COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-Chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males?

 ,
,  ,
, 
Abstract
1. Introduction
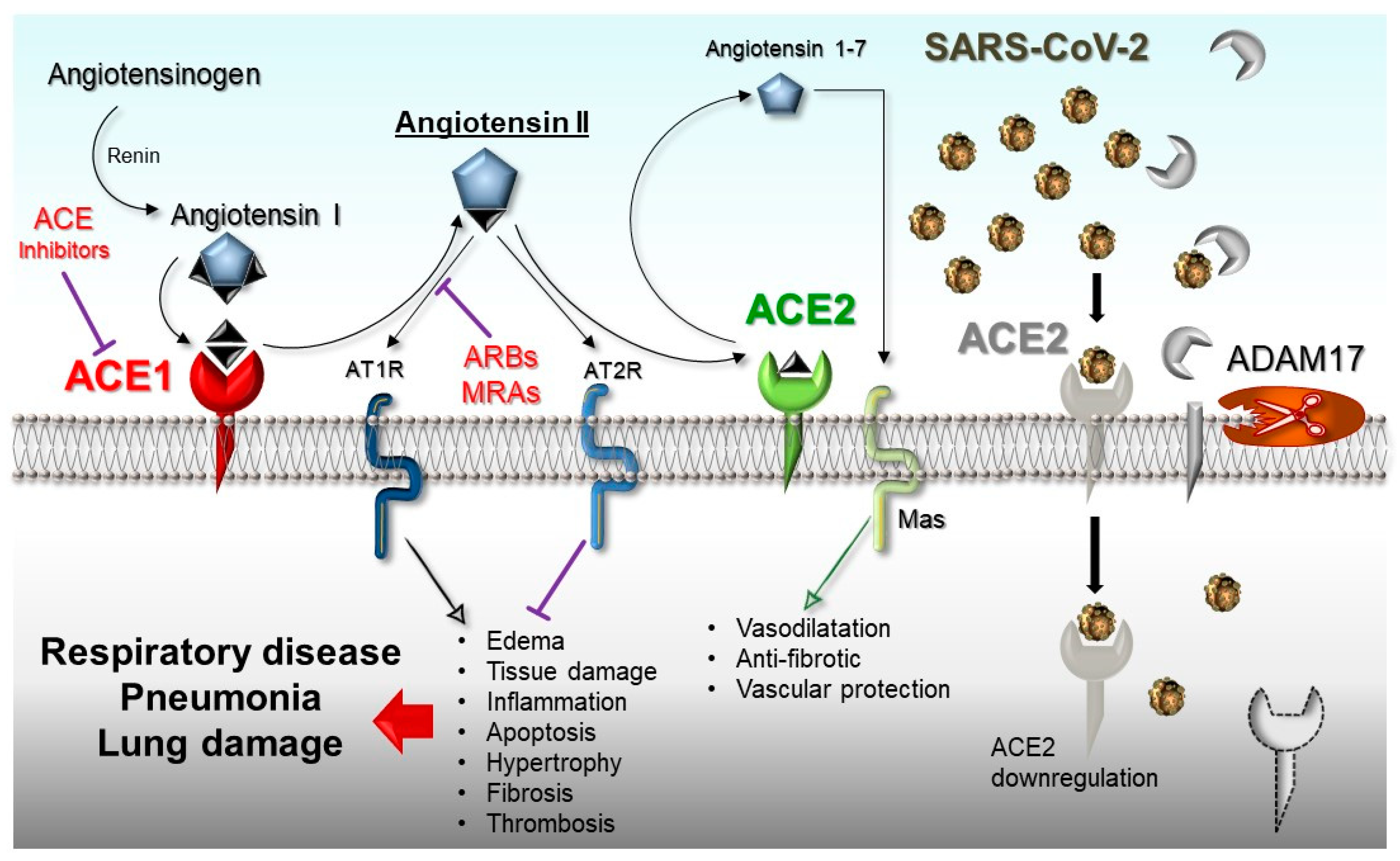
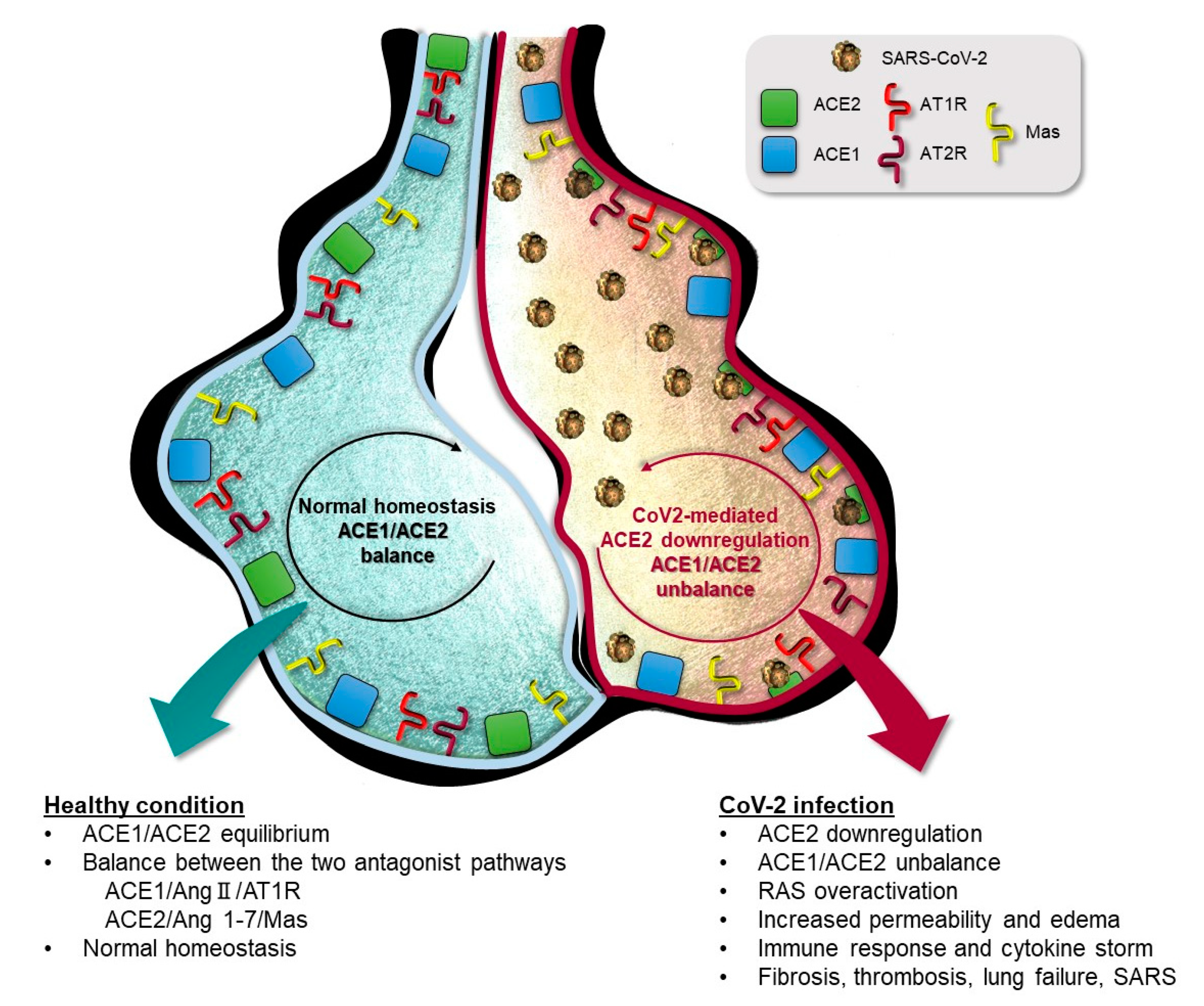
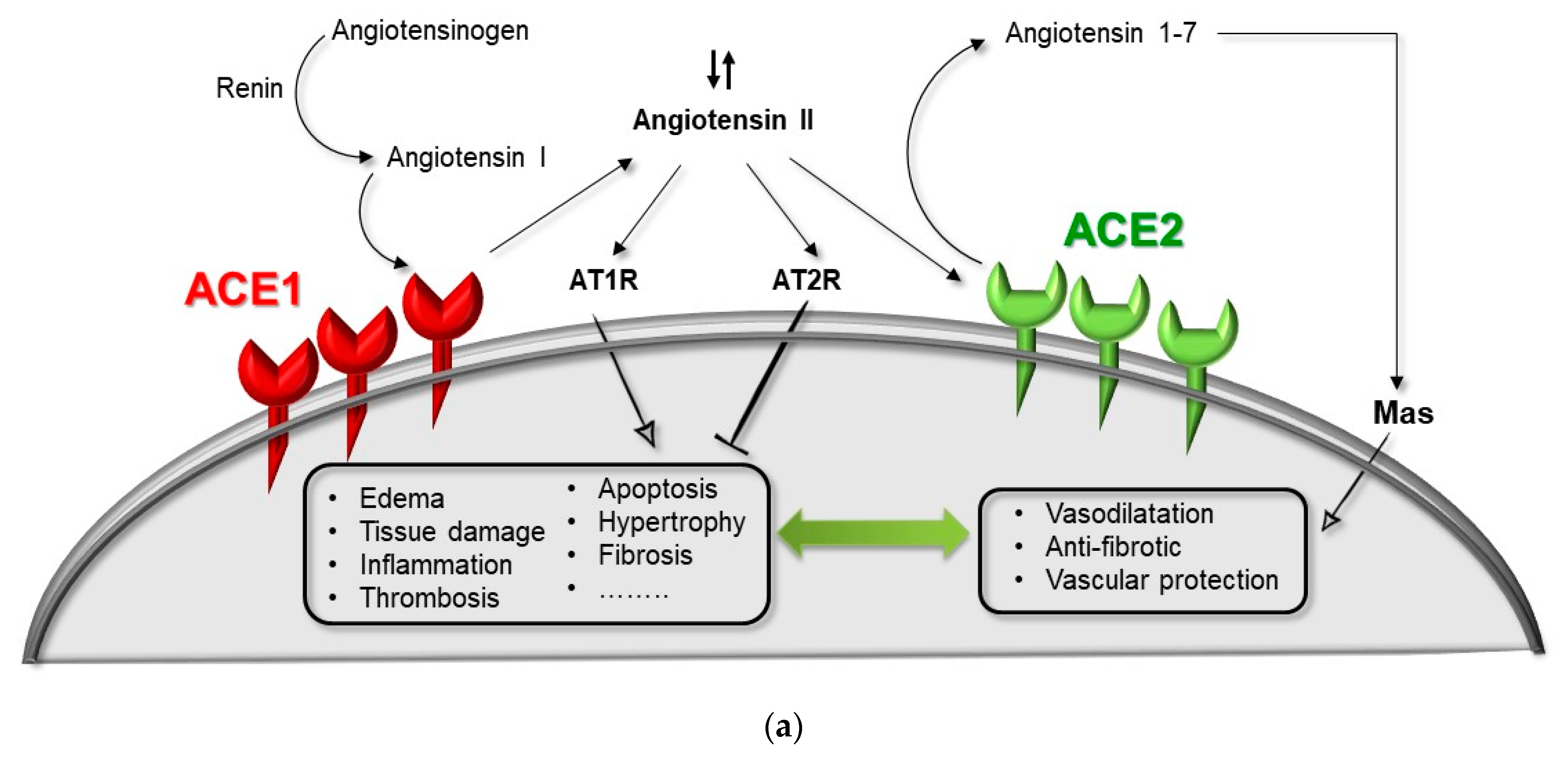
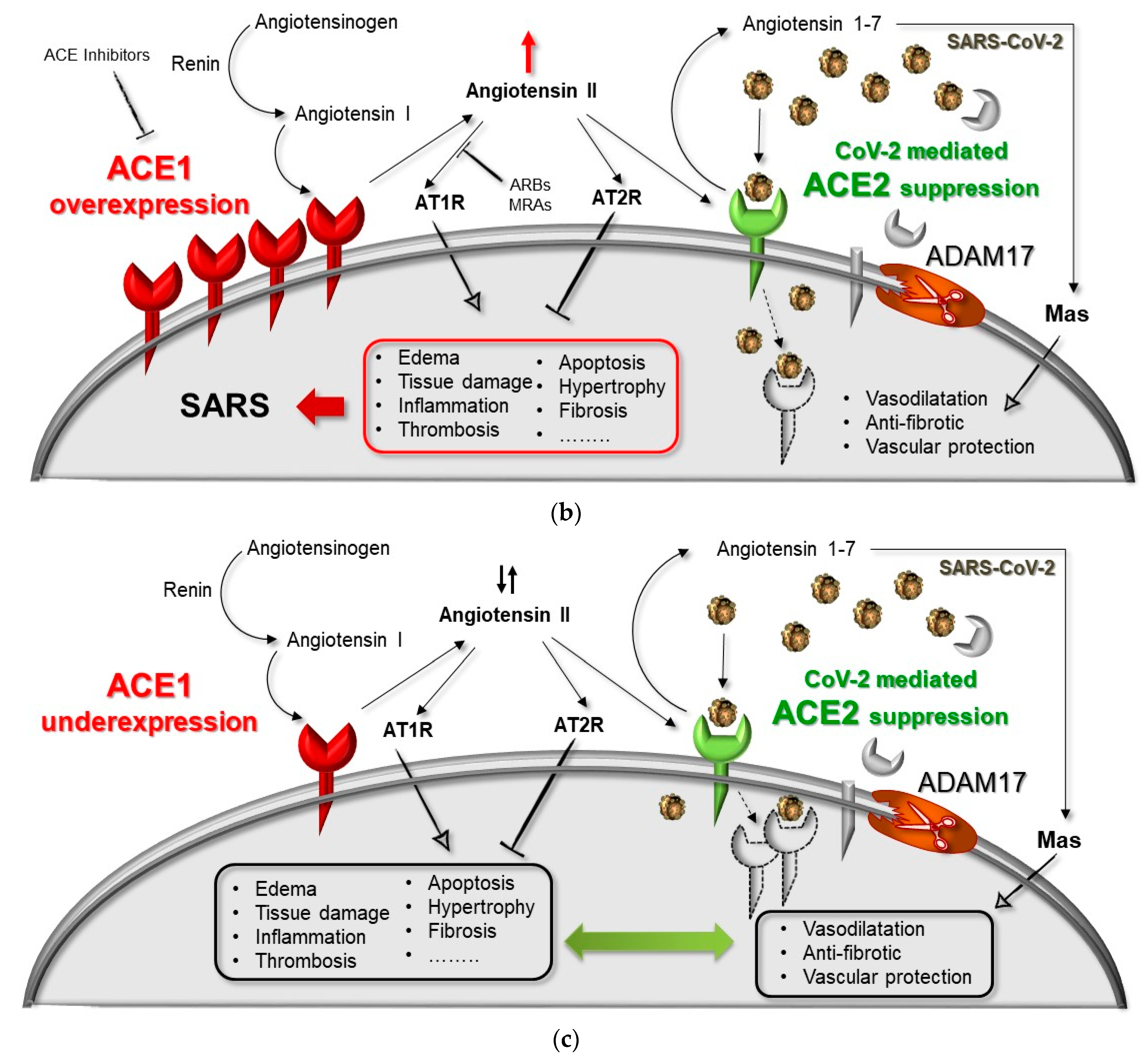
2. ACE1/ACE2 Pathway and Acute Respiratory Distress Syndrome (ARDS)
ACE1 and ACE2 Genes
3. Immune Processes: An X-Related View
4. Inflammatory Processes: An X-Related View
5. Habits, Gender and Environmental Related Risks
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Peiris, J.S.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L., Jr.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Alagaili, A.N.; Briese, T.; Mishra, N.; Kapoor, V.; Sameroff, S.C.; Burbelo, P.D.; de Wit, E.; Munster, V.J.; Hensley, L.E.; Zalmout, I.S.; et al. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. mBio 2014, 5, e00884-14. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Gunther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, T.; Fouchier, R.A.; Schutten, M.; Rimmelzwaan, G.F.; van Amerongen, G.; van Riel, D.; Laman, J.D.; de Jong, T.; van Doornum, G.; Lim, W.; et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 2003, 362, 263–270. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fett, C.; Mack, M.; Ten Eyck, P.P.; Meyerholz, D.K.; Perlman, S. Sex-Based Differences in Susceptibility to Severe Acute Respiratory Syndrome Coronavirus Infection. J. Immunol. 2017, 198, 4046–4053. [Google Scholar] [CrossRef]
- Casimir, G.J.; Lefevre, N.; Corazza, F.; Duchateau, J. Sex and inflammation in respiratory diseases: A clinical viewpoint. Biol. Sex Differ. 2013, 4, 16. [Google Scholar] [CrossRef]
- Nicholls, J.M.; Poon, L.L.; Lee, K.C.; Ng, W.F.; Lai, S.T.; Leung, C.Y.; Chu, C.M.; Hui, P.K.; Mak, K.L.; Lim, W.; et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet 2003, 361, 1773–1778. [Google Scholar] [CrossRef]
- Chen, J.; Subbarao, K. The Immunobiology of SARS*. Annu. Rev. Immunol. 2007, 25, 443–472. [Google Scholar] [CrossRef]
- Su, L.; Ma, X.; Yu, H.; Zhang, Z.; Bian, P.; Han, Y.; Sun, J.; Liu, Y.; Yang, C.; Geng, J.; et al. The different clinical characteristics of corona virus disease cases between children and their families in China—The character of children with COVID-19. Emerg. Microbes Infect. 2020, 9, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Lam, C.W.; Li, A.M.; Wong, C.K.; Cheng, F.W.; Leung, T.F.; Hon, E.K.; Chan, I.H.; Li, C.K.; Fung, K.S.; et al. Inflammatory cytokine profile in children with severe acute respiratory syndrome. Pediatrics 2004, 113, e7–e14. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Serino, M.L.; Tognazzo, S.; Ongaro, A.; Moratelli, S.; Gilli, G.; Forini, E.; De Mattei, M.; Scapoli, G.L. The reduced sensitivity of the ProC Global test in protein S deficient subjects reflects a reduction in the associated thrombotic risk. Blood Coagul. Fibrinolysis 2001, 12, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Serino, M.L.; Moratelli, S.; Tognazzo, S.; Ongaro, A.; Scapoli, G.L. Coexistence of factor V G1691A and factor II G20210A gene mutations in a thrombotic family is associated with recurrence and early onset of venous thrombosis. Haemostasis 2001, 31, 99–105. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Moratelli, S.; Mari, R.; Ballerini, G.; Scapoli, G.L. Coexistence of antithrombin deficiency, factor V Leiden and hyperhomocysteinemia in a thrombotic family. Blood Coagul. Fibrinolysis 1998, 9, 173–176. