Connecting RNA-Modifying Similarities of TDP-43, FUS, and SOD1 with MicroRNA Dysregulation Amidst A Renewed Network Perspective of Amyotrophic Lateral Sclerosis Proteinopathy



Abstract
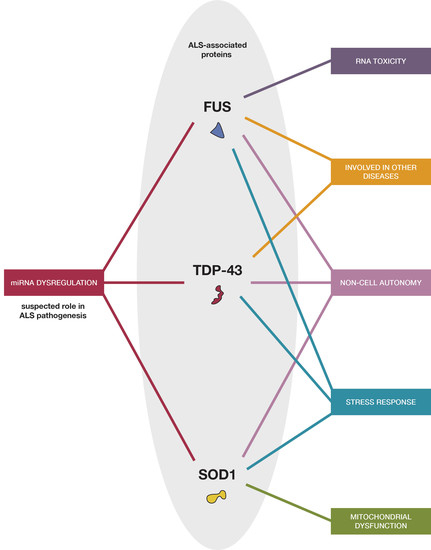
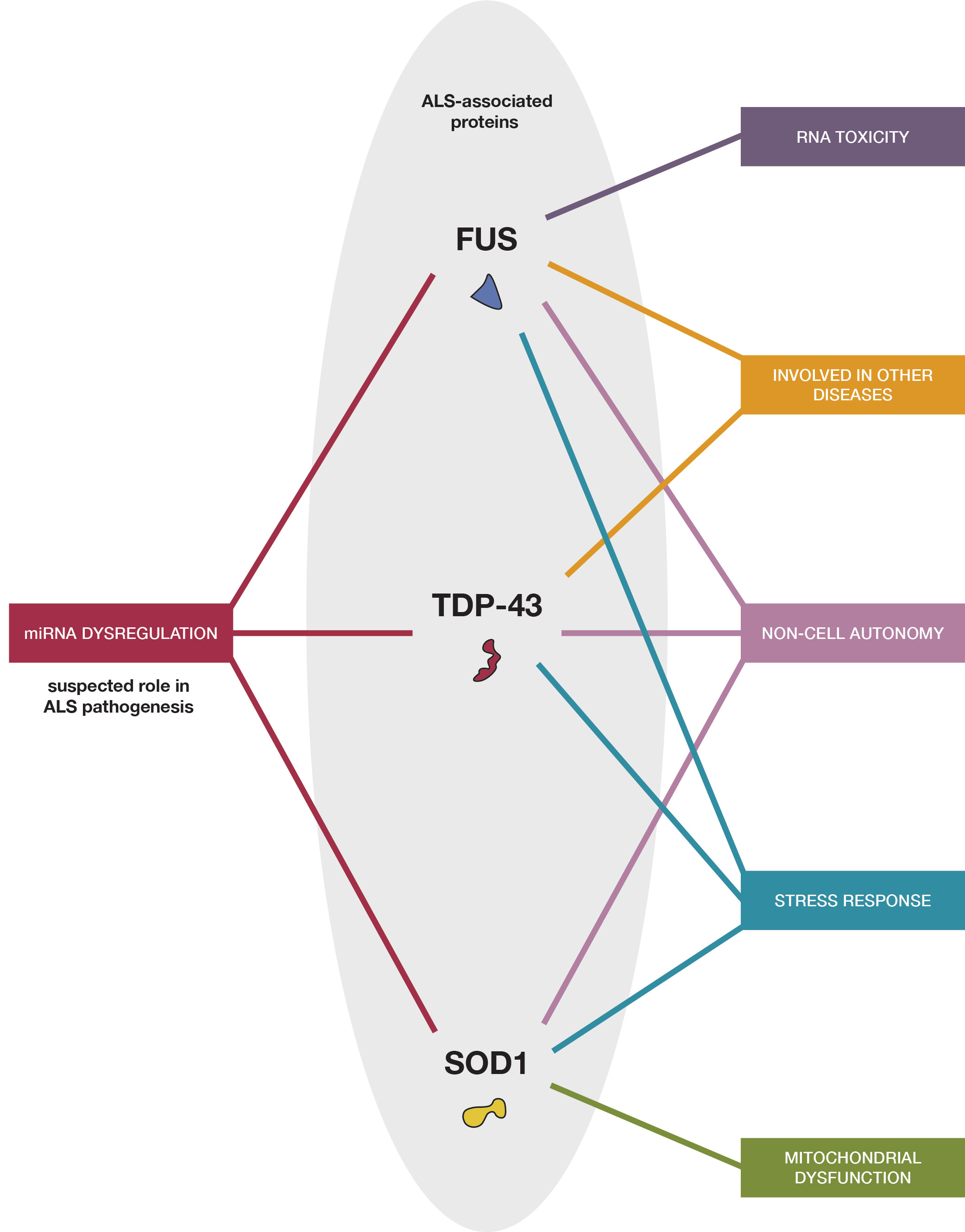{kind=link}
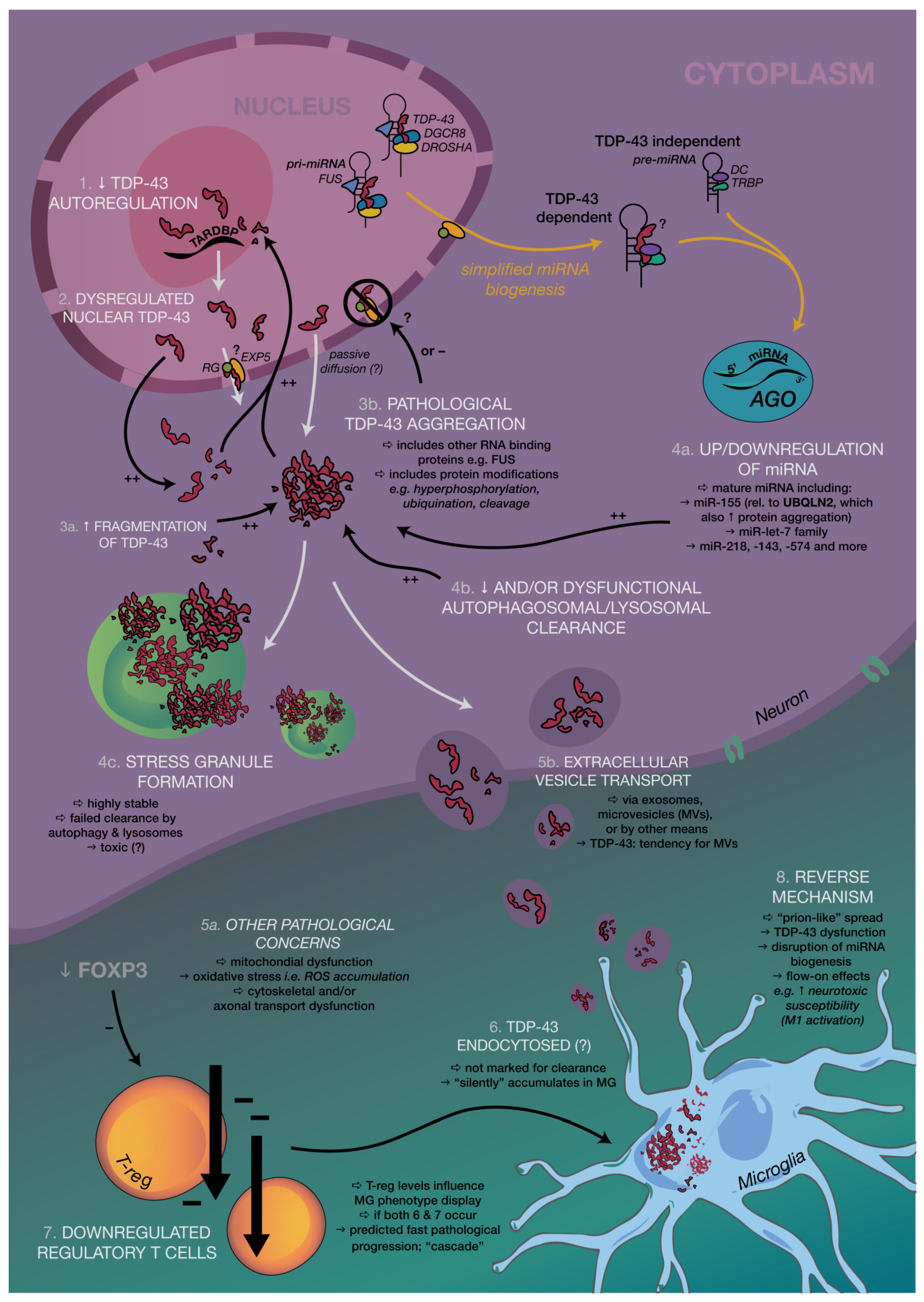{kind=link}
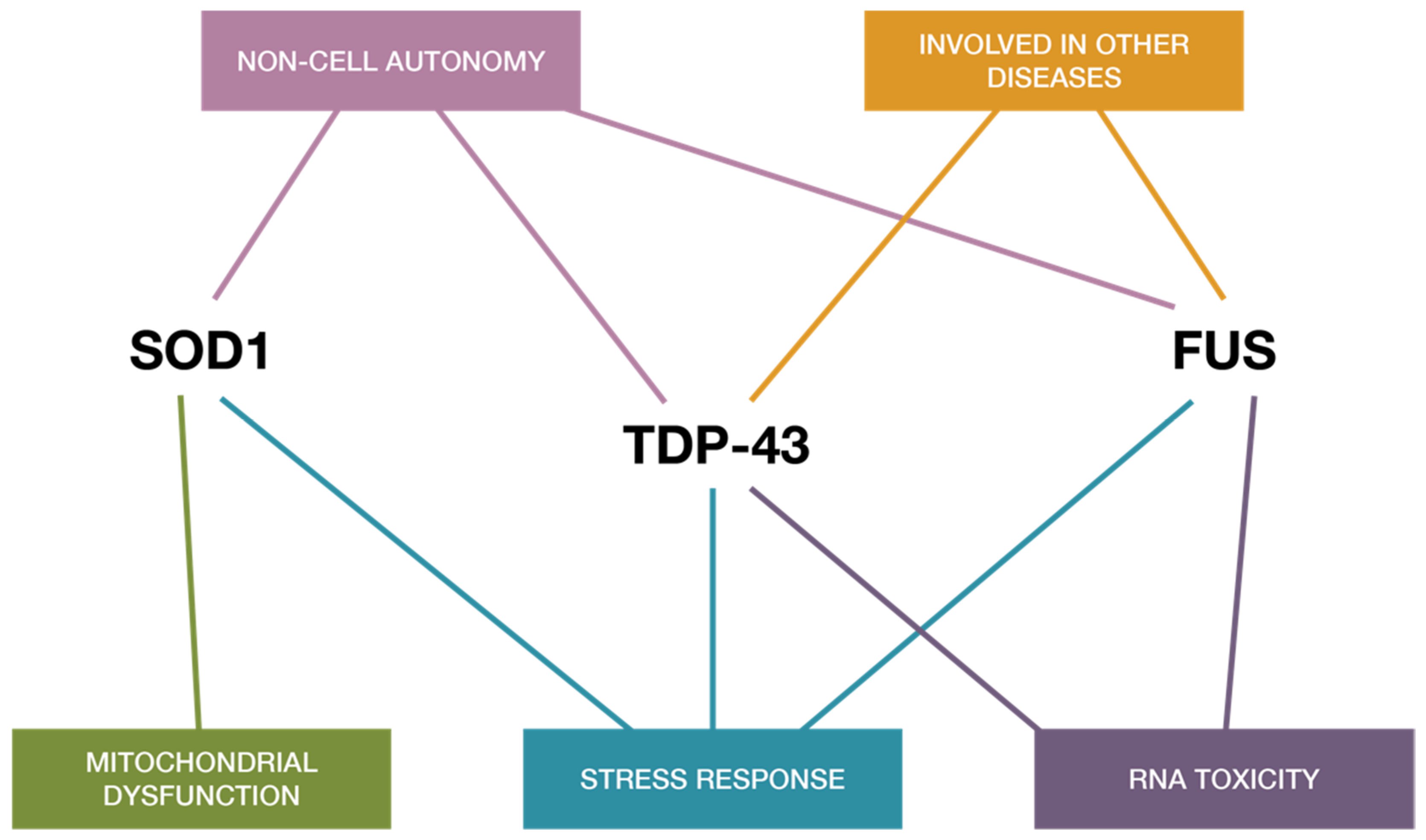{kind=link}
