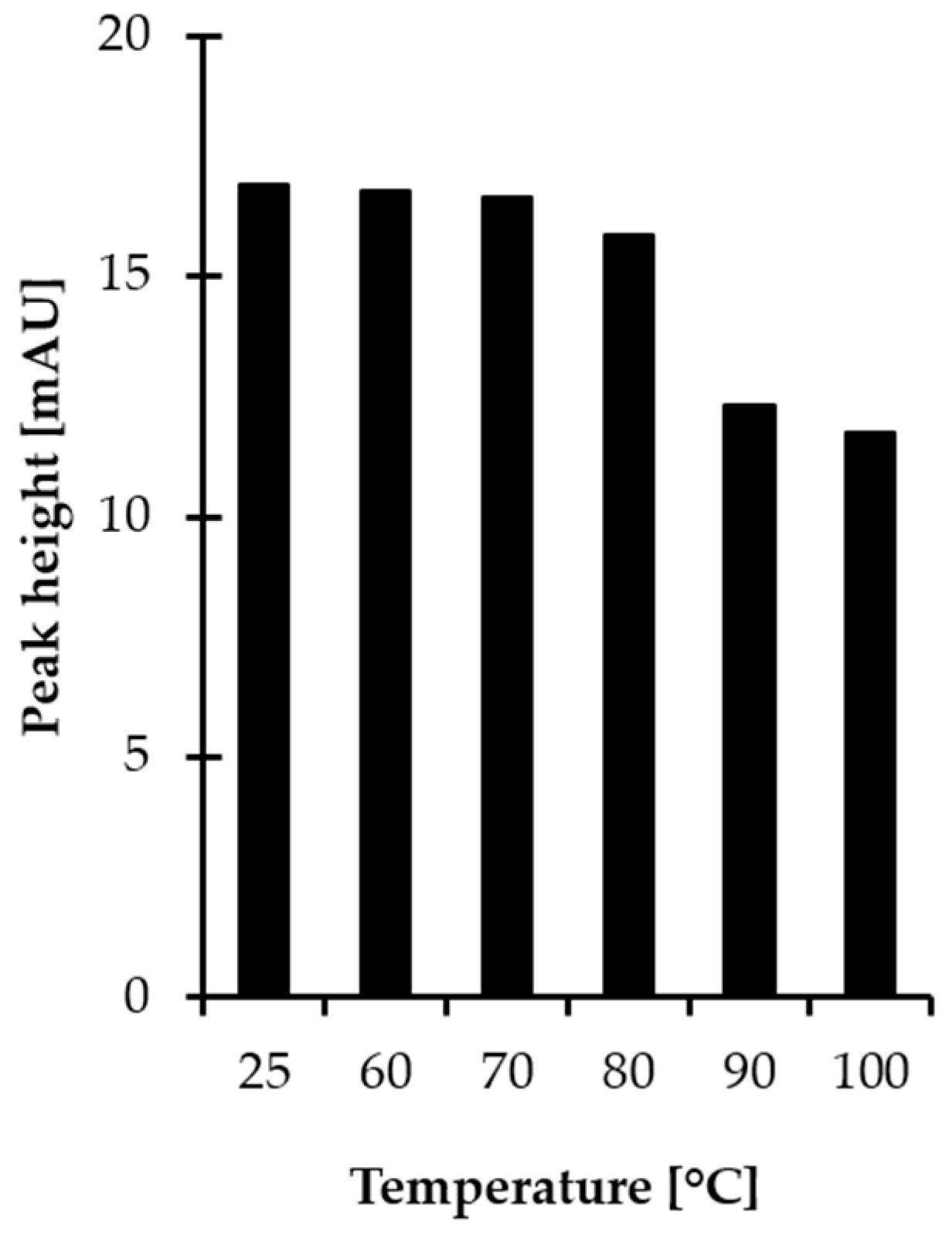
2.1.3. Derivatization
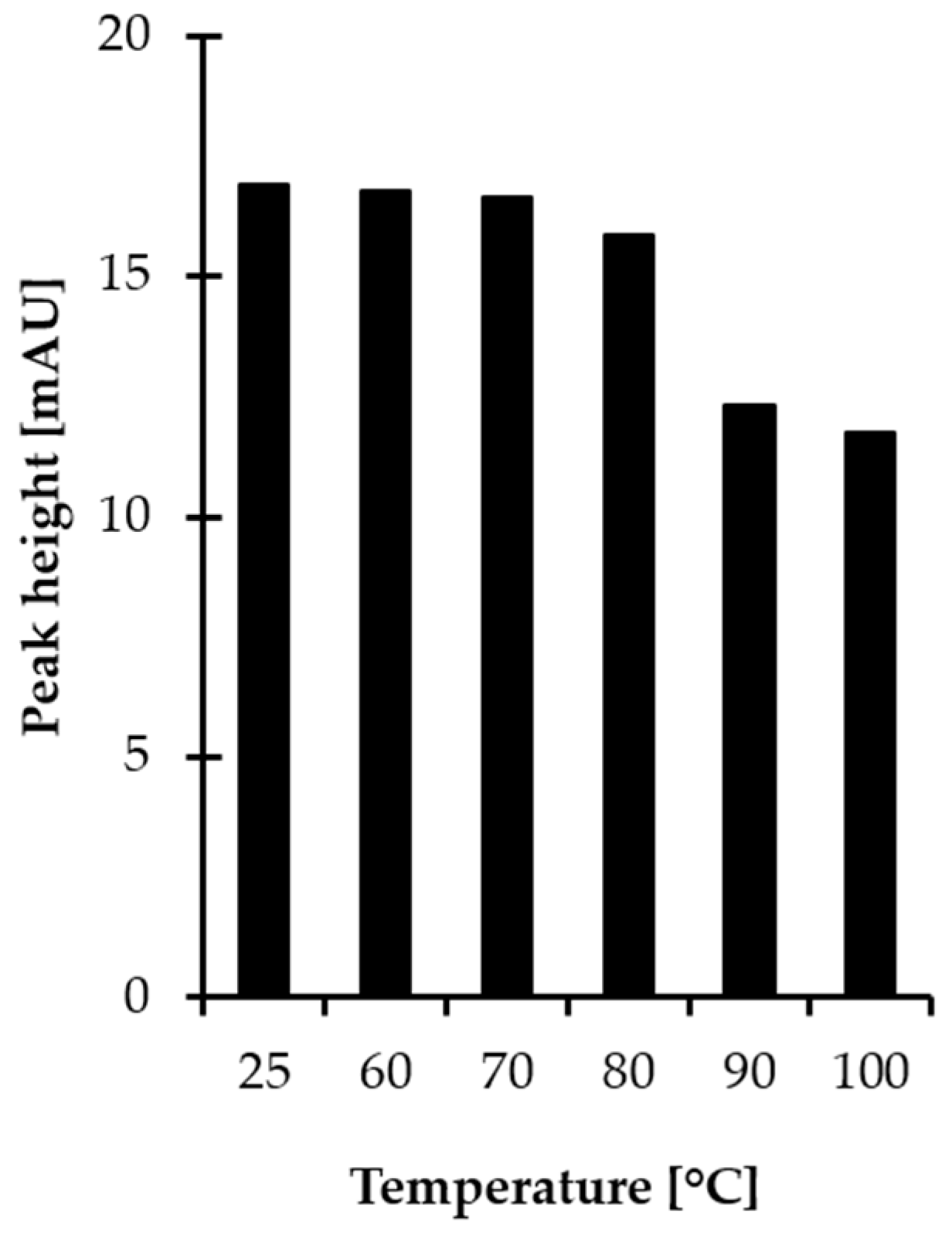
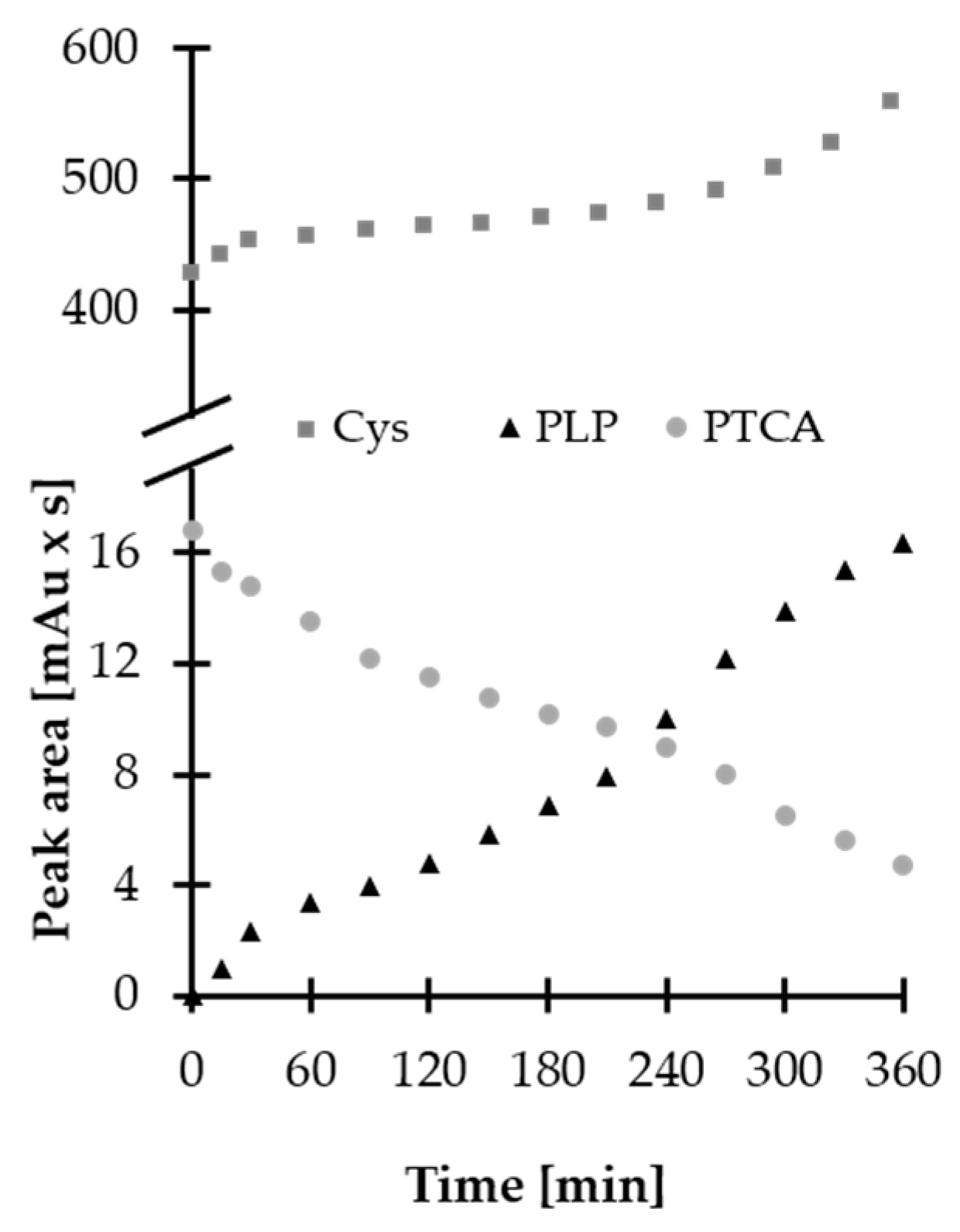
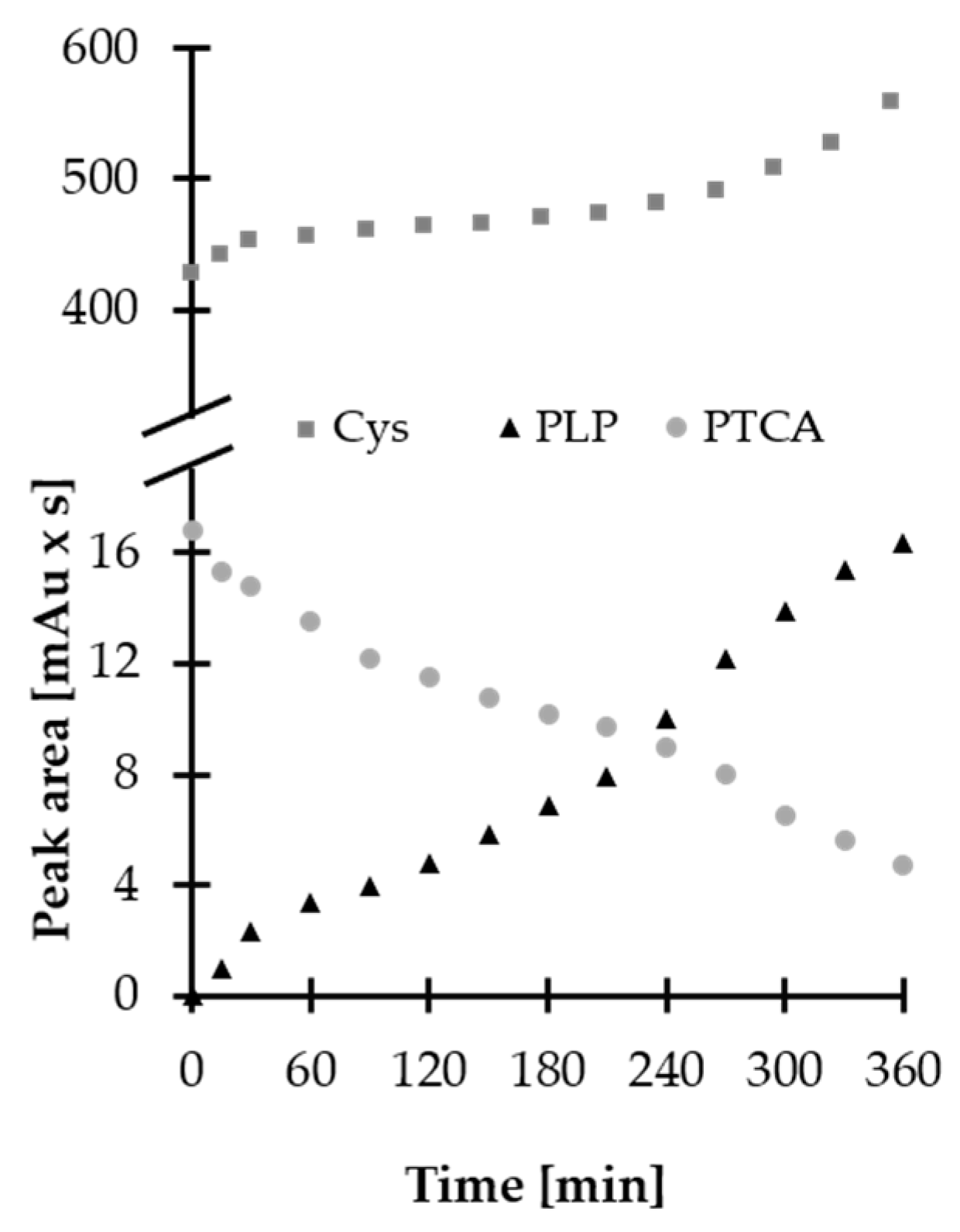
2.2. Stability of HPPTCA
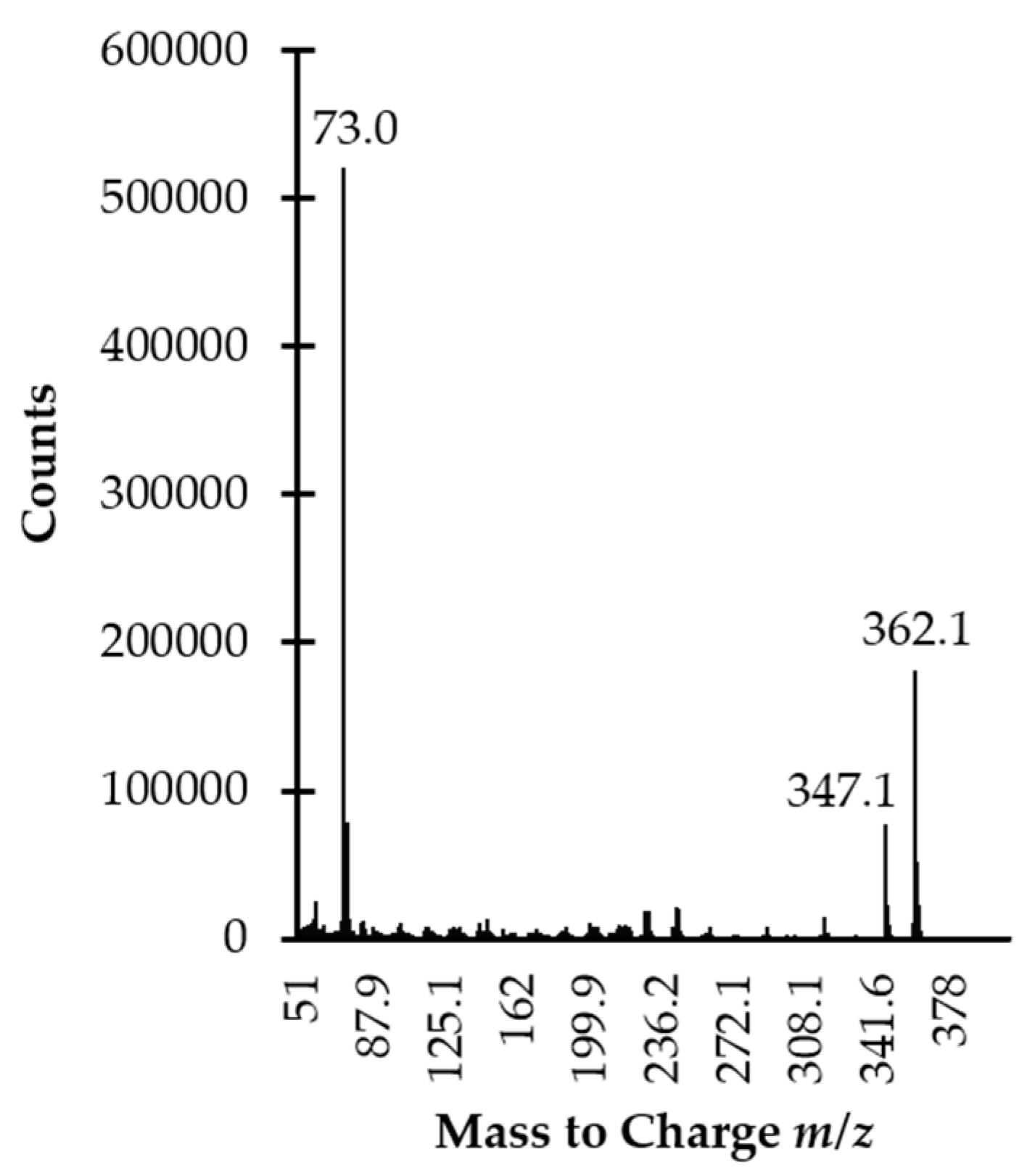
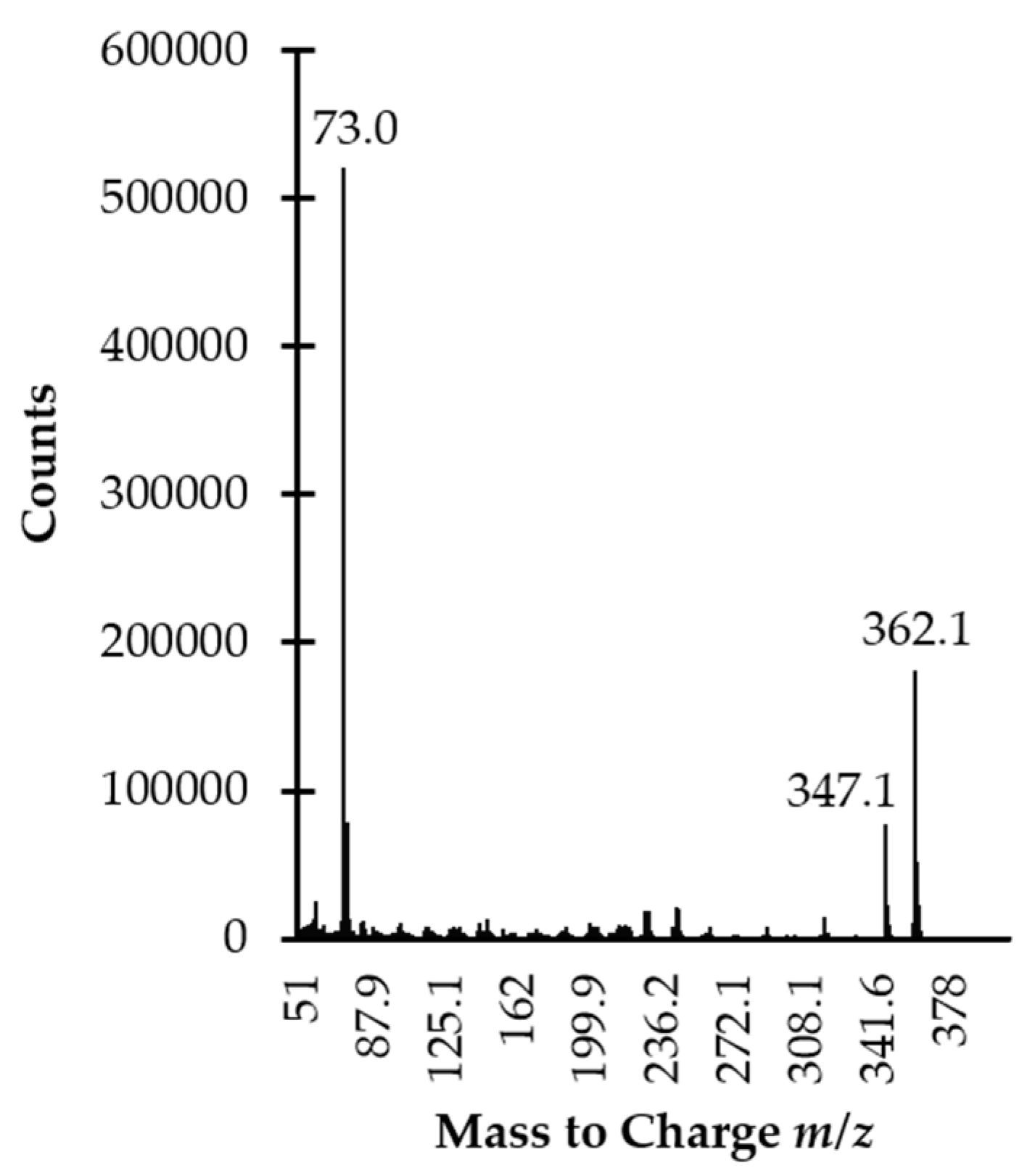
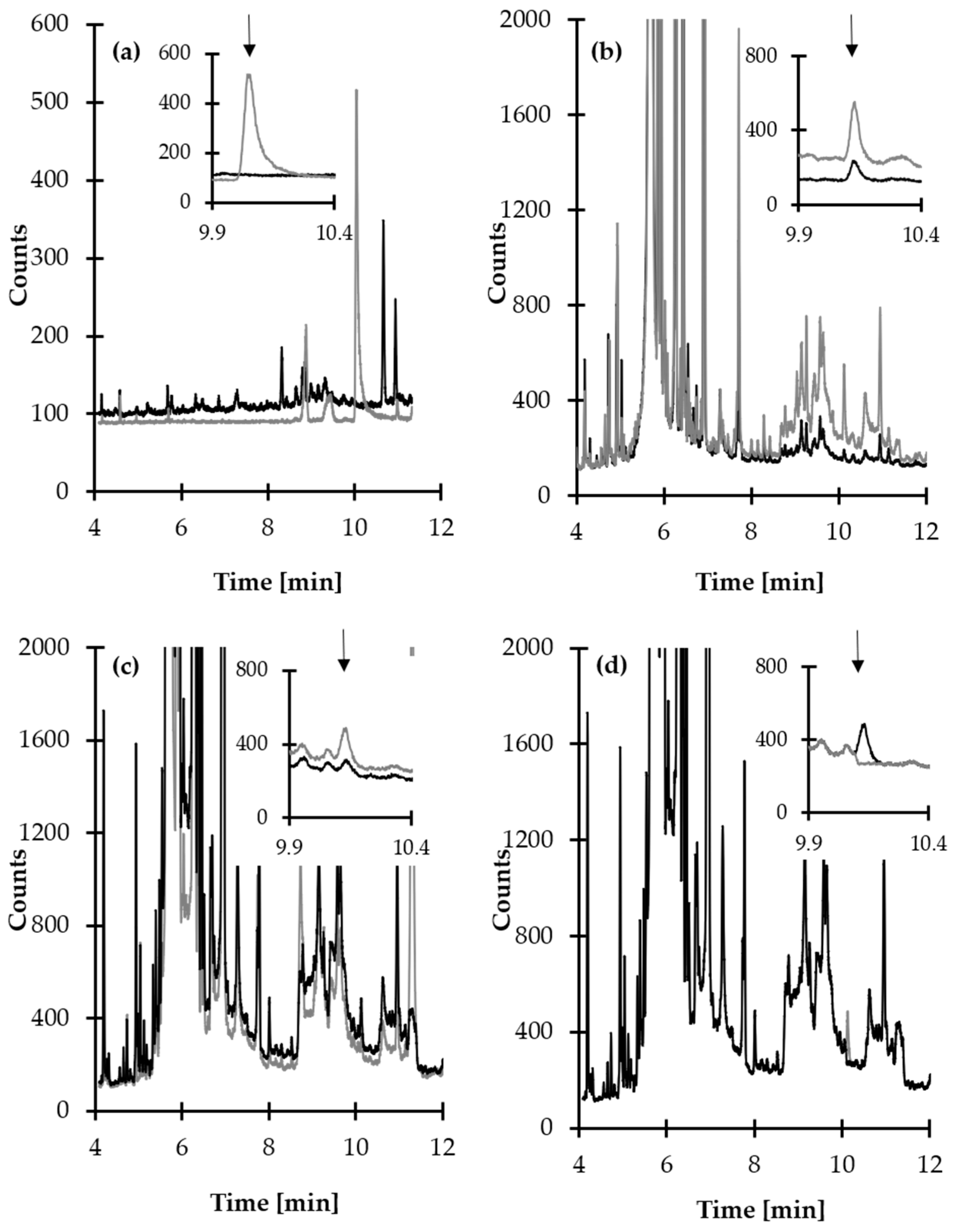
2.3. GC Separation and MS Detection
2.4. Validation of Analytical Method
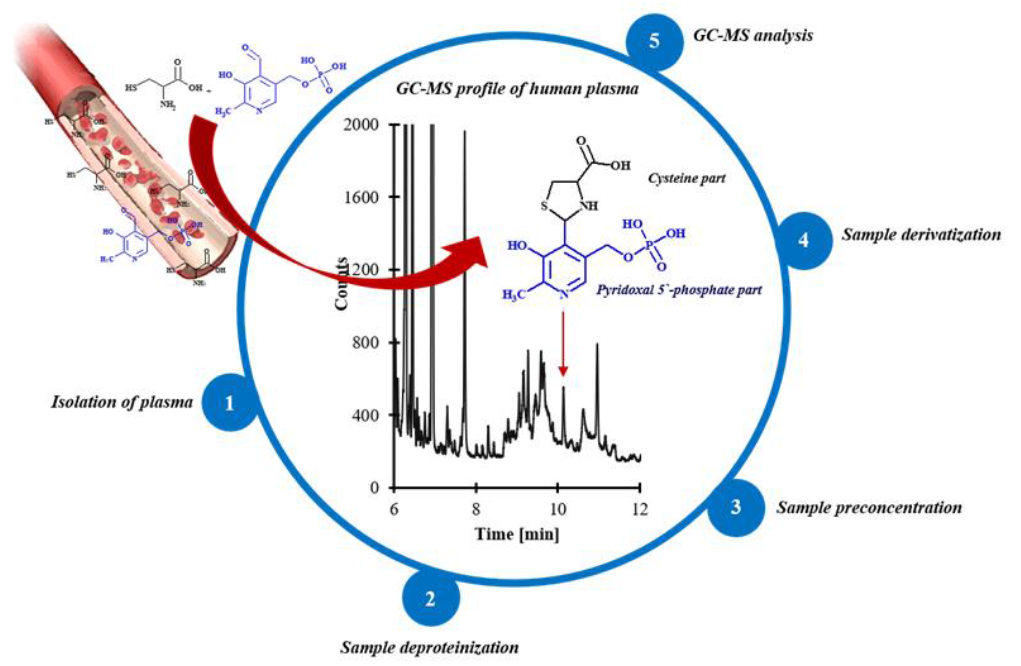
2.5. Application of the Method
3. Materials and Methods
3.1. Reagents and Materials
3.2. Instrumentation
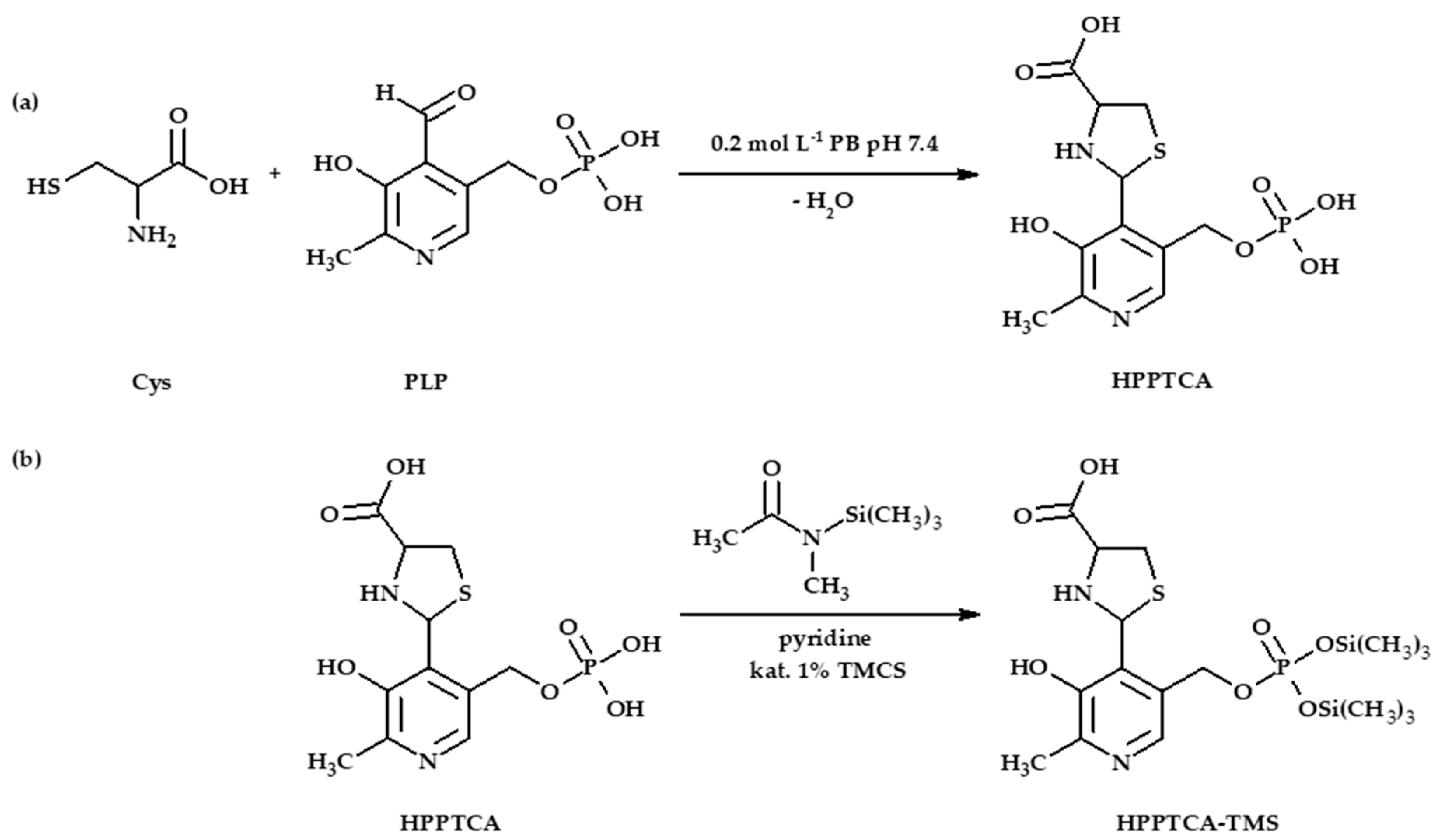
3.3. Synthesis of HPPTCA
3.4. Stock Solution of HPPTCA
3.5. Biological Samples Collection
3.6. Plasma Specimen Preparation for HPPTCA Quantification by GC-MS
3.7. GC-MS Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CMLT | 2-chloro-1-methyllepidinium tetrafluoroborate |
| Cys | cysteine |
| Cys2 | cystine |
| CV | coefficient of variation |
| DAD | diode-array detector |
| EDTA | disodium ethylenediaminetetraacetate |
| EtOH | ethanol |
| GC | gas chromatography |
| Hcy | homocysteine |
| HILIC | hydrophilic interaction chromatography |
| HPLC | high performance liquid chromatography |