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Verzola, I.; Mari, R.; Moratelli, S.; Ballerini, G. Resistance to activated protein C and low levels of protein S activity in nine thrombophilic families: A correct diagnosis. Blood Coagul. Fibrinolysis 1997, 8, 118–123. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Mari, R.; Verzola, I.; Moratelli, S.; Ballerini, G. Different anticoagulant response to activated protein C (APC test) and to Agkistrodon contortix venom (ACV test) in a family with FV-R506Q substitution. Clin. Appl. Thromb. Hemost. 1997, 3, 168–173. [Google Scholar] [CrossRef]
- Yin, S.; Huang, M.; Li, D.; Tang, N. Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J. Thromb. Thrombolysis 2020. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-up. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef]
- Thachil, J. The versatile heparin in COVID-19. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Karlberg, J.; Chong, D.S.; Lai, W.Y. Do men have a higher case fatality rate of severe acute respiratory syndrome than women do? Am. J. Epidemiol. 2004, 159, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.N.; Earnest, A.; Lim, H.H.; Chin, C.F.; Tan, C.; Puhaindran, M.E.; Tan, A.; Chen, M.I.; Leo, Y.S. SARS in Singapore--predictors of disease severity. Ann. Acad. Med. Singap. 2006, 35, 326–331. [Google Scholar] [PubMed]
- Alghamdi, I.G.; Hussain, I.I.; Almalki, S.S.; Alghamdi, M.S.; Alghamdi, M.M.; El-Sheemy, M.A. The pattern of Middle East respiratory syndrome coronavirus in Saudi Arabia: A descriptive epidemiological analysis of data from the Saudi Ministry of Health. Int. J. Gen. Med. 2014, 7, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Tzoran, I.; Hoffman, R.; Monreal, M. Hemostasis and Thrombosis in the Oldest Old. Semin. Thromb. Hemost. 2018, 44, 624–631. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Hannah, M.F.; Bajic, V.B.; Klein, S.L. Sex differences in the recognition of and innate antiviral responses to Seoul virus in Norway rats. Brain Behav. Immun. 2008, 22, 503–516. [Google Scholar] [CrossRef]
- Robinson, D.P.; Hall, O.J.; Nilles, T.L.; Bream, J.H.; Klein, S.L. 17beta-estradiol protects females against influenza by recruiting neutrophils and increasing virus-specific CD8 T cell responses in the lungs. J. Virol. 2014, 88, 4711–4720. [Google Scholar] [CrossRef]
- Robinson, D.P.; Huber, S.A.; Moussawi, M.; Roberts, B.; Teuscher, C.; Watkins, R.; Arnold, A.P.; Klein, S.L. Sex chromosome complement contributes to sex differences in coxsackievirus B3 but not influenza A virus pathogenesis. Biol. Sex Differ. 2011, 2, 8. [Google Scholar] [CrossRef]
- Rettew, J.A.; Huet-Hudson, Y.M.; Marriott, I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol. Reprod. 2008, 78, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H. The complex role of estrogens in inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef]
- Malkin, C.J.; Pugh, P.J.; Jones, R.D.; Kapoor, D.; Channer, K.S.; Jones, T.H. The effect of testosterone replacement on endogenous inflammatory cytokines and lipid profiles in hypogonadal men. J. Clin. Endocrinol. Metab. 2004, 89, 3313–3318. [Google Scholar] [CrossRef] [PubMed]