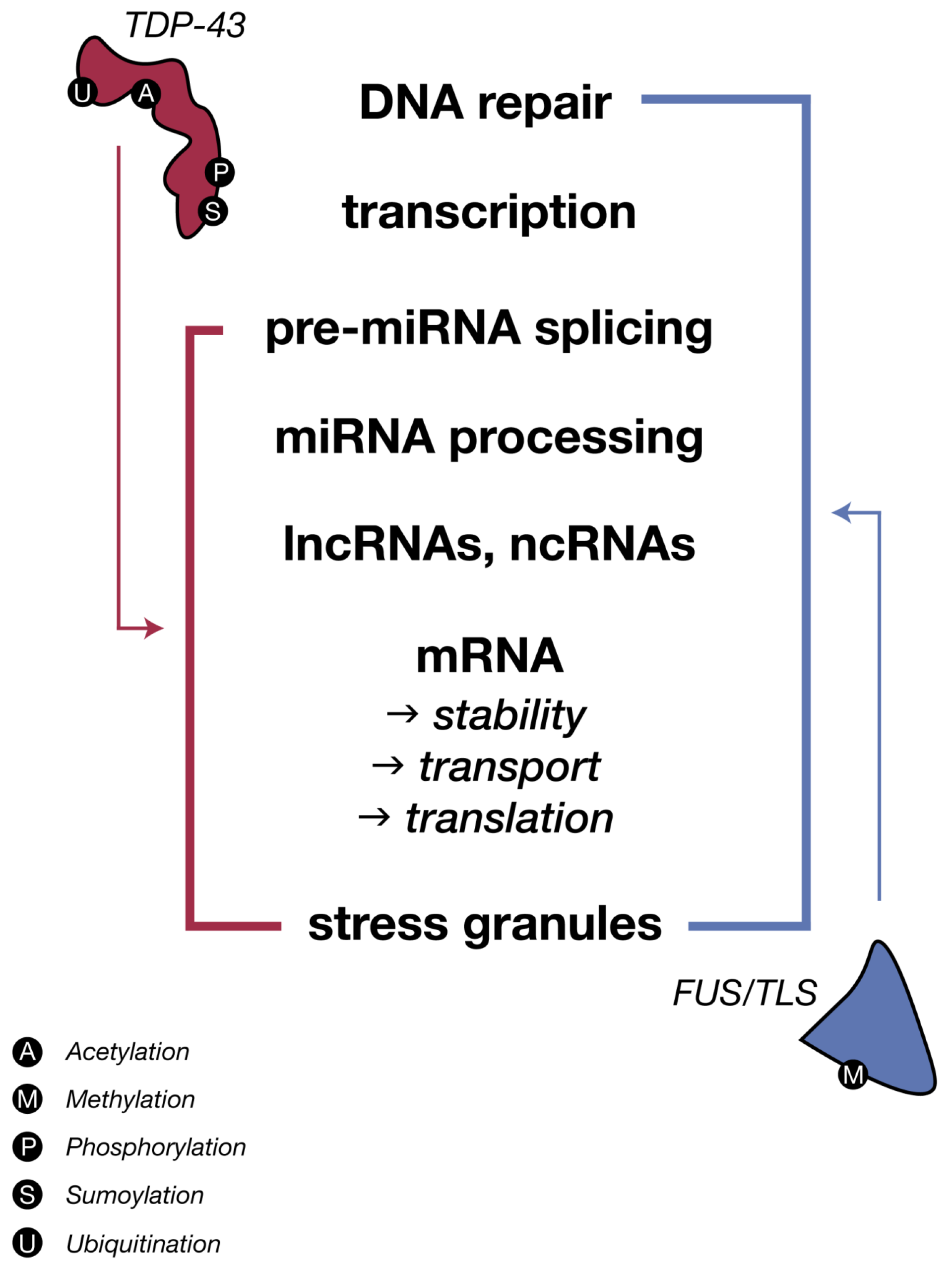{kind=link}
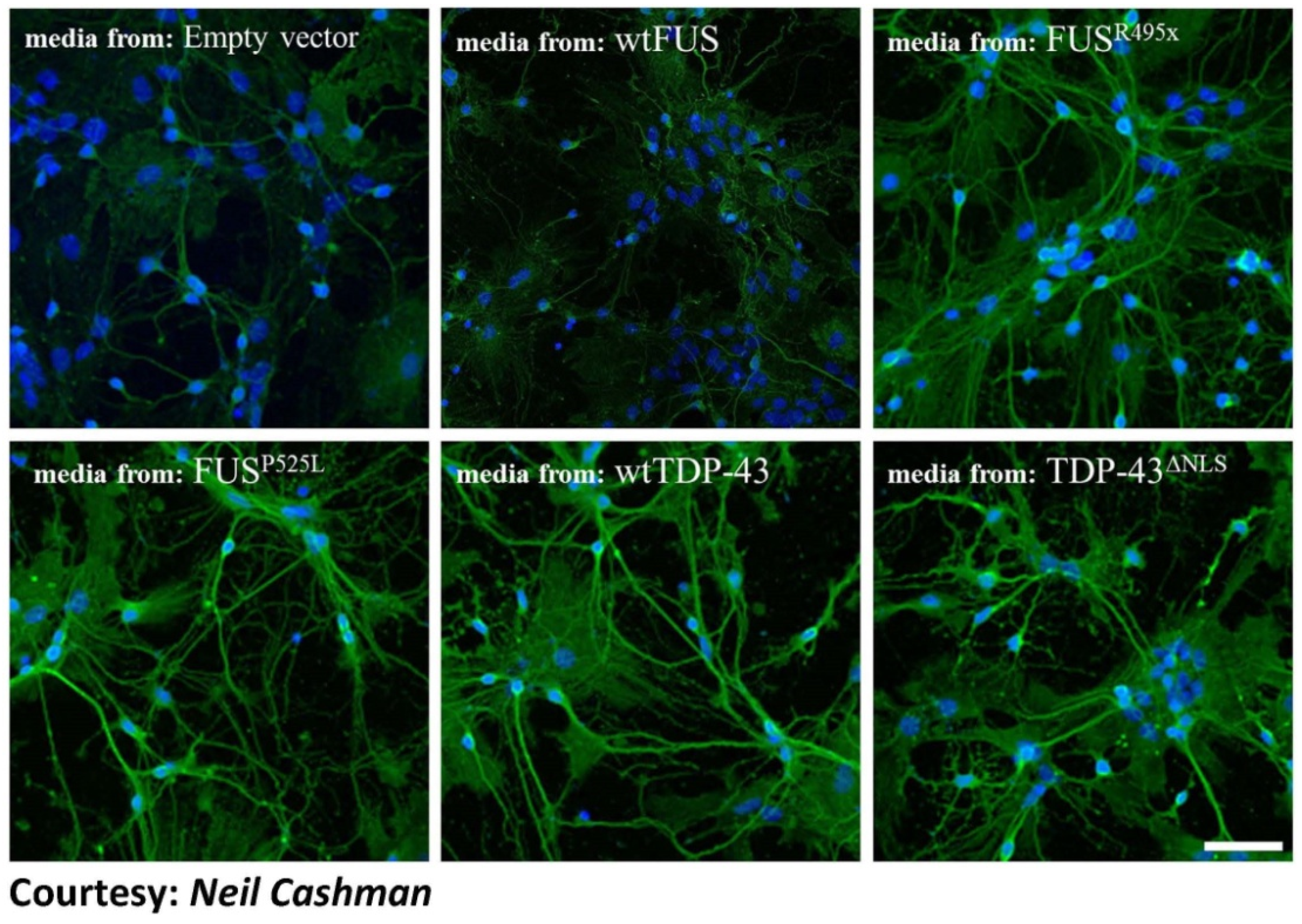{kind=link}
1. Introduction
Examination of Amyotrophic Lateral Sclerosis (ALS) with a Focus on Proteostasis and microRNAs