| HPPTCA | 2-(3-hydroxy-5-phosphonooxymethyl-2-methyl-4-pyridyl)-1,3-thiazolidine-4-carboxylic acid |
| i-PrOH | 2-propanol |
| LOQ | limit of quantification |
| MeCN | acetonitrile |
| MeOH | methanol |
| MS | mass spectrometry |
| MSTFA | N-trimethylsilyl-N-methyl trifluoroacetamide |
| NMR | nuclear magnetic resonance spectroscopy |
| PB | phosphate buffer |
| PCA | perchloric acid |
| PES | polyethersulfone membrane |
| PL | pyridoxal |
| PLP | pyridoxal 5′-phosphate |
| PN | pyridoxine |
| R | correlation coefficient |
| TCA | trichloroacetic acid |
| TFA | trifluoroacetic acid |
| TMCS | trimethylchlorosilane |
| TMS | trimethylsilyl group |
| UV | spectrophotometric detection |
References
- Oliveira, P.; Laurindo, F. Implications of plasma thiol redox in disease. Clin. Sci. 2018, 132, 1257–1280. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Caruso, P. The controversial role of homocysteine in neurology: From labs to clinical practice. Int. J. Mol. Sci. 2019, 20, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, T.; Arora, R.; Bansal, A.; Bhattacharya, R.; Sharma, G.; Singh, L. Disturbed homocysteine metabolism is associated with cancer. Exp. Mol. Med. 2019, 51, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Chrysant, S.G.; Chrysant, G.S. The current status of homocysteine as a risk factor for cardiovascular disease: A mini review. Expert Rev. Cardiovasc. 