- Peretz, J.; Pekosz, A.; Lane, A.P.; Klein, S.L. Estrogenic compounds reduce influenza A virus replication in primary human nasal epithelial cells derived from female, but not male, donors. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L415–L425. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020. [Google Scholar] [CrossRef]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020. [Google Scholar] [CrossRef]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pohlmann, S.; Soilleux, E.J. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Brojakowska, A.; Narula, J.; Shimony, R.; Bander, J. Clinical Implications of SARS-Cov2 Interaction with Renin Angiotensin System. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef]
- Zhou, M.; Dai, W.; Cui, Y.; Li, Y. Estrogen downregulates gp130 expression in HUVECs by regulating ADAM10 and ADAM17 via the estrogen receptor. Biochem. Biophys. Res. Commun. 2020, 523, 753–758. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.W.; Tang, N.L.; Hui, D.S.; Chung, G.T.; Chim, S.S.; Chan, K.C.; Sung, Y.M.; Chan, L.Y.; Tong, Y.K.; Lee, W.S.; et al. ACE2 gene polymorphisms do not affect outcome of severe acute respiratory syndrome. Clin. Chem. 2004, 50, 1683–1686. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Asselta, R.; Paraboschi, E.M.; Mantovani, A.; Duga, S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. medRxiv 2020. [Google Scholar] [CrossRef]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhou, J.; To, K.K.; Chu, H.; Li, C.; Wang, D.; Yang, D.; Zheng, S.; Hao, K.; Bosse, Y.; et al. Identification of TMPRSS2 as a Susceptibility Gene for Severe 2009 Pandemic A(H1N1) Influenza and A(H7N9) Influenza. J. Infect. Dis 2015, 212, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Baena, E.; Shao, Z.; Linn, D.E.; Glass, K.; Hamblen, M.J.; Fujiwara, Y.; Kim, J.; Nguyen, M.; Zhang, X.; Godinho, F.J.; et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013, 27, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef] [PubMed]
- Crowley, S.D.; Gurley, S.B.; Oliverio, M.I.; Pazmino, A.K.; Griffiths, R.; Flannery, P.J.; Spurney, R.F.; Kim, H.S.; Smithies, O.; Le, T.H.; et al. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J. Clin. Investig. 2005, 115, 1092–1099. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol. Metab. 2004, 15, 166–169. [Google Scholar] [CrossRef]
- Olkowicz, M.; Chlopicki, S.; Smolenski, R.T. Perspectives for angiotensin profiling with liquid chromatography/mass spectrometry to evaluate ACE/ACE2 balance in endothelial dysfunction and vascular pathologies. Pharm. Rep. 2015, 67, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Fraga-Silva, R.A.; Sorg, B.S.; Wankhede, M.; Dedeugd, C.; Jun, J.Y.; Baker, M.B.; Li, Y.; Castellano, R.K.; Katovich, M.J.; Raizada, M.K.; et al. ACE2 activation promotes antithrombotic activity. Mol. Med. 2010, 16, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kang, Z.; Gong, H.; Xu, D.; Wang, J.; Li, Z.; Cui, X.; Xiao, J.; Meng, T.; Zhou, W.; et al. The digestive system is a potential route of 2019-nCov infection: A bioinformatics analysis based on single-cell transcriptomes. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. BioRxiv 2020. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Vicenzi, E.; Canducci, F.; Pinna, D.; Mancini, N.; Carletti, S.; Lazzarin, A.; Bordignon, C.; Poli, G.; Clementi, M. Coronaviridae and SARS-associated coronavirus strain HSR1. Emerg. Infect. Dis 2004, 10, 413–418. [Google Scholar] [CrossRef]
- De Haan, C.A.; Li, Z.; te Lintelo, E.; Bosch, B.J.; Haijema, B.J.; Rottier, P.J. Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J. Virol. 2005, 79, 14451–14456. [Google Scholar] [CrossRef]
- Madu, I.G.; Chu, V.C.; Lee, H.; Regan, A.D.; Bauman, B.E.; Whittaker, G.R. Heparan sulfate is a selective attachment factor for the avian coronavirus infectious bronchitis virus Beaudette. Avian Dis. 2007, 51, 45–51. [Google Scholar] [CrossRef]