2. MicroRNA: Biogenesis, Regulation, and Protein-Related Dysfunction
Biogenesis of miRNA and Gene Regulation
Canonical MicroRNA Biogenesis
3. TDP-43, FUS, and SOD1 Relations with miRNA in ALS
3.1. TDP-43 and Regulation of miRNA Biogenesis
TDP-43 and Associated miRNAs: Beyond Biogenesis
3.2. FUS-TLS: Functional and Pathological Comparisons to TDP-43 in ALS
Repercussions of FUS Dysfunctions on miRNA Biogenesis
3.3. SOD1 and miRNA: Aggregation, Dysfunction, and Other Pathological Similarities to RNA-Binding Proteins in ALS
Repercussions of SOD1 Dysfunction on miRNA Biogenesis and Regulation
4. Defining Interrelationships and the Plausibility of a Two-Way Interaction between RNA-Modifying Proteins and miRNAs
4.1. Autoregulation in Propelling a ‘Doomed’ RBP and miRNA Relationship
4.2. On Stress Response and Prion-Like Similarities: for Better or for Worse?
4.2.1. On Functional Aggregation: Considering Subnuclear Paraspeckles (SNPs)
4.2.2. Further Studies on SOD1 and RNA-Binding Proteins Interrelationships and Their Implications for Future Research
4.3. Connecting microRNA into a Complex Proteomic Picture
5. Discussion
5.1. Examining miRNA Regulation as a Potential Diagnostic Tool
5.2. Examining miRNA Regulation as a Potential Avenue for Future Treatment
5.3. Beyond Motor Neurons: Applying Connections between miRNA and RNA-Modifying Proteins to Holistically Understand Pathogenesis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AGO | Argonaute |
| ALS | amyotrophic lateral sclerosis |
| ATXN2 | Ataxin-2 |
| dsRNA | double stranded RNA |
| EIF2 | Eukaryotic initiation factor 2 |
| EWSR1 | EWS RNA-binding protein 1 |
| EXO | exosome |
| EXP | exportin 5 |
| fALS | familial ALS |
| FMRP | fragile X mental retardation protein |
| FTLD | frontotemporal lobular dementia |
| FUS | fused in sarcoma |
| FUS/TLS | fused in sarcoma/ translocated in liposarcoma |
| hnRNP | heterogeneous nuclear ribonucleoproteins |
| HuWtSOD1 | human wild-type SOD1 |
| iPSC | induced pluripotent stem cell |
| LC | low-complexity |
| MATR3 | Matrin-3 |
| miRISC | miRNA-induced silencing complex |
| miRNA | micro RNA |
| MN | motor neuron |
| MV | microvesicle |
| ND | neurodegenerative disease |
| NSC-34 | Neuroblastoma/spinal cord hybrid |
| PD | Parkinson’s disease |
| pre-miRNA | precursor miRNA |
| pri-miRNA | primary miRNA |
| PRNP | Prion protein |
| PY-NLS | PY nuclear localization signal |
| RACK1 | ribosomal receptor for activated C kinase 1 |
| RAN | Ras-related nuclear protein |
| RBP | RNA-binding protein |
| RISC | RNA-induced silencing complex |
| RNP | ribonucleoprotein particle |
| RRM | RNA recognition motif |
| sALS | sporadic ALS |
| SEDI | SOD1 exposed dimer interface |
| SG | stress granule |
| SMA | Spinal muscular atrophy |
| SMN | survival of motor neuron |
| SNP | subnuclear paraspeckle |
| SOD1 | superoxide dismutase-1 |
| STMN2 | Stathmin-2 |
| TA | tibialis anterior |
| TAF15 | TATA-box binding protein associated factor 15 |
| TARDBP | TAR DNA-binding protein |
| TDP-43 | transactive response DNA-binding protein |
| TIA1 | TIA1 cytotoxic granule associated RNA-binding protein |
| UBQLN2 | Ubiquilin-2 |
| UTR | Untranslated region |
| WT | wild-type |
Appendix A
References
- Conlon, E.G.; Fagegaltier, D.; Agius, P.; Davis-Porada, J.; Gregory, J.; Hubbard, I.; Kang, K.; Kim, D.; Phatnani, H.; Shneider, N.A. Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism. Elife 2018, 7, e37754. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Models Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Cali, C.P.; Lee, E.B. RNA metabolism in neurodegenerative disease. Dis. Models Mech. 2017, 10, 509–518. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kim, J.R.; Van Bruggen, R.; Park, J. RNA-binding proteins in amyotrophic lateral sclerosis. Mol. Cells 2018, 41, 818. [Google Scholar]
- Jeon, G.S.; Shim, Y.-M.; Lee, D.-Y.; Kim, J.-S.; Kang, M.; Ahn, S.H.; Shin, J.-Y.; Geum, D.; Hong, Y.H.; Sung, J.-J. Pathological modification of TDP-43 in amyotrophic lateral sclerosis with SOD1 mutations. Mol. Neurobiol. 2019, 56, 2007–2021. [Google Scholar] [CrossRef]
- Sumi, H.; Kato, S.; Mochimaru, Y.; Fujimura, H.; Etoh, M.; Sakoda, S. Nuclear TAR DNA binding protein 43 expression in spinal cord neurons correlates with the clinical course in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2009, 68, 37–47. [Google Scholar] [CrossRef]
- Okamoto, Y.; Ihara, M.; Urushitani, M.; Yamashita, H.; Kondo, T.; Tanigaki, A.; Oono, M.; Kawamata, J.; Ikemoto, A.; Kawamoto, Y. An autopsy case of SOD1-related ALS with TDP-43 positive inclusions. Neurology 2011, 77, 1993–1995. [Google Scholar] [CrossRef]
- Sabatelli, M.; Zollino, M.; Conte, A.; Del Grande, A.; Marangi, G.; Lucchini, M.; Mirabella, M.; Romano, A.; Piacentini, R.; Bisogni, G. Primary fibroblasts cultures reveal TDP-43 abnormalities in amyotrophic lateral sclerosis patients with and without SOD1 mutations. Neurobiol. Aging 2015, 36, 2005.e5–2005.e13. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Hergesheimer, R.C.; Chami, A.A.; De Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Smethurst, P.; Sidle, K.C.L.; Hardy, J. Prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS). Neuropathol. Appl. Neurobiol. 2015, 41, 578–597. [Google Scholar] [CrossRef]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Cook, C.; Zhang, Y.-J.; Xu, Y.-F.; Dickson, D.W.; Petrucelli, L. TDP-43 in neurodegenerative disorders. Expert Opin. Biol. Ther. 2008, 8, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef]
- Parisi, C.; Arisi, I.; D’Ambrosi, N.; Storti, A.; Brandi, R.; D’Onofrio, M.; Volonte, C. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013, 4, e959. [Google Scholar] [CrossRef]
- Mathis, S.; Le Masson, G. RNA-targeted therapies and amyotrophic lateral sclerosis. Biomedicines 2018, 6, 9. [Google Scholar] [CrossRef]
- Eitan, C.; Hornstein, E. Vulnerability of microRNA biogenesis in FTD–ALS. Brain Res. 2016, 1647, 105–111. [Google Scholar] [CrossRef]
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y.Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2010, 1803, 1231–1243. [Google Scholar] [CrossRef]
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.; Enayat, Z.; Chioza, B.; Al-Chalabi, A.; Radunovic, A.; Powell, J.; Leigh, P. Mutations in all five exons of SOD-1 may cause ALS. Ann. Neurol. 1998, 43, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Traynor, B.J.; Lombardo, F.; Fimognari, M.; Calvo, A.; Ghiglione, P.; Mutani, R.; Restagno, G. Prevalence of SOD1 mutations in the Italian ALS population. Neurology 2008, 70, 533. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Weskamp, K.; Barmada, S.J. TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Res. 2018, 1693, 67–74. [Google Scholar] [CrossRef]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef]