2018, 16, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and dementia: An international consensus statement. J. Alzheimer’s Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martí-Carvajal, A.; Solà, I.; Lathyris, D.; Dayer, M. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst. Rev. 2015, 1, CD006612. [Google Scholar] [CrossRef]
- Smith, A.D.; Refsum, H.; Homocysteine, B. Vitamins, and cognitive impairment. Annu. Rev. Nutr. 2016, 36, 211–239. [Google Scholar] [CrossRef]
- Azzini, E.; Ruggeri, S.; Polito, A. Homocysteine: Its possible emerging role in at-risk population groups. Int. J. Mol. Sci. 2020, 21, 1421. [Google Scholar] [CrossRef] [Green Version]
- Midttun, Ø.; Hustad, S.; Schneede, J.; Vollset, S.E.; Ueland, P.M. Plasma vitamin B-6 forms and their relation to transsulfuration metabolites in a large, population-based study. Am. J. Clin. Nutr. 2007, 86, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, H.; Mooney, S. Vitamin B6: A molecule for human health? Molecules 2010, 15, 442–459. [Google Scholar] [CrossRef] [Green Version]
- da Silva, V.R.; Russell, K.A.; Gregory, J.F. Vitamin B6. In Present Knowledge in Nutrition; Erdman, J., Macdonald, I., Zeisel, S., Eds.; Wiley-Blackwell: Oxford, UK, 2012; pp. 307–320. ISBN 9780470959176. [Google Scholar]
- Matsuo, Y. Formation of schiff bases of pyridoxal phosphate. Reaction with metal ions. J. Am. Chem. Soc. 1957, 79, 2011–2015. [Google Scholar] [CrossRef]
- Buell, M.V.; Hansen, R.E. Reaction of pyridoxal-5-phosphate with aminothiols. J. Am. Chem. Soc. 1960, 82, 6042–6049. [Google Scholar] [CrossRef]
- Bergel, F.; Harrap, K. Interaction between carbonyl groups and biologically essential substituents. Part 111 The formation of a thiazolidine derivative in aqueous solution from pyridoxal phosphate and l-cysteine. J. Chem. Soc. 1961, 789, 4051–4056. [Google Scholar] [CrossRef]
- Mackay, D. The mechanism of the reaction of cysteine with pyridoxal 5′-phosphate. Arch. Biochem. Biophys. 1962, 99, 93–100. [Google Scholar] [CrossRef]
- Mackay, D.; Shepherd, D.M. Inhibition of histidine decarboxylases and interaction of the inhibitors with pyridoxal 5′-phosphate. Biochim. Biophys. Acta 1962, 59, 553–561. [Google Scholar] [CrossRef]
- Pestaña, A.; Sandoval, I.; Sols, A. Inhibition by homocysteine of serine dehydratase and other pyridoxal 5′-phosphate enzymes of the rat through cofactor blockage. Arch. Biochem. Biophys. 1971, 146, 373–379. [Google Scholar] [CrossRef]