- Lang, J.; Yang, N.; Deng, J.; Liu, K.; Yang, P.; Zhang, G.; Jiang, C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE 2011, 6, e23710. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ohishi, M.; Katsuya, T.; Ito, N.; Ikushima, M.; Kaibe, M.; Tatara, Y.; Shiota, A.; Sugano, S.; Takeda, S.; et al. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension 2006, 47, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Herzenberg, A.M.; Kassiri, Z.; Wong, D.; Reich, H.; Khokha, R.; Crackower, M.A.; Backx, P.H.; Penninger, J.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am. J. Pathol. 2006, 168, 1808–1820. [Google Scholar] [CrossRef]
- Zhong, J.; Guo, D.; Chen, C.B.; Wang, W.; Schuster, M.; Loibner, H.; Penninger, J.M.; Scholey, J.W.; Kassiri, Z.; Oudit, G.Y. Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension 2011, 57, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Nakamura, K.; Koibuchi, N.; Nishimatsu, H.; Higashikuni, Y.; Hirata, Y.; Kugiyama, K.; Nagai, R.; Sata, M. Candesartan ameliorates cardiac dysfunction observed in angiotensin-converting enzyme 2-deficient mice. Hypertens. Res. 2008, 31, 1953–1961. [Google Scholar] [CrossRef]
- Haschke, M.; Schuster, M.; Poglitsch, M.; Loibner, H.; Salzberg, M.; Bruggisser, M.; Penninger, J.; Krahenbuhl, S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin. Pharm. 2013, 52, 783–792. [Google Scholar] [CrossRef]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef]
- Zhang, H.; Baker, A. Recombinant human ACE2: Acing out angiotensin II in ARDS therapy. Crit. Care 2017, 21, 305. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Deng, W.; Tong, Z.; Liu, Y.X.; Zhang, L.F.; Zhu, H.; Gao, H.; Huang, L.; Liu, Y.L.; Ma, C.M.; et al. Mice transgenic for human angiotensin-converting enzyme 2 provide a model for SARS coronavirus infection. Comp. Med. 2007, 57, 450–459. [Google Scholar] [PubMed]
- Zhang, R.; Pan, Y.; Fanelli, V.; Wu, S.; Luo, A.A.; Islam, D.; Han, B.; Mao, P.; Ghazarian, M.; Zeng, W.; et al. Mechanical Stress and the Induction of Lung Fibrosis via the Midkine Signaling Pathway. Am. J. Respir. Crit. Care Med. 2015, 192, 315–323. [Google Scholar] [CrossRef]
- Wosten-van Asperen, R.M.; Lutter, R.; Specht, P.A.; Moll, G.N.; van Woensel, J.B.; van der Loos, C.M.; van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Jarcho, J.A.; Ingelfinger, J.R.; Hamel, M.B.; D’Agostino, R.B., Sr.; Harrington, D.P. Inhibitors of the Renin-Angiotensin-Aldosterone System and Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Araujo, F.C.; Milsted, A.; Watanabe, I.K.; Del Puerto, H.L.; Santos, R.A.; Lazar, J.; Reis, F.M.; Prokop, J.W. Similarities and differences of X and Y chromosome homologous genes, SRY and SOX3, in regulating the renin-angiotensin system promoters. Physiol. Genom. 2015, 47, 177–186. [Google Scholar] [CrossRef]
- Yuan, X.; Lu, M.L.; Li, T.; Balk, S.P. SRY interacts with and negatively regulates androgen receptor transcriptional activity. J. Biol. Chem. 2001, 276, 46647–46654. [Google Scholar] [CrossRef]
- Hubert, C.; Houot, A.M.; Corvol, P.; Soubrier, F. Structure of the angiotensin I-converting enzyme gene. Two alternate promoters correspond to evolutionary steps of a duplicated gene. J. Biol. Chem. 1991, 266, 15377–15383. [Google Scholar]
- ClinVar Genomic Variation as It Relates to Human Health. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/18061/ (accessed on 28 February 2020).