- Medina, P.P.; Slack, F.J. microRNAs and cancer: An overview. Cell Cycle 2008, 7, 2485–2492. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Hur, J.; Lunn, J.S.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of microRNAs in human post-mortem amyotrophic lateral sclerosis spinal cords provides insight into disease mechanisms. Mol. Cell. Neurosci. 2016, 71, 34–45. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Meyer, J.; Borkhardt, A.; Tuschl, T. New microRNAs from mouse and human. RNA 2003, 9, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Glasner, M.E.; Yekta, S.; Burge, C.B.; Bartel, D.P. Vertebrate microRNA genes. Science 2003, 299, 1540. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Partin, A.C.; Ngo, T.D.; Herrell, E.; Jeong, B.-C.; Hon, G.; Nam, Y. Heme enables proper positioning of Drosha and DGCR8 on primary microRNAs. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.-P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.-J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef]
- Carroll, A.P.; Tooney, P.A.; Cairns, M.J. Context-specific microRNA function in developmental complexity. J. Mol. Cell Biol. 2013, 5, 73–84. [Google Scholar] [CrossRef]
- Sephton, C.F.; Cenik, B.; Cenik, B.K.; Herz, J.; Yu, G. TDP-43 in central nervous system development and function: Clues to TDP-43-associated neurodegeneration. Biol. Chem. 2012, 393, 589–594. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Kitamura, A. TDP-43 depletion: Mechanism of neuronal cell death in ALS. Future Neurol. 2018, 13, 143–149. [Google Scholar] [CrossRef]
- Mori, F.; Tada, M.; Kon, T.; Miki, Y.; Tanji, K.; Kurotaki, H.; Tomiyama, M.; Ishihara, T.; Onodera, O.; Kakita, A.; et al. Phosphorylated TDP-43 aggregates in skeletal and cardiac muscle are a marker of myogenic degeneration in amyotrophic lateral sclerosis and various conditions. Acta Neuropathol. Commun. 2019, 7, 165. [Google Scholar] [CrossRef]
- Cykowski, M.D.; Dickson, D.W.; Powell, S.Z.; Arumanayagam, A.S.; Rivera, A.L.; Appel, S.H. Dipeptide repeat (DPR) pathology in the skeletal muscle of ALS patients with C9ORF72 repeat expansion. Acta Neuropathol 2019, 138, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kwong, L.K.; Lee, E.B.; Kremmer, E.; Flatley, A.; Xu, Y.; Forman, M.S.; Troost, D.; Kretzschmar, H.A.; Trojanowski, J.Q. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol 2009, 117, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Berning, B.A.; Walker, A.K. The pathobiology of TDP-43 C-terminal fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef]
- Di Carlo, V.; Grossi, E.; Laneve, P.; Morlando, M.; Modigliani, S.D.; Ballarino, M.; Bozzoni, I.; Caffarelli, E. TDP-43 regulates the microprocessor complex activity during in vitro neuronal differentiation. Mol. Neurobiol. 2013, 48, 952–963. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Xu, Y.-F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.-L.; Tong, J.; Castanedes-Casey, M.; Ash, P. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef]
- Nonaka, T.; Kametani, F.; Arai, T.; Akiyama, H.; Hasegawa, M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 2009, 18, 3353–3364. [Google Scholar] [CrossRef]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Cushman, M.; Johnson, B.S.; King, O.D.; Gitler, A.D.; Shorter, J. Prion-like disorders: Blurring the divide between transmissibility and infectivity. J. Cell Sci. 2010, 123, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 2012, 3, 1307. [Google Scholar] [CrossRef] [PubMed]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; De Conti, L.; Stuani, C.; Romano, M.; Baralle, M.; Baralle, F. Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J. 2010, 277, 2268–2281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Almeida, S.; Lu, Y.; Nishimura, A.L.; Peng, L.; Sun, D.; Wu, B.; Karydas, A.M.; Tartaglia, M.C.; Fong, J.C. Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS ONE 2013, 8, e76055. [Google Scholar] [CrossRef]
- Dolinar, A.; Ravnik-Glavač, M.; Glavač, D. Epigenetic mechanisms in amyotrophic lateral sclerosis: A short review. Mech. Ageing Dev. 2018, 174, 103–110. [Google Scholar] [CrossRef]
- Emde, A.; Eitan, C.; Liou, L.L.; Libby, R.T.; Rivkin, N.; Magen, I.; Reichenstein, I.; Oppenheim, H.; Eilam, R.; Silvestroni, A. Dysregulated miRNA biogenesis downstream of cellular stress and ALS-causing mutations: A new mechanism for ALS. EMBO J. 2015, 34, 2633–2651. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.; Leclerc, A.; Tamrazian, E.; Vanderburg, C.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.; Munsat, T. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Winton, M.; Igaz, L.; Wong, M.; Kwong, L.; Trojanowski, J.; Lee, V. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef]
- Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Leung, A.Y.; Newcombe, C.G.; O’Meally, R.N.; Cole, R.N.; Shewmaker, F.P. The prionlike domain of FUS is multiphosphorylated following DNA damage without altering nuclear localization. Mol. Biol. Cell 2018, 29, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Dangoumau, A.; Veyrat-Durebex, C.; Blasco, H.; Praline, J.; Corcia, P.; Andres, C.R.; Vourc’h, P. Protein SUMOylation, an emerging pathway in amyotrophic lateral sclerosis. Int. J. Neurosci. 2013, 123, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Sobue, G. Importance of functional loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Masuda, A.; Takeda, J.-I.; Okuno, T.; Okamoto, T.; Ohkawara, B.; Ito, M.; Ishigaki, S.; Sobue, G.; Ohno, K. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev. 2015, 29, 1045–1057. [Google Scholar] [CrossRef]
- Udagawa, T.; Fujioka, Y.; Tanaka, M.; Honda, D.; Yokoi, S.; Riku, Y.; Ibi, D.; Nagai, T.; Yamada, K.; Watanabe, H.; et al. FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun. 2015, 6, 7098. [Google Scholar] [CrossRef]
- Yokoi, S.; Udagawa, T.; Fujioka, Y.; Honda, D.; Okado, H.; Watanabe, H.; Katsuno, M.; Ishigaki, S.; Sobue, G. 3′ UTR length-dependent control of SynGAP isoform α2 mRNA by FUS and ELAV-like proteins promotes dendritic spine maturation and cognitive function. Cell Rep. 2017, 20, 3071–3084. [Google Scholar] [CrossRef]
- Tateishi, T.; Hokonohara, T.; Yamasaki, R.; Miura, S.; Kikuchi, H.; Iwaki, A.; Tashiro, H.; Furuya, H.; Nagara, Y.; Ohyagi, Y.; et al. Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol. 2010, 119, 355–364. [Google Scholar] [CrossRef]