- Tunnicliff, G.; Ngo, T.T. The mode of action of homocysteine on mouse brain glutamic decarboxylase and γ-aminobutyrate aminotransferase. Can. J. Biochem. 1977, 55, 1013–1018. [Google Scholar] [CrossRef]
- Griffiths, R.; Williams, D.; O’Neill, C.; Dewhurst, I.; Ekuwem, C.; Sinclair, C. Synergistic inhibition of [3H]muscimol binding to calf-brain synaptic membranes in the presence of l-homocysteine and pyridoxal 5′-phosphate. Eur. J. Biochem. 1983, 137, 467–478. [Google Scholar] [CrossRef]
- Terzuoli, L.; Leoncini, R.; Pagani, R.; Guerranti, R.; Vannoni, D.; Ponticelli, F.; Marinello, E. Some chemical properties and biological role of thiazolidine compounds. Life Sci. 1998, 63, 1251–1267. [Google Scholar] [CrossRef]
- Głowacki, R.; Stachniuk, J.; Borowczyk, K.; Jakubowski, H. Quantification of homocysteine and cysteine by derivatization with pyridoxal 5′-phosphate and hydrophilic interaction liquid chromatography. Anal. Bioanal. Chem. 2016, 408, 1935–1941. [Google Scholar] [CrossRef]
- Stachniuk, J.; Kubalczyk, P.; Furmaniak, P.; Głowacki, R. A versatile method for analysis of saliva, plasma and urine for total thiols using HPLC with UV detection. Talanta 2016, 155. [Google Scholar] [CrossRef] [PubMed]