- Zhong, W.G.; Wang, Y.; Zhu, H.; Zhao, X. Meta analysis of angiotensin-converting enzyme I/D polymorphism as a risk factor for preeclampsia in Chinese women. Genet. Mol. Res. 2012, 11, 2268–2276. [Google Scholar] [CrossRef]
- Mizuiri, S.; Hemmi, H.; Kumanomidou, H.; Iwamoto, M.; Miyagi, M.; Sakai, K.; Aikawa, A.; Ohara, T.; Yamada, K.; Shimatake, H.; et al. Angiotensin-converting enzyme (ACE) I/D genotype and renal ACE gene expression. Kidney Int. 2001, 60, 1124–1130. [Google Scholar] [CrossRef]
- Itoyama, S.; Keicho, N.; Quy, T.; Phi, N.C.; Long, H.T.; Ha, L.D.; Ban, V.V.; Ohashi, J.; Hijikata, M.; Matsushita, I.; et al. ACE1 polymorphism and progression of SARS. Biochem. Biophys. Res. Commun. 2004, 323, 1124–1129. [Google Scholar] [CrossRef]
- Marshall, R.P.; Webb, S.; Bellingan, G.J.; Montgomery, H.E.; Chaudhari, B.; McAnulty, R.J.; Humphries, S.E.; Hill, M.R.; Laurent, G.J. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2002, 166, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Jerng, J.S.; Yu, C.J.; Wang, H.C.; Chen, K.Y.; Cheng, S.L.; Yang, P.C. Polymorphism of the angiotensin-converting enzyme gene affects the outcome of acute respiratory distress syndrome. Crit. Care Med. 2006, 34, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.; Tang, N.L.; Hui, D.S.; Chung, G.T.; Wu, A.K.; Chim, S.S.; Chiu, R.W.; Lee, N.; Choi, K.W.; Sung, Y.M.; et al. Absence of association between angiotensin converting enzyme polymorphism and development of adult respiratory distress syndrome in patients with severe acute respiratory syndrome: A case control study. BMC Infect. Dis. 2005, 5, 26. [Google Scholar] [CrossRef]
- Oruc, N.; Lamb, J.; Whitcomb, D.J.; Sass, D.A. The ACE gene I/D polymorphism does not affect the susceptibility to or prognosis of PBC. Turk. J. Gastroenterol. 2008, 19, 250–253. [Google Scholar]
- Zhang, Y.F.; Cheng, Q.; Tang, N.L.; Chu, T.T.; Tomlinson, B.; Liu, F.; Kwok, T.C. Gender difference of serum angiotensin-converting enzyme (ACE) activity in DD genotype of ACE insertion/deletion polymorphism in elderly Chinese. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 547–552. [Google Scholar] [CrossRef]
- Bayoglu, B.; Cengiz, M.; Karacetin, G.; Uysal, O.; Kocabasoglu, N.; Bayar, R.; Balcioglu, I. Genetic polymorphism of angiotensin I-converting enzyme (ACE), but not angiotensin II type I receptor (ATr1), has a gender-specific role in panic disorder. Psychiatry Clin. Neurosci. 2012, 66, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Srivastava, A.; Mittal, B. Angiotensin I-converting enzyme insertion/deletion polymorphism and increased risk of gall bladder cancer in women. DNA Cell Biol. 2010, 29, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.Q.; He, F.Z.; Luo, Z.Y.; Wen, J.G.; Wang, L.Y.; Sun, N.L.; Tang, G.F.; Li, Q.; Guo, D.; Liu, Z.Q.; et al. Rs495828 polymorphism of the ABO gene is a predictor of enalapril-induced cough in Chinese patients with essential hypertension. Pharm. Genom. 2014, 24, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Yamagata University Genomic Cohort Consortium (YUGCC). Pleiotropic effect of common variants at ABO Glycosyltranferase locus in 9q32 on plasma levels of pancreatic lipase and angiotensin converting enzyme. PLoS ONE 2014, 9, e55903. [Google Scholar] [CrossRef]
- Terao, C.; Bayoumi, N.; McKenzie, C.A.; Zelenika, D.; Muro, S.; Mishima, M.; Connell, J.M.; Vickers, M.A.; Lathrop, G.M.; Farrall, M.; et al. Quantitative variation in plasma angiotensin-I converting enzyme activity shows allelic heterogeneity in the ABO blood group locus. Ann. Hum. Genet. 2013, 77, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Guillon, P.; Clement, M.; Sebille, V.; Rivain, J.G.; Chou, C.F.; Ruvoen-Clouet, N.; Le Pendu, J. Inhibition of the interaction between the SARS-CoV spike protein and its cellular receptor by anti-histo-blood group antibodies. Glycobiology 2008, 18, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.J.; Canadas-Garre, M.; Cappa, R.C.; Maxwell, A.P.; McKnight, A.J. Genetic associations between genes in the renin-angiotensin-aldosterone system and renal disease: A systematic review and meta-analysis. BMJ Open 2019, 9, e026777. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.B.; Yang, J.K. Meta-analysis of association of ACE2 G8790A polymorphism with Chinese Han essential hypertension. J. Renin Angiotensin Aldosterone Syst. 2009, 10, 31–34. [Google Scholar] [CrossRef]
- Patel, S.K.; Velkoska, E.; Freeman, M.; Wai, B.; Lancefield, T.F.; Burrell, L.M. From gene to protein-experimental and clinical studies of ACE2 in blood pressure control and arterial hypertension. Front. Physiol. 2014, 5, 227. [Google Scholar] [CrossRef]
- Pinheiro, D.S.; Santos, R.S.; Jardim, P.; Silva, E.G.; Reis, A.A.S.; Pedrino, G.R.; Ulhoa, C.J. The combination of ACE I/D and ACE2 G8790A polymorphisms revels susceptibility to hypertension: A genetic association study in Brazilian patients. PLoS ONE 2019, 14, e0221248. [Google Scholar] [CrossRef]
- Wu, Y.H.; Li, J.Y.; Wang, C.; Zhang, L.M.; Qiao, H. The ACE2 G8790A Polymorphism: Involvement in Type 2 Diabetes Mellitus Combined with Cerebral Stroke. J. Clin. Lab. Anal. 2017, 31. [Google Scholar] [CrossRef]
- Yang, M.; Zhao, J.; Xing, L.; Shi, L. The association between angiotensin-converting enzyme 2 polymorphisms and essential hypertension risk: A meta-analysis involving 14,122 patients. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 1240–1244. [Google Scholar] [CrossRef]
- Li, Y.Y. Lack of Association of ACE2 G8790A Gene Mutation with Essential Hypertension in the Chinese Population: A Meta-Analysis Involving 5260 Subjects. Front. Physiol. 2012, 3, 364. [Google Scholar] [CrossRef]
- Benjafield, A.V.; Wang, W.Y.; Morris, B.J. No association of angiotensin-converting enzyme 2 gene (ACE2) polymorphisms with essential hypertension. Am. J. Hypertens. 2004, 17, 624–628. [Google Scholar] [CrossRef]
- Huang, W.; Yang, W.; Wang, Y.; Zhao, Q.; Gu, D.; Chen, R. Association study of angiotensin-converting enzyme 2 gene (ACE2) polymorphisms and essential hypertension in northern Han Chinese. J. Hum. Hypertens. 2006, 20, 968–971. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Imai, Y.; Kuba, K.; Penninger, J.M. [Lessons from SARS: A new potential therapy for acute respiratory distress syndrome (ARDS) with angiotensin converting enzyme 2 (ACE2)]. Masui 2008, 57, 302–310. [Google Scholar] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Zhou, Q. Structure of dimeric full-length human ACE2 in complex with B0AT1. BioRxiv 2020. [Google Scholar] [CrossRef]
- NCBI. dbSNP Short Genetic Variations. Available online: https://www.ncbi.nlm.nih.gov/snp/ (accessed on 8 May 2020).