- Suzuki, M.; Mikami, H.; Watanabe, T.; Yamano, T.; Yamazaki, T.; Nomura, M.; Yasui, K.; Ishikawa, H.; Ono, S. Increased expression of TDP-43 in the skin of amyotrophic lateral sclerosis. Acta Neurol. Scand. 2010, 122, 367–372. [Google Scholar] [CrossRef]
- Morlando, M.; Modigliani, S.D.; Torrelli, G.; Rosa, A.; Di Carlo, V.; Caffarelli, E.; Bozzoni, I. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 2012, 31, 4502–4510. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Y.-C.; Mullane, P.; Ji, Y.J.; Liu, H.; He, L.; Arora, A.; Hwang, H.-Y.; Alessi, A.F.; Niaki, A.G. FUS regulates activity of MicroRNA-mediated gene silencing. Mol. Cell 2018, 69, 787–801.e8. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Skelt, L.; Notaro, A.; Highley, J.R.; Fox, A.H.; La Bella, V.; Buchman, V.L.; Shelkovnikova, T.A. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol. Commun. 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- De Santis, R.; Santini, L.; Colantoni, A.; Peruzzi, G.; De Turris, V.; Alfano, V.; Bozzoni, I.; Rosa, A. FUS mutant human motoneurons display altered transcriptome and microRNA pathways with implications for ALS pathogenesis. Stem Cell Rep. 2017, 9, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Bhinge, A.; Namboori, S.C.; Bithell, A.; Soldati, C.; Buckley, N.J.; Stanton, L.W. Mir-375 is essential for human spinal motor neuron development and may be involved in motor neuron degeneration. Stem Cells 2016, 34, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Ajmone-Cat, M.A.; Onori, A.; Toselli, C.; Stronati, E.; Morlando, M.; Bozzoni, I.; Monni, E.; Kokaia, Z.; Lupo, G.; Minghetti, L. Increased FUS levels in astrocytes leads to astrocyte and microglia activation and neuronal death. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in amyotrophic lateral sclerosis:“ambivalent” behavior connected to the disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef]
- Münch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef]
- Grad, L.I.; Pokrishevsky, E.; Silverman, J.M.; Cashman, N.R. Exosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfolding. Prion 2014, 8, 331–335. [Google Scholar] [CrossRef]
- Polymenidou, M.; Cleveland, D.W. Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med. 2012, 209, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Hare, D.J.; Double, K.L. A proposed mechanism for neurodegeneration in movement disorders characterized by metal dyshomeostasis and oxidative stress. Cell Chem. Biol. 2018, 25, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 2017, 134, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Fifita, J.A.; Freckleton, S.E.; Hare, D.J.; Lewis, S.J.; Halliday, G.M.; Blair, I.P.; Double, K.L. Accumulation of dysfunctional SOD1 protein in Parkinson’s disease is not associated with mutations in the SOD1 gene. Acta Neuropathol. 2018, 135, 155–156. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Boca, M.; Girotto, S.; Martinelli, M.; Valentine, J.S.; Vieru, M. SOD1 and amyotrophic lateral sclerosis: Mutations and oligomerization. PLoS ONE 2008, 3, e1677. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Durazo, A.; Girotto, S.; Gralla, E.B.; Martinelli, M.; Valentine, J.S.; Vieru, M.; Whitelegge, J.P. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: A possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. USA 2007, 104, 11263–11267. [Google Scholar] [CrossRef]
- Furukawa, Y.; O’Halloran, T.V. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo-and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 2005, 280, 17266–17274. [Google Scholar] [CrossRef]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.-F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef]
- Schmitt, N.D.; Agar, J.N. Parsing disease-relevant protein modifications from epiphenomena: Perspective on the structural basis of SOD1-mediated ALS. J. Mass Spectrom. 2017, 52, 480–491. [Google Scholar] [CrossRef]
- Keller, B.A.; Volkening, K.; Droppelmann, C.A.; Ang, L.C.; Rademakers, R.; Strong, M.J. Co-aggregation of RNA binding proteins in ALS spinal motor neurons: Evidence of a common pathogenic mechanism. Acta Neuropathol. 2012, 124, 733–747. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.; Saccon, R.A.; Fratta, P.; Fisher, E.M. SOD1 function and its implications for amyotrophic lateral sclerosis pathology: New and renascent themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Butti, Z.; Patten, S.A. RNA dysregulation in amyotrophic lateral sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and oxidative stress in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J.-I. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Waller, R.; Goodall, E.F.; Milo, M.; Cooper-Knock, J.; Da Costa, M.; Hobson, E.; Kazoka, M.; Wollff, H.; Heath, P.R.; Shaw, P.J. Serum miRNAs miR-206, 143–3p and 374b-5p as potential biomarkers for amyotrophic lateral sclerosis (ALS). Neurobiol. Aging 2017, 55, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, L.; Lu, Y.; Zhang, M.; Zhang, Z.; Wang, K.; Lv, J. Down-regulation of microRNA-142-5p attenuates oxygen-glucose deprivation and reoxygenation-induced neuron injury through up-regulating Nrf2/ARE signaling pathway. Biomed. Pharmacother. 2017, 89, 1187–1195. [Google Scholar] [CrossRef]
- Koval, E.D.; Shaner, C.; Zhang, P.; Du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef]
- Paladino, S.; Conte, A.; Caggiano, R.; Pierantoni, G.M.; Faraonio, R. Nrf2 pathway in age-related neurological disorders: Insights into MicroRNAs. Cell. Physiol. Biochem. 2018, 47, 1951–1976. [Google Scholar] [CrossRef]
- Ba, Q.; Cui, C.; Wen, L.; Feng, S.; Zhou, J.; Yang, K. Schisandrin B shows neuroprotective effect in 6-OHDA-induced Parkinson’s disease via inhibiting the negative modulation of miR-34a on Nrf2 pathway. Biomed. Pharmacother. 2015, 75, 165–172. [Google Scholar] [CrossRef]
- Xiong, R.; Wang, Z.; Zhao, Z.; Li, H.; Chen, W.; Zhang, B.; Wang, L.; Wu, L.; Li, W.; Ding, J. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol. Aging 2014, 35, 705–714. [Google Scholar] [CrossRef]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Jain, M.R.; Li, H.; Junn, E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic. Biol. Med. 2015, 89, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef] [PubMed]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet. 2007, 16, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef]
- Kovanda, A.; Leonardis, L.; Zidar, J.; Koritnik, B.; Dolenc-Groselj, L.; Kovacic, S.R.; Curk, T.; Rogelj, B. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Raheja, R.; Regev, K.; Healy, B.C.; Mazzola, M.A.; Beynon, V.; Von Glehn, F.; Paul, A.; Diaz-Cruz, C.; Gholipour, T.; Glanz, B.I. Correlating serum micrornas and clinical parameters in amyotrophic lateral sclerosis. Muscle Nerve 2018, 58, 261–269. [Google Scholar] [CrossRef]