- Turitto, V.; Slack, S. Blood and related fluids. In Handbook of Biomaterial Properties; Murphy, W., Black, J., Hastings, G., Eds.; Springer: New York, NY, USA, 2016; pp. 115–124. ISBN 9781493933051. [Google Scholar]
- Halket, J.; Zaikin, V. Derivatization in mass spectrometry—1. Silylation. Eur. J. Mass Spectrom. 2003, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zaikin, V.; Halket, J. Derivatization in mass spectrometry—2. Acylation. Eur. J. Mass Spectrom. 2003, 9, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Halket, J.M.; Zaikin, V.G. Zaikin derivatization in mass spectrometry—3. Alkylation (arylation). Eur. J. Mass Spectrom. 2004, 10, 1–19. [Google Scholar] [CrossRef]
- Hušek, P. Chromatography: Gas derivatization. In Encyclopedia of Separation Science; Poole, C., Cooke, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2000; pp. 434–443. ISBN 9780080917795. [Google Scholar]
- Sakuda, S.; Tanaka, S.; Mizuno, K.; Sukcharoen, O.; Nihira, T.; Yamada, Y. Biosynthetic studies on virginiae butanolide A, a butyrolactone autoregulator from Streptomyces. Part 2. Preparation of possible biosynthetic intermediates and conversion experiments in a cell-free system. J. Chem. Soc. Perkin Trans. 1993, 19, 2309–2315. [Google Scholar] [CrossRef]