- Gemmati, D.; Varani, K.; Bramanti, B.; Piva, R.; Bonaccorsi, G.; Trentini, A.; Manfrinato, M.C.; Tisato, V.; Care, A.; Bellini, T. “Bridging the Gap” Everything that Could Have Been Avoided If We Had Applied Gender Medicine, Pharmacogenetics and Personalized Medicine in the Gender-Omics and Sex-Omics Era. Int. J. Mol. Sci. 2019, 21, 296. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef]
- Zamboni, P.; Gemmati, D. Clinical implications of gene polymorphisms in venous leg ulcer: A model in tissue injury and reparative process. Thromb. Haemost. 2007, 98, 131–137. [Google Scholar] [PubMed]
- Gemmati, D.; Federici, F.; Campo, G.; Tognazzo, S.; Serino, M.L.; De Mattei, M.; Valgimigli, M.; Malagutti, P.; Guardigli, G.; Ferraresi, P.; et al. Factor XIIIA-V34L and factor XIIIB-H95R gene variants: Effects on survival in myocardial infarction patients. Mol. Med. 2007, 13, 112–120. [Google Scholar] [CrossRef]
- Tognazzo, S.; Gemmati, D.; Palazzo, A.; Catozzi, L.; Carandina, S.; Legnaro, A.; Tacconi, G.; Scapoli, G.L.; Zamboni, P. Prognostic role of factor XIII gene variants in nonhealing venous leg ulcers. J. Vasc. Surg. 2006, 44, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Tognazzo, S.; Catozzi, L.; Federici, F.; De Palma, M.; Gianesini, S.; Scapoli, G.L.; De Mattei, M.; Liboni, A.; Zamboni, P. Influence of gene polymorphisms in ulcer healing process after superficial venous surgery. J. Vasc. Surg. 2006, 44, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Tognazzo, S.; Serino, M.L.; Fogato, L.; Carandina, S.; De Palma, M.; Izzo, M.; De Mattei, M.; Ongaro, A.; Scapoli, G.L.; et al. Factor XIII V34L polymorphism modulates the risk of chronic venous leg ulcer progression and extension. Wound Repair Regen. 2004, 12, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Occhionorelli, S.; Tisato, V.; Vigliano, M.; Longo, G.; Gonelli, A.; Sibilla, M.G.; Serino, M.L.; Zamboni, P. Inherited genetic predispositions in F13A1 and F13B genes predict abdominal adhesion formation: Identification of gender prognostic indicators. Sci. Rep. 2018, 8, 16916. [Google Scholar] [CrossRef] [PubMed]
- Tisato, V.; Muggeo, P.; Lupiano, T.; Longo, G.; Serino, M.L.; Grassi, M.; Arcamone, E.; Secchiero, P.; Zauli, G.; Santoro, N.; et al. Maternal Haplotypes in DHFR Promoter and MTHFR Gene in Tuning Childhood Acute Lymphoblastic Leukemia Onset-Latency: Genetic/Epigenetic Mother/Child Dyad Study (GEMCDS). Genes 2019, 10, 634. [Google Scholar] [CrossRef] [PubMed]
- Syrett, C.M.; Anguera, M.C. When the balance is broken: X-linked gene dosage from two X chromosomes and female-biased autoimmunity. J. Leukoc. Biol. 2019, 106, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L. Sex influences immune responses to viruses, and efficacy of prophylaxis and treatments for viral diseases. Bioessays 2012, 34, 1050–1059. [Google Scholar] [CrossRef]
- Klein, S.L.; Jedlicka, A.; Pekosz, A. The Xs and Y of immune responses to viral vaccines. Lancet Infect. Dis. 2010, 10, 338–349. [Google Scholar] [CrossRef]
- Fish, E.N. The X-files in immunity: Sex-based differences predispose immune responses. Nat. Rev. Immunol. 2008, 8, 737–744. [Google Scholar] [CrossRef]
- Ubeda, F.; Jansen, V.A. The evolution of sex-specific virulence in infectious diseases. Nat. Commun. 2016, 7, 13849. [Google Scholar] [CrossRef]
- Klein, S.L.; Marriott, I.; Fish, E.N. Sex-based differences in immune function and responses to vaccination. Trans. R. Soc. Trop. Med. Hyg. 2015, 109, 9–15. [Google Scholar] [CrossRef]
- Schurz, H.; Salie, M.; Tromp, G.; Hoal, E.G.; Kinnear, C.J.; Moller, M. The X chromosome and sex-specific effects in infectious disease susceptibility. Hum. Genom. 2019, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Carter, A.C.; Chang, H.Y. Mechanistic insights in X-chromosome inactivation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Zito, A.; Davies, M.N.; Tsai, P.C.; Roberts, S.; Andres-Ejarque, R.; Nardone, S.; Bell, J.T.; Wong, C.C.Y.; Small, K.S. Heritability of skewed X-inactivation in female twins is tissue-specific and associated with age. Nat. Commun. 2019, 10, 5339. [Google Scholar] [CrossRef] [PubMed]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef]
- Wang, J.; Syrett, C.M.; Kramer, M.C.; Basu, A.; Atchison, M.L.; Anguera, M.C. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc. Natl. Acad. Sci. USA 2016, 113, E2029–E2038. [Google Scholar] [CrossRef]
- Cai, H. Sex difference and smoking predisposition in patients with COVID-19. Lancet Respir. Med. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Ji, H.; Zheng, W.; Wu, X.; Zhu, J.J.; Arnold, A.P.; Sandberg, K. Sex differences in renal angiotensin converting enzyme 2 (ACE2) activity are 17beta-oestradiol-dependent and sex chromosome-independent. Biol Sex Differ. 2010, 1, 6. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Q.; Xia, X.; Liu, K.; Yu, Z.; Tao, W.; Gong, W.; Han, J.J. Individual Variation of the SARS-CoV2 Receptor ACE2 Gene Expression and Regulation. Preprints 2020, 2020030191. Available online: https://www.preprints.org/manuscript/202003.0191/v1 (accessed on 26 March 2020).