- Russell, A.P.; Ghobrial, L.; Ngo, S.; Yerbury, J.; Zacharewicz, E.; Chung, R.; Lamon, S. Dysregulation of microRNA biogenesis machinery and microRNA/RNA ratio in skeletal muscle of amyotrophic lateral sclerosis mice. Muscle Nerve 2018, 57, 838–847. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O. Targeting mi R-155 restores abnormal microglia and attenuates disease in SOD 1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef]
- Hoye, M.L.; Koval, E.D.; Wegener, A.J.; Hyman, T.S.; Yang, C.; O’Brien, D.R.; Miller, R.L.; Cole, T.; Schoch, K.M.; Shen, T. MicroRNA profiling reveals marker of motor neuron disease in ALS models. J. Neurosci. 2017, 37, 5574–5586. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Zetterström, P.; Andersen, P.M.; Brännström, T.; Marklund, S.L. Misfolded superoxide dismutase-1 in CSF from amyotrophic lateral sclerosis patients. J. Neurochem. 2011, 117, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Gal, J.; Kuang, L.; Barnett, K.R.; Zhu, B.Z.; Shissler, S.C.; Korotkov, K.V.; Hayward, L.J.; Kasarskis, E.J.; Zhu, H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016, 132, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.; Keon, M.; Liu, B.; Su, Z.; Saksena, N.K. Panoramic visualization of circulating microRNAs across neurodegenerative diseases in humans. Mol. Neurobiol. 2019, 56, 7380–7407. [Google Scholar] [CrossRef] [PubMed]
- King, O.D.; Gitler, A.D.; Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum. Genet. 2017, 136, 1193–1214. [Google Scholar] [CrossRef] [PubMed]
- Treiber, T.; Treiber, N.; Plessmann, U.; Harlander, S.; Daiß, J.-L.; Eichner, N.; Lehmann, G.; Schall, K.; Urlaub, H.; Meister, G. A compendium of RNA-binding proteins that regulate microRNA biogenesis. Mol. Cell 2017, 66, 270–284.e13. [Google Scholar] [CrossRef]
- Ling, S.-C.; Albuquerque, C.P.; Han, J.S.; Lagier-Tourenne, C.; Tokunaga, S.; Zhou, H.; Cleveland, D.W. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. USA 2010, 107, 13318–13323. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Wickner, R.B.; Shewmaker, F. FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell 2011, 2, 223–236. [Google Scholar] [CrossRef]
- Pokrishevsky, E.; Grad, L.I.; Cashman, N.R. TDP-43 or FUS-induced misfolded human wild-type SOD1 can propagate intercellularly in a prion-like fashion. Sci. Rep. 2016, 6, 22155. [Google Scholar] [CrossRef]
- Lin, G.; Mao, D.; Bellen, H. Amyotrophic lateral sclerosis pathogenesis converges on defects in protein homeostasis associated with TDP-43 mislocalization and proteasome-mediated degradation overload. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 121, pp. 111–171. [Google Scholar]
- Kabashi, E.; Bercier, V.; Lissouba, A.; Liao, M.; Brustein, E.; Rouleau, G.A.; Drapeau, P. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 2011, 7, e1002214. [Google Scholar] [CrossRef]
- Jiang, P.; Coller, H. Functional interactions between microRNAs and RNA binding proteins. MicroRNA 2012, 1, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Wienholds, E.; Kloosterman, W.P.; Miska, E.; Alvarez-Saavedra, E.; Berezikov, E.; De Bruijn, E.; Horvitz, H.R.; Kauppinen, S.; Plasterk, R.H. MicroRNA expression in zebrafish embryonic development. Science 2005, 309, 310–311. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.; Liu, G.; Öztürk, A.; Hicks, G.G. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013, 9, e1003895. [Google Scholar] [CrossRef] [PubMed]
- Sugai, A.; Kato, T.; Koyama, A.; Koike, Y.; Kasahara, S.; Konno, T.; Ishihara, T.; Onodera, O. Robustness and vulnerability of the autoregulatory system that maintains nuclear TDP-43 levels: A trade-off hypothesis for als pathology based on in silico data. Front. Neurosci. 2018, 12, 28. [Google Scholar] [CrossRef]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef]
- Leibiger, C.; Deisel, J.; Aufschnaiter, A.; Ambros, S.; Tereshchenko, M.; Verheijen, B.M.; Büttner, S.; Braun, R.J. Endolysosomal pathway activity protects cells from neurotoxic TDP-43. Microb. Cell 2018, 5, 212–214. [Google Scholar] [CrossRef]
- Filimonenko, M.; Stuffers, S.; Raiborg, C.; Yamamoto, A.; Malerød, L.; Fisher, E.M.; Isaacs, A.; Brech, A.; Stenmark, H.; Simonsen, A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007, 179, 485–500. [Google Scholar] [CrossRef]
- Scotter, E.L.; Chen, H.-J.; Shaw, C.E. TDP-43 proteinopathy and ALS: Insights into disease mechanisms and therapeutic targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef]
- Ravits, J.M.; La Spada, A.R. ALS motor phenotype heterogeneity, focality, and spread: Deconstructing motor neuron degeneration. Neurology 2009, 73, 805–811. [Google Scholar] [CrossRef]
- Buchan, J.R. mRNP granules: Assembly, function, and connections with disease. RNA Biol. 2014, 11, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Dansithong, W.; Figueroa, K.P.; Scoles, D.R.; Pulst, S.M. Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A. Pathological proteins are transported by extracellular vesicles of sporadic amyotrophic lateral sclerosis patients. Front. Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Kukharsky, M.S.; An, H.; Dimasi, P.; Alexeeva, S.; Shabir, O.; Heath, P.R.; Buchman, V.L. Protective paraspeckle hyper-assembly downstream of TDP-43 loss of function in amyotrophic lateral sclerosis. Mol. Neurodegener. 2018, 13, 30. [Google Scholar] [CrossRef]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef]
- Lee, S.; Kim, H.-J. Prion-like mechanism in amyotrophic lateral sclerosis: Are protein aggregates the key? Exp. Neurobiol. 2015, 24, 1–7. [Google Scholar] [CrossRef]
- Bond, C.S.; Fox, A.H. Paraspeckles: Nuclear bodies built on long noncoding RNA. J. Cell Biol. 2009, 186, 637–644. [Google Scholar] [CrossRef]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, K.S.; Ho, D.; Newcombe, E.A. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Neumann, M. Reappraisal of TDP-43 pathology in FTLD-U subtypes. Acta Neuropathol. 2017, 134, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B. Regulated protein aggregation: Stress granules and neurodegeneration. Mol. Neurodegener. 2012, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shao, C.; Wu, Q.-J.; Chen, G.; Zhou, J.; Yang, B.; Li, H.; Gou, L.-T.; Zhang, Y.; Wang, Y. NEAT1 scaffolds RNA-binding proteins and the microprocessor to globally enhance pri-miRNA processing. Nat. Struct. Mol. Biol. 2017, 24, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, S.-B.; Wang, M.-R.; Yao, R.-W.; Wu, D.; Yang, L.; Chen, L.-L. Genome-wide screening of NEAT1 regulators reveals cross-regulation between paraspeckles and mitochondria. Nat. Cell Biol. 2018, 20, 1145–1158. [Google Scholar] [CrossRef]