- Piechocka, J.; Wrońska, M.; Chwatko, G.; Jakubowski, H.; Głowacki, R. Quantification of homocysteine thiolactone in human saliva and urine by gas chromatography-mass spectrometry. J. Chromatogr. B 2020, in press. [Google Scholar] [CrossRef]
- FDA. Bioanalytical Method Validation Guidance for Industry; FDA: Rockville, MD, USA, 2018.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acceptance Criteria | Value |
|---|---|
| CV of retention time ≤ 1% | 0.04% |
| Assymetry 0.8–1.5 | 0.94 |
| Number of theoretical plates ≥ 2000 | 8306 |
| Regression Equation | R | CV Slope (%) | Linear Range (µmol L−1) | Intermediate Precision (%) | Intermediate Accuracy (%) | LOQ (µmol L−1) | ||
|---|---|---|---|---|---|---|---|---|
| Min | Max | Min | Max | |||||
| y = 0.9321x + 2.7134 | 0.9984 | 3.1 | 1–20 | 2.58 | 14.35 | 94.85 | 104.83 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piechocka, J.; Wrońska, M.; Głowacka, I.E.; Głowacki, R. 2-(3-Hydroxy-5-phosphonooxymethyl-2-methyl-4-pyridyl)-1,3-thiazolidine-4-carboxylic Acid, Novel Metabolite of Pyridoxal 5′-Phosphate and Cysteine Is Present in Human Plasma—Chromatographic Investigations. Int. J. Mol. Sci. 2020, 21, 3548. https://doi.org/10.3390/ijms21103548