- Jaillon, S.; Berthenet, K.; Garlanda, C. Sexual Dimorphism in Innate Immunity. Clin. Rev. Allergy Immunol. 2019, 56, 308–321. [Google Scholar] [CrossRef]
- Jespersen, L.; Tarnow, I.; Eskesen, D.; Morberg, C.M.; Michelsen, B.; Bugel, S.; Dragsted, L.O.; Rijkers, G.T.; Calder, P.C. Effect of Lactobacillus paracasei subsp. paracasei, L. casei 431 on immune response to influenza vaccination and upper respiratory tract infections in healthy adult volunteers: A randomized, double-blind, placebo-controlled, parallel-group study. Am. J. Clin. Nutr. 2015, 101, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Angele, M.K.; Pratschke, S.; Hubbard, W.J.; Chaudry, I.H. Gender differences in sepsis: Cardiovascular and immunological aspects. Virulence 2014, 5, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Casimir, G.J.; Lefevre, N.; Corazza, F.; Duchateau, J.; Chamekh, M. The Acid-Base Balance and Gender in Inflammation: A Mini-Review. Front. Immunol. 2018, 9, 475. [Google Scholar] [CrossRef]
- Postma, D.S. Gender differences in asthma development and progression. Gend. Med. 2007, 4 (Suppl. B), S133–S146. [Google Scholar] [CrossRef]
- Townsend, E.A.; Miller, V.M.; Prakash, Y.S. Sex differences and sex steroids in lung health and disease. Endocr. Rev. 2012, 33, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Badawi, A.; Ryoo, S.G. Prevalence of comorbidities in the Middle East respiratory syndrome coronavirus (MERS-CoV): A systematic review and meta-analysis. Int. J. Infect. Dis. 2016, 49, 129–133. [Google Scholar] [CrossRef]
- World Health Organization. Consensus Document on the Epidemiology of Severe Acute Respiratory Syndrome (SARS). Available online: https://apps.who.int/iris/bitstream/handle/10665/70863/WHO_CDS_CSR_GAR_2003.11_eng.pdf (accessed on 26 March 2020).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef]
- NIH. Clinical Trials.gov. Available online: https://clinicaltrials.gov/ (accessed on 26 March 2020).
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef]
- Casimir, G.J.; Mulier, S.; Hanssens, L.; Knoop, C.; Ferster, A.; Hofman, B.; Duchateau, J. Chronic inflammatory diseases in children are more severe in girls. Shock 2010, 34, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Casimir, G.J.; Heldenbergh, F.; Hanssens, L.; Mulier, S.; Heinrichs, C.; Lefevre, N.; Desir, J.; Corazza, F.; Duchateau, J. Gender differences and inflammation: An in vitro model of blood cells stimulation in prepubescent children. J. Inflamm. 2010, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Casimir, G.J.; Mulier, S.; Hanssens, L.; Zylberberg, K.; Duchateau, J. Gender differences in inflammatory markers in children. Shock 2010, 33, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Meester, I.; Manilla-Munoz, E.; Leon-Cachon, R.B.R.; Paniagua-Frausto, G.A.; Carrion-Alvarez, D.; Ruiz-Rodriguez, C.O.; Rodriguez-Rangel, X.; Garcia-Martinez, J.M. SeXY chromosomes and the immune system: Reflections after a comparative study. Biol. Sex Differ. 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.T.; Grafham, D.V.; Coffey, A.J.; Scherer, S.; McLay, K.; Muzny, D.; Platzer, M.; Howell, G.R.; Burrows, C.; Bird, C.P.; et al. The DNA sequence of the human X chromosome. Nature 2005, 434, 325–337. [Google Scholar] [CrossRef]
- Spolarics, Z.; Pena, G.; Qin, Y.; Donnelly, R.J.; Livingston, D.H. Inherent X-Linked Genetic Variability and Cellular Mosaicism Unique to Females Contribute to Sex-Related Differences in the Innate Immune Response. Front. Immunol. 2017, 8, 1455. [Google Scholar] [CrossRef]
- Lefevre, N.; Corazza, F.; Valsamis, J.; Delbaere, A.; De Maertelaer, V.; Duchateau, J.; Casimir, G. The Number of X Chromosomes Influences Inflammatory Cytokine Production Following Toll-Like Receptor Stimulation. Front. Immunol. 2019, 10, 1052. [Google Scholar] [CrossRef]
- Spolarics, Z. The X-files of inflammation: Cellular mosaicism of X-linked polymorphic genes and the female advantage in the host response to injury and infection. Shock 2007, 27, 597–604. [Google Scholar] [CrossRef]
- Berletch, J.B.; Yang, F.; Xu, J.; Carrel, L.; Disteche, C.M. Genes that escape from X inactivation. Hum. Genet. 2011, 130, 237–245. [Google Scholar] [CrossRef]
- Oghumu, S.; Varikuti, S.; Stock, J.C.; Volpedo, G.; Saljoughian, N.; Terrazas, C.A.; Satoskar, A.R. Cutting Edge: CXCR3 Escapes X Chromosome Inactivation in T Cells during Infection: Potential Implications for Sex Differences in Immune Responses. J. Immunol. 2019, 203, 789–794. [Google Scholar] [CrossRef]
- Gausman, J.; Langer, A. Sex and Gender Disparities in the COVID-19 Pandemic. J. Womens Health (Larchmt) 2020, 29, 465–466. [Google Scholar] [CrossRef] [PubMed]
- Hefler, M.; Gartner, C.E. The tobacco industry in the time of COVID-19: Time to shut it down? Tob. Control 2020, 29, 245–246. [Google Scholar] [CrossRef]
- Society, E.R. European Respiratory Society. Available online: https://www.ersnet.org/covid-19-blog/covid-19--propelled-by-smoking--could-destroy-entire-nations. (accessed on 8 May 2020).
- Liu, W.; Tao, Z.W.; Wang, L.; Yuan, M.L.; Liu, K.; Zhou, L.; Wei, S.; Deng, Y.; Liu, J.; Liu, H.G.; et al. Analysis of factors associated with disease outcomes in hospitalized patients with 2019 novel coronavirus disease. Chin. Med. J. 2020, 133, 1032–1038. [Google Scholar] [CrossRef]
- Murin, S.; Bilello, K.S. Respiratory tract infections: Another reason not to smoke. Cleve Clin. J. Med. 2005, 72, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Simons, D.; Brown, J. Covid-19: The role of smoking cessation during respiratory virus epidemics. BMJ Opin. 2020. Available online: https://blogs.bmj.com/bmj/2020/03/20/covid-19-the-role-of-smoking-cessation-during-respiratory-virus-epidemics/ (accessed on 8 May 2020).