- Pokrishevsky, E.; Grad, L.I.; Yousefi, M.; Wang, J.; Mackenzie, I.R.; Cashman, N.R. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e35050. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M. FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Urushitani, M.; Kurisu, J.; Tateno, M.; Hatakeyama, S.; Nakayama, K.I.; Kato, S.; Takahashi, R. CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J. Neurochem. 2004, 90, 231–244. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef]
- Zeineddine, R.; Farrawell, N.E.; Lambert-Smith, I.A.; Yerbury, J.J. Addition of exogenous SOD1 aggregates causes TDP-43 mislocalisation and aggregation. Cell Stress Chaperones 2017, 22, 893–902. [Google Scholar] [CrossRef]
- Rizzuti, M.; Filosa, G.; Melzi, V.; Calandriello, L.; Dioni, L.; Bollati, V.; Bresolin, N.; Comi, G.P.; Barabino, S.; Nizzardo, M. MicroRNA expression analysis identifies a subset of downregulated miRNAs in ALS motor neuron progenitors. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Castanotto, D.; Zhang, X.; Alluin, J.; Zhang, X.; Rüger, J.; Armstrong, B.; Rossi, J.; Riggs, A.; Stein, C. A stress-induced response complex (SIRC) shuttles miRNAs, siRNAs, and oligonucleotides to the nucleus. Proc. Natl. Acad. Sci. USA 2018, 115, E5756–E5765. [Google Scholar] [CrossRef]
- Kanata, E.; Thüne, K.; Xanthopoulos, K.; Ferrer, I.; Dafou, D.; Zerr, I.; Sklaviadis, T.; Llorens, F. MicroRNA alterations in the brain and body fluids of humans and animal prion disease models: Current status and perspectives. Front. Aging Neurosci. 2018, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Majer, A.; Medina, S.J.; Niu, Y.; Abrenica, B.; Manguiat, K.J.; Frost, K.L.; Philipson, C.S.; Sorensen, D.L.; Booth, S.A. Early mechanisms of pathobiology are revealed by transcriptional temporal dynamics in hippocampal CA1 neurons of prion infected mice. PLoS Pathog. 2012, 8, e1003002. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.J.; Cansizoglu, A.E.; Süel, K.E.; Louis, T.H.; Zhang, Z.; Chook, Y.M. Rules for nuclear localization sequence recognition by karyopherinβ2. Cell 2006, 126, 543–558. [Google Scholar] [CrossRef]
- Si, W.; Ye, S.; Ren, Z.; Liu, X.; Wu, Z.; Li, Y.; Zhou, J.; Zhang, S.; Li, Y.; Deng, R.; et al. miR-335 promotes stress granule formation to inhibit apoptosis by targeting ROCK2 in acute ischemic stroke. Int. J. Mol. Med. 2019, 43, 1452–1466. [Google Scholar] [CrossRef]
- De Luna, N.; Turon-Sans, J.; Cortes-Vicente, E.; Carrasco-Rozas, A.; Illán-Gala, I.; Dols-Icardo, O.; Clarimón, J.; Lleó, A.; Gallardo, E.; Illa, I.; et al. Downregulation of miR-335-5P in Amyotrophic Lateral Sclerosis Can Contribute to Neuronal Mitochondrial Dysfunction and Apoptosis. Sci. Rep. 2020, 10, 4308. [Google Scholar] [CrossRef]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; De Mieri, G.; Cotrufo, R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2012, 508, 35–40. [Google Scholar] [CrossRef]
- De Felice, B.; Manfellotto, F.; Fiorentino, G.; Annunziata, A.; Biffali, E.; Pannone, R.; Federico, A. Wide-ranging analysis of MicroRNA profiles in sporadic amyotrophic lateral sclerosis using next-generation sequencing. Front. Genet. 2018, 9, 310. [Google Scholar] [CrossRef] [PubMed]
- Joilin, G.; Leigh, P.N.; Newbury, S.F.; Hafezparast, M. An overview of MicroRNAs as biomarkers of ALS. Front. Neurol. 2019, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.; Marzocchi, C.; Battistini, S. MicroRNAs as biomarkers in amyotrophic lateral sclerosis. Cells 2018, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA sequencing of sporadic amyotrophic lateral sclerosis cerebrospinal fluid reveals differentially expressed miRNAs related to neural and glial activity. Front. Neurosci. 2018, 11, 731. [Google Scholar] [CrossRef] [PubMed]
- Klim, J.R.; Williams, L.A.; Limone, F.; San Juan, I.G.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Farrawell, N.E.; Lambert-Smith, I.A.; Warraich, S.T.; Blair, I.P.; Saunders, D.N.; Hatters, D.M.; Yerbury, J.J. Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep. 2015, 5, 13416. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Sun, H.; Stock, R.; Blackwood, M.; Brown, R.H.; Mueller, C. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci. Transl. Med. 2018, 10, eaau6414. [Google Scholar] [CrossRef]
- Pozzi, S.; Thammisetty, S.S.; Codron, P.; Rahimian, R.; Plourde, K.V.; Soucy, G.; Bareil, C.; Phaneuf, D.; Kriz, J.; Gravel, C. Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J. Clin. Investig. 2019, 129, 1581–1595. [Google Scholar] [CrossRef]
- Liu, H.-N.; Tjostheim, S.; DaSilva, K.; Taylor, D.; Zhao, B.; Rakhit, R.; Brown, M.; Chakrabartty, A.; McLaurin, J.; Robertson, J. Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J. Neurosci. 2012, 32, 8791–8799. [Google Scholar] [CrossRef]
- Im, H.-I.; Kenny, P.J. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012, 35, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Zhao, W.; Appel, S.H. The role of regulatory T lymphocytes in amyotrophic lateral sclerosis. JAMA Neurol. 2018, 75, 656–658. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, J.; Keon, M.; Brennan, S.; Saksena, N. Connecting RNA-Modifying Similarities of TDP-43, FUS, and SOD1 with MicroRNA Dysregulation Amidst A Renewed Network Perspective of Amyotrophic Lateral Sclerosis Proteinopathy. Int. J. Mol. Sci. 2020, 21, 3464. https://doi.org/10.3390/ijms21103464
Pham J, Keon M, Brennan S, Saksena N. Connecting RNA-Modifying Similarities of TDP-43, FUS, and SOD1 with MicroRNA Dysregulation Amidst A Renewed Network Perspective of Amyotrophic Lateral Sclerosis Proteinopathy. International Journal of Molecular Sciences. 2020; 21(10):3464. https://doi.org/10.3390/ijms21103464
Chicago/Turabian StylePham, Jade, Matt Keon, Samuel Brennan, and Nitin Saksena. 2020. "Connecting RNA-Modifying Similarities of TDP-43, FUS, and SOD1 with MicroRNA Dysregulation Amidst A Renewed Network Perspective of Amyotrophic Lateral Sclerosis Proteinopathy" International Journal of Molecular Sciences 21, no. 10: 3464. https://doi.org/10.3390/ijms21103464
APA StylePham, J., Keon, M., Brennan, S., & Saksena, N. (2020). Connecting RNA-Modifying Similarities of TDP-43, FUS, and SOD1 with MicroRNA Dysregulation Amidst A Renewed Network Perspective of Amyotrophic Lateral Sclerosis Proteinopathy. International Journal of Molecular Sciences, 21(10), 3464. https://doi.org/10.3390/ijms21103464