Piechocka J, Wrońska M, Głowacka IE, Głowacki R. 2-(3-Hydroxy-5-phosphonooxymethyl-2-methyl-4-pyridyl)-1,3-thiazolidine-4-carboxylic Acid, Novel Metabolite of Pyridoxal 5′-Phosphate and Cysteine Is Present in Human Plasma—Chromatographic Investigations. International Journal of Molecular Sciences. 2020; 21(10):3548. https://doi.org/10.3390/ijms21103548
Chicago/Turabian StylePiechocka, Justyna, Monika Wrońska, Iwona E. Głowacka, and Rafał Głowacki. 2020. "2-(3-Hydroxy-5-phosphonooxymethyl-2-methyl-4-pyridyl)-1,3-thiazolidine-4-carboxylic Acid, Novel Metabolite of Pyridoxal 5′-Phosphate and Cysteine Is Present in Human Plasma—Chromatographic Investigations" International Journal of Molecular Sciences 21, no. 10: 3548. https://doi.org/10.3390/ijms21103548
APA StylePiechocka, J., Wrońska, M., Głowacka, I. E., & Głowacki, R. (2020). 2-(3-Hydroxy-5-phosphonooxymethyl-2-methyl-4-pyridyl)-1,3-thiazolidine-4-carboxylic Acid, Novel Metabolite of Pyridoxal 5′-Phosphate and Cysteine Is Present in Human Plasma—Chromatographic Investigations. International Journal of Molecular Sciences, 21(10), 3548. https://doi.org/10.3390/ijms21103548