- Wu, W.; Patel, K.B.; Booth, J.L.; Zhang, W.; Metcalf, J.P. Cigarette smoke extract suppresses the RIG-I-initiated innate immune response to influenza virus in the human lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L821–L830. [Google Scholar] [CrossRef] [PubMed]
- Halim, A.A.; Alsayed, B.; Embarak, S.; Yaseen, T.; Dabbous, S. Clinical characteristics and outcome of ICU admitted MERS corona virus infected patients. Egypt J. Chest Dis. Tuberc. 2016, 65, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Yang, L.; Chan, K.P.; Chan, W.M.; Song, L.; Lai, H.K.; Thach, T.Q.; Ho, L.M.; Chan, K.H.; Lam, T.H.; et al. Cigarette smoking as a risk factor for influenza-associated mortality: Evidence from an elderly cohort. Influenza Other Respir. Viruses 2013, 7, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Atto, B.; Eapen, M.S.; Sharma, P.; Frey, U.; Ammit, A.J.; Markos, J.; Chia, C.; Larby, J.; Haug, G.; Weber, H.C.; et al. New therapeutic targets for the prevention of infectious acute exacerbations of COPD: Role of epithelial adhesion molecules and inflammatory pathways. Clin. Sci. 2019, 133, 1663–1703. [Google Scholar] [CrossRef]
- Eurosurveillance Editorial Team. Updated rapid risk assessment from ECDC on coronavirus disease 2019 (COVID-19) pandemic: Increased transmission in the EU/EEA and the UK. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef]
- Garufi, G.; Carbognin, L.; Orlandi, A.; Tortora, G.; Bria, E. Smoking habit and hospitalization for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-related pneumonia: The unsolved paradox behind the evidence. Eur. J. Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.P.; Amoura, Z.; Rey, F.; Miyara, M. A nicotinic hypothesis for Covid-19 with preventive and therapeutic implications. Qeios 2020. [Google Scholar] [CrossRef]
- Zheng, Y.; Xiong, C.; Liu, Y.; Qian, X.; Tang, Y.; Liu, L.; Leung, E.L.; Wang, M. Epidemiological and Clinical Characteristics Analysis of COVID-19 in the Surrounding Areas of Wuhan, Hubei Province in 2020. Pharm. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Bonassi, S.; Giacconi, R.; Malavolta, M.; Tomino, C.; Maggi, F. COVID-19 and Smoking. Is Nicotine the Hidden Link? Eur. Respir. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Peng, F.; Xu, B.; Zhao, J.; Liu, H.; Peng, J.; Li, Q.; Jiang, C.; Zhou, Y.; Liu, S.; et al. Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J. Infect. 2020. [Google Scholar] [CrossRef]
- Brake, S.J.; Barnsley, K.; Lu, W.; McAlinden, K.D.; Eapen, M.S.; Sohal, S.S. Smoking Upregulates Angiotensin-Converting Enzyme-2 Receptor: A Potential Adhesion Site for Novel Coronavirus SARS-CoV-2 (Covid-19). J. Clin. Med. 2020, 9, 841. [Google Scholar] [CrossRef]
- Cai, G.; Bosse, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco Smoking Increases the Lung Gene Expression of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef]
- Barcelo, D. An Environmental and Health Perspective for COVID-19 Outbreak: Meteorology and Air Quality Influence, Sewage Epidemiology Indicator, Hospitals Disinfection, Drug Therapies and Recommendations. J. Environ. Chem. Eng. 2020. [Google Scholar] [CrossRef]
- Coccia, M. Factors determining the diffusion of COVID-19 and suggested strategy to prevent future accelerated viral infectivity similar to COVID. Sci. Total Environ. 2020, 729, 138474. [Google Scholar] [CrossRef]
- Daughton, C. The international imperative to rapidly and inexpensively monitor community-wide Covid-19 infection status and trends. Sci. Total Environ. 2020, 726, 138149. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Chen, K.; Zhou, J.; Deng, S.; Wang, Y. Shelter hospital mode: How to prevent novel coronavirus infection 2019 (COVID-19) hospital-acquired infection? Infect. Control Hosp. Epidemiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Ma, S.; Wang, Y.; Cai, Z.; Hu, J.; Wei, N.; Wu, J.; Du, H.; Chen, T.; Li, R.; et al. Factors Associated with Mental Health Outcomes Among Health Care Workers Exposed to Coronavirus Disease 2019. JAMA Netw. Open 2020, 3, e203976. [Google Scholar] [CrossRef] [PubMed]
- Dipartimento della Protezione Civile COVID-19 Italia—Monitoraggio della Situazione. Available online: http://opendatadpc.maps.arcgis.com/apps/opsdashboard/index.html#/b0c68bce2cce478eaac82fe38d4138b1 (accessed on 7 May 2020).
- Napoli, P.E.; Nioi, M. Global Spread of Coronavirus Disease 2019 and Malaria: An Epidemiological Paradox in the Early Stage of A Pandemic. J. Clin. Med. 2020, 9, 1138. [Google Scholar] [CrossRef]
- Gallego-Delgado, J.; Rodriguez, A. Malaria and hypertension. Another co-evolutionary adaptation? Front. Cell Infect. Microbiol. 2014, 4, 121. [Google Scholar] [CrossRef]
- Driss, A.; Hibbert, J.M.; Wilson, N.O.; Iqbal, S.A.; Adamkiewicz, T.V.; Stiles, J.K. Genetic polymorphisms linked to susceptibility to malaria. Malar J. 2011, 10, 271. [Google Scholar] [CrossRef]
- Thomas, M.R.; Lip, G.Y. Novel Risk Markers and Risk Assessments for Cardiovascular Disease. Circ. Res. 2017, 120, 133–149. [Google Scholar] [CrossRef]
- Gemmati, D.; Zeri, G.; Orioli, E.; Mari, R.; Moratelli, S.; Vigliano, M.; Marchesini, J.; Grossi, M.E.; Pecoraro, A.; Cuneo, A.; et al. Factor XIII-A dynamics in acute myocardial infarction: A novel prognostic biomarker? Thromb. Haemost. 2015, 114, 123–132. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | HGNC ID | Name | Locus |
|---|---|---|---|
| AGT | 333 | Angiotensinogen | 1q42.2 |
| REN | 9958 | Renin | 1q32.1 |
| ACE1 | 2707 | Angiotensin I converting enzyme | 17q23.3 |
| ACE2 | 13557 | Angiotensin I converting enzyme 2 | Xp22.2 |
| AGTR1 (AT1) | 336 | Angiotensin II receptor type 1 | 3q24 |
| AGTR2 (AT2) | 338 | Angiotensin II receptor type 2 | Xq23 |
| MAS1 | 6899 | MAS1 proto-oncogene | 6q25.3 |
| ABO | 79 | α 1-3-N-acetylgalactosaminyltransferase α 1-3-galactosyltransferase | 9q34.2 |
| ADAM17 | 195 | Metallopeptidase domain 17 TNFα-converting enzyme (TACE) | 2p25.1 |
| SRY | 11311 | Sex determining region Y | Yp11.2 |
| SOX3 | 11199 | SRY-box transcription factor 3 | Xq27.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gemmati, D.; Bramanti, B.; Serino, M.L.; Secchiero, P.; Zauli, G.; Tisato, V. COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-Chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? Int. J. Mol. Sci. 2020, 21, 3474. https://doi.org/10.3390/ijms21103474
Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli G, Tisato V. COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-Chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? International Journal of Molecular Sciences. 2020; 21(10):3474. https://doi.org/10.3390/ijms21103474
Chicago/Turabian StyleGemmati, Donato, Barbara Bramanti, Maria Luisa Serino, Paola Secchiero, Giorgio Zauli, and Veronica Tisato. 2020. "COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-Chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males?" International Journal of Molecular Sciences 21, no. 10: 3474. https://doi.org/10.3390/ijms21103474
APA StyleGemmati, D., Bramanti, B., Serino, M. L., Secchiero, P., Zauli, G., & Tisato, V. (2020). COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-Chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? International Journal of Molecular Sciences, 21(10), 3474. https://doi.org/10.3390/ijms